


Abstract
The CD20 cell surface antigen is expressed at high levels by over 90% of B-cell non-Hodgkin lymphomas (NHL) and is the target of the anti-CD20 monoclonal antibody rituximab. To provide more sensitive, tumor-specific PET imaging of NHL, we sought to develop PET agents targeting CD20. Methods: Two recombinant anti-CD20 rituximab fragments, a minibody (scFv-CH3 dimer; 80 kDa) and a modified scFv-Fc fragment (105 kDa), designed to clear rapidly, were generated. Both fragments were radiolabeled with 124I, and the minibody was additionally labeled with 64Cu (radiometal) after conjugation to 1,4,7,10-tetraazacyclododecane-N,N′,N″,N′′′-tetraacetic acid (DOTA). The radioiodinated fragments and the radiometal-labeled minibody were evaluated in mice as small-animal PET imaging agents for the in vivo imaging of human CD20–expressing lymphomas. Results: Rapid and specific localization to CD20-positive tumors was observed with the radioiodinated fragments. However, the tumor uptake levels and blood activities differed, resulting in different levels of contrast in the images. The better candidate was the minibody, with superior uptake (2-fold higher than that obtained with scFv-Fc) in CD20-positive tumors and low uptake in CD20-negative tumors. Ratios of CD20-positive tumors to CD20-negative tumors at 21 h were 7.0 ± 3.1 (mean ± SD) and 3.9 ± 0.7 for the minibody and scFv-Fc, respectively. The ratio achieved with the 64Cu-DOTA-minibody at 19 h was about 5-fold lower because of higher residual background activity in CD20-negative tumors. Conclusion: A radioiodinated minibody and a radioiodinated scFv-Fc fragment produced excellent, high-contrast images in vivo. These new immunoPET agents may prove useful for imaging CD20-positive lymphomas in preclinical models and in humans with NHL.
Non-Hodgkin lymphoma (NHL) is the fifth most common malignancy in the United States, accounting for approximately 4% of all cancers. Over 90% of NHL tumors are derived from B cells and express the B-cell differentiation antigen CD20. CD20 is an integral transmembrane protein expressed exclusively by cells in the B lineage, from normal pre–B-cell precursors through mature B cells, but not by terminally differentiated plasma cells (1). Although the precise functions of CD20 remain unknown, it is thought that CD20 plays a role in B-cell activation events, the regulation of cell growth, and transmembrane calcium flux (2). This 33- to 37-kDa nonglycosylated, hydrophobic, phosphorylated protein is not shed and undergoes very slow modulation or internalization in response to antibody binding, making it an ideal target for exogenous antibody therapies (2).
The chimeric (mouse–human) anti-CD20 antibody rituximab C2B8 (Rituxan; Genentech/Biogen-IDEC) has become a mainstay in the treatment of B-cell NHL, achieving high response rates for low-grade B-cell lymphomas (3) and improvements in survival for both low-grade and aggressive lymphomas when combined with chemotherapy (4,5). The mechanism of action of rituximab in vivo appears to be mediated by antibody-dependent cell-mediated cytotoxicity, complement-mediated cell lysis, and the induction of apoptosis in tumor cells (6). The efficacy of anti-CD20 antibodies against lymphoma has been further enhanced through combinations with therapeutic radionuclides, such as 131I (tositumomab [Bexxar]; Smithkline Beecham) and 90Y (ibritumomab [Zevalin]; IDEC Pharmaceuticals Corp.) (7,8).
The tracer 18F-FDG is currently standard in clinical PET for many malignancies, but its utility for lymphomas can be limited in cases of indolent disease with low metabolic activity. The rate of tumor detection by 18F-FDG PET in low-grade small lymphocytic and marginal-zone lymphomas can be as low as 50% (9,10). An imaging agent directed against a cell surface target could provide more sensitive, tumor-specific imaging. Recently, there has been a renewed interest in using antibodies for imaging malignancies (immunoPET). Antibodies radiolabeled with 124I, 64Cu, and 89Zr have been evaluated in patients with tumors (11). However, despite promising results, these PET tracers were all based on intact antibodies, and as a result, days were required for the activity levels to drop sufficiently to allow acceptable target-to-background ratios.
Redesigning antibodies without compromising their specificity by reducing their size results in rapid clearance from the blood, a desirable property for an imaging agent. We previously generated engineered antibody fragments, including diabodies (dimers of single-chain Fv [scFv]; 55 kDa) (12), minibodies (dimers of scFv-CH3; 80 kDa) (13), and scFv-Fc fragments (dimers of scFv-Fc; 105 kDa), with pharmacokinetics optimized for imaging in vivo (14). Small-animal PET imaging with 124I- or 64Cu-labeled fragments has demonstrated rapid, high-level tumor targeting to tumor-specific surface molecules, such as carcinoembryonic antigen (CEA) (colon carcinoma), HER2 (breast cancer), and prostate-stem cell antigen (PSCA) (prostate cancer), in tumor-bearing mice (14–18). The major advantage of using nonresidualizing labels (i.e.,124I) over residualizing labels (i.e., 64Cu) is the low background activity obtained in normal organs (e.g., liver and kidneys) with radioiodinated proteins. This difference is a result of differences in metabolism and clearance of activity after administration: metabolites (e.g., iodide or iodotyrosines) of radioiodinated proteins are quickly released from the cells and excreted via the kidneys, whereas metabolites of radiometal-chelated proteins are trapped in the cells, leading to increased retention of activity over time. When the anti-CEA T84.66 minibody was labeled with both labels and evaluated by small-animal PET imaging in tumor-bearing mice, the tumor-to-background ratios were 11 with the 124I-labeled minibody at 18 h and almost 2-fold lower (6.1) with the 64Cu-labeled minibody at 24 h (16,17).
Here we describe the generation of 2 anti-CD20 rituximab fragments, a minibody and a scFv-Fc fragment with mutations in 2 residues (H310A and H435Q) in the Fc region that have been shown to interfere with binding to the rodent neonatal Fc receptor (FcRn) (19). The fragments were radiolabeled with 124I and evaluated as small-animal PET imaging agents for the in vivo imaging of human CD20–expressing lymphomas. Rapid and specific localization to CD20-positive tumors was observed. The tumor uptake levels and blood activities in the 2 fragments differed, resulting in different levels of contrast in the images. The best candidate was the minibody because of its superior uptake in CD20-positive tumors and rapid blood clearance, producing high-contrast images. The minibody was also radiolabeled with 64Cu and evaluated in the same tumor model as the radioiodinated fragments. However, the minibody labeled with the stable or residualizing radiometal produced a high background signal in CD20-negative tumors and organs, resulting in relatively poor image contrast. These immunoPET agents may prove useful for imaging CD20-positive lymphomas in preclinical models and in humans with NHL.
MATERIALS AND METHODS
Design and Gene Assembly of Anti-CD20 Antibody Fragments
A polymerase chain reaction (PCR) with splice overlap extension (SOE-PCR) was used to create fully synthetic anti-CD20 variable (V) genes based on the V gene sequences of murine 2B8 (20) as described previously (21). Full-length 2B8 VL and VH genes were then assembled by SOE-PCR to produce scFv with an 18-residue linker (Whitlow 218 linker; GSTSGSGKPGSGEGSTKG) (22) in the VL-VH orientation. After SOE-PCR, which also included a signal peptide at the 5′-end (upstream) to enable secretion, the construct was cloned into the pCR-2.1-TOPO vector (Invitrogen Corp.) and confirmed by sequencing.
The human IgG4 Fc including the hinge (h-CH2-CH3) was amplified by PCR and fused to 2B8 scFv by another SOE-PCR to make chimeric scFv-Fc. With a Quick-Change Site-Directed Mutagenesis Kit (Stratagene), specific mutations in the CH2 and CH3 domains (H310A and H435Q; EU index numbering) were introduced to eliminate binding to FcRn, creating the so-called double-mutant construct (scFv-Fc DM), as described previously (23). The minibody was assembled by initial amplification of the human IgG4 hinge and the CH3 domain, which were joined by SOE-PCR and fused to 2B8 scFv by another SOE-PCR to produce a full-length minibody construct. For correction of the deficiency of IgG4 in forming inter–heavy-chain bonds (24), a single mutation (S228P) was introduced into the core hinge region (CPSC; residues 226–229) of scFv-Fc DM and the minibody.
Finally, the minibody and scFv-Fc DM fragments were both inserted into the mammalian expression vector pEE12 at the XbaI and EcoRI sites as described previously (14). This vector contains the human cytomegalovirus promoter and the glutamine synthetase gene for selection.
Expression, Selection, and Purification
A total of 2 × 106 NS0 mouse myeloma cells were transfected with 10 μg of linearized (cut with SalI) vector DNA by electroporation and selected in glutamine-deficient medium (JHR Biosciences) as described previously (23). After 2–3 wk, supernatants were screened for expression by an enzyme-linked immunosorbent assay and analyzed for size by Western blotting as described previously (23). The minibody and scFv-Fc DM were captured by goat antihuman Fc-specific antibodies and detected by alkaline phosphatase–conjugated goat antihuman Fc-specific antibodies (both from Jackson ImmunoResearch Laboratories). The highest-expressing clones were expanded and brought to terminal cultures. Supernatants were passed over a protein L–agarose column (Thermo Fisher Scientific Inc.), and bound proteins were eluted with 5, 10, and 5 column volumes of 30%, 50%, and 70% 0.2 M citrate buffer (pH 2.1) in phosphate-buffered saline (PBS), respectively, into 80% (v/v) 1 M Tris base (pH 8.2). The eluted fractions containing the desired protein were dialyzed against PBS with Slide-A-Lyzer Dialysis Cassettes (Thermo Fisher Scientific Inc.) and then concentrated to 0.5–1.0 mL with Vivaspin 20 (Vivascience AG). The final concentration of purified protein was determined by measuring the absorbance at 280 nm with the extinction coefficient 1.4 mg/mL.
Biochemical Characterization of Purified Anti-CD20 Antibody Fragments
Samples of purified proteins were analyzed by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) under nonreducing or reducing (1 mM dithiothreitol) conditions. Samples were also subjected to size exclusion high-pressure liquid chromatography with a Superdex-200 HR 10/30 column (GE Healthcare Bio-Sciences Corp.), a flow rate of 0.5 mL/min, and 50 mM Na3PO4−0.15 M NaCl (pH 7.0) buffer. Retention times were compared with standards of intact anti-CEA cT84.66 antibody, minibody, and diabody as described previously (14).
The binding of fragments to human CD20 was assessed by indirect immunofluorescence with the murine B-cell lymphoma line 38C13-huCD20 (described later). Cells (5 × 105) were incubated for 1 h on ice with 500 μL of minibody or scFv-Fc at 5 μg/mL in PBS plus 1% fetal bovine serum. Cells were washed and stained with phycoerythrin (PE)-conjugated goat antihuman (Fc-specific) antibodies (Jackson ImmunoResearch Laboratories) at a 1:100 dilution for detection. Rituximab was used as a positive control.
Radioiodination with 124I
Purified rituximab minibody and scFv-Fc DM (0.2 mg each) were radioiodinated with 17.1 MBq (0.461 mCi) and 15.6 MBq (0.422 mCi), respectively, of the positron-emitting isotope 124I (sodium iodide in 0.02 M NaOH; radionuclide purity, >99%) (IBA Molecular) by the IODO-GEN (Thermo Scientific) method as described previously (14,23). Instant thin-layer chromatography with a Monoclonal Antibody ITLC Strips Kit (Biodex Medical Systems) was used to determine labeling efficiency as described previously (23). Immunoreactivity was assayed by incubating radioiodinated protein with an excess amount of 38C13-huCD20 cells for 1 h, centrifuging the cells, and counting the activity remaining in the supernatant. Labeling efficiencies for scFv-Fc DM and the minibody, respectively, were 84.9% and 80.9%, and immunoreactivities were 32.8% and 37.7%.
Minibody Conjugation and Radiolabeling with 64Cu
Purified minibody was conjugated to 1,4,7,10-tetraazacyclododecane-N,N′,N″,N″′-tetraacetic acid (DOTA; Macrocyclics) by the water-soluble N-hydroxysuccinimide method as described previously (25). The extent of modification was evaluated by isoelectric focusing (26).
The positron-emitting isotope 64Cu (copper chloride in 0.1 M HCl; radionuclide purity, >99%) was provided by the Mallinckrodt Institute of Radiology (Washington University School of Medicine). The DOTA-conjugated anti-CD20 minibody (200 μg) was incubated with 13.0 MBq (0.35 mCi) of 64Cu in 0.1 M NH4 citrate (pH 5.5) for 50 min at 43°C. The labeling efficiency was 65.4%, as determined with the Monoclonal Antibody ITLC Strips Kit.
Syngeneic Human CD20-Expressing Murine Lymphoma Model
The murine B-cell lymphoma line 38C13 (27) was stably transduced with the human CD20 gene by use of lentiviral vector pRRLsin.hEF1α.CD20.Wpre, which was provided by Josie Golay (28). Human CD20–positive cells were selected with anti-CD20 MACS Beads (Miltenyi Biotec), and then flow cytometric sorting was used to obtain a population of cells with uniformly high CD20 expression (38C13-huCD20). These cells maintained a high level of human CD20 expression during in vitro culturing and passage in immunocompetent C3H mice. 38C13 and 38C13-huCD20 tumor cells were maintained in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 2 mM l-glutamine, penicillin at 100 U/mL, streptomycin at 100 μg/mL, and 50 μM 2-mercaptoethanol. All media and supplements were obtained from Invitrogen. Six- to 8-wk-old female C3Hf/Sed/Kam mice were bred and housed at the UCLA Defined Pathogen Colony according to a protocol approved by the UCLA Chancellor Animal Research Committee. Tumors were established by subcutaneous injection of 5 × 103 cells above each shoulder as described previously (14).
Human CD20 Surface Expression by 38C13-huCD20 Cells
CD20 levels on 38C13-huCD20 were quantitated, and 3 human B-cell lymphoma lines were used extensively for CD20 xenograft studies (Daudi, Raji, and Ramos) with flow cytometry. Cell lines were stained with PE-labeled anti–human CD20 antibody or κ-PE–labeled control mouse IgG1 antibody (BD Biosciences). Samples were run on a FACSCalibur flow cytometer (BD Biosciences), and data were analyzed with FCS Express V3 software (De Novo Software). For confirmation of human CD20 expression in vivo, mice were inoculated subcutaneously with 5 × 103 38C13-huCD20 tumors on day 0. Some mice were injected intravenously with 1 mg of rituximab 4 h before tumor collection. On day 14, tumors were removed, placed in formalin for 8 h, transferred to 70% ethanol, and embedded in paraffin. The sections were deparaffinized with xylene and rehydrated through graded ethanol. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide in methanol for 10 min. Heat-induced antigen retrieval (HIER) was performed for all sections with 0.01 M sodium citrate buffer (pH 6.0) in a vegetable steamer at 95°C for 25 min. Mouse monoclonal antibody to human CD20 (Dako Corp.) and rabbit polyclonal antibody to human IgG1 for the detection of rituximab were applied at dilutions of 1:1,000 and 1:300, respectively, for 45 min at room temperature. The signal was detected with horseradish peroxidase EnVision kits (Dako Corp.) and visualized with the diaminobenzidine reaction. The sections were counterstained with hematoxylin.
Small-Animal PET Imaging
Before imaging studies were conducted, thyroid uptake was blocked with Lugol solution (Sigma-Aldrich) and stomach uptake was blocked by gastric lavage with potassium perchlorate as previously described (16). Mice were serially imaged under anesthesia at about 4 and 20 h after intravenous injection of radioactivity, using a Focus 220 microPET scanner (Siemens Preclinical Solutions). The acquisition time was 10 min (one bed position), and images were reconstructed with a filtered backprojection algorithm (29,30). After the last scan, mice were euthanized, tumors and organs were excised and weighed, and activity was counted with a Wallac WIZARD automatic γ-counter (Perkin-Elmer Life and Analytic Sciences Inc.). The injected dose was corrected for labeling efficiency and immunoreactivity, and the percentage injected dose per gram of tissue (%ID/g), along with the SD, was calculated and decay corrected. Images were displayed with AMIDE software (31), and regions of interest (ROIs) were drawn as described previously (16) and quantitated by use of a cylinder with a known weight and radioactivity to determine a calibration factor (MBq per voxel). ROIs were also drawn to calculate ratios for CD20-positive tumors, CD20-negative tumors, and soft tissues. All significance testing was done at the 0.05 level with a one-tailed Student t test.
RESULTS
In Vitro Characterization of Engineered Anti-CD20 Antibody Fragments
Expression and SDS-PAGE
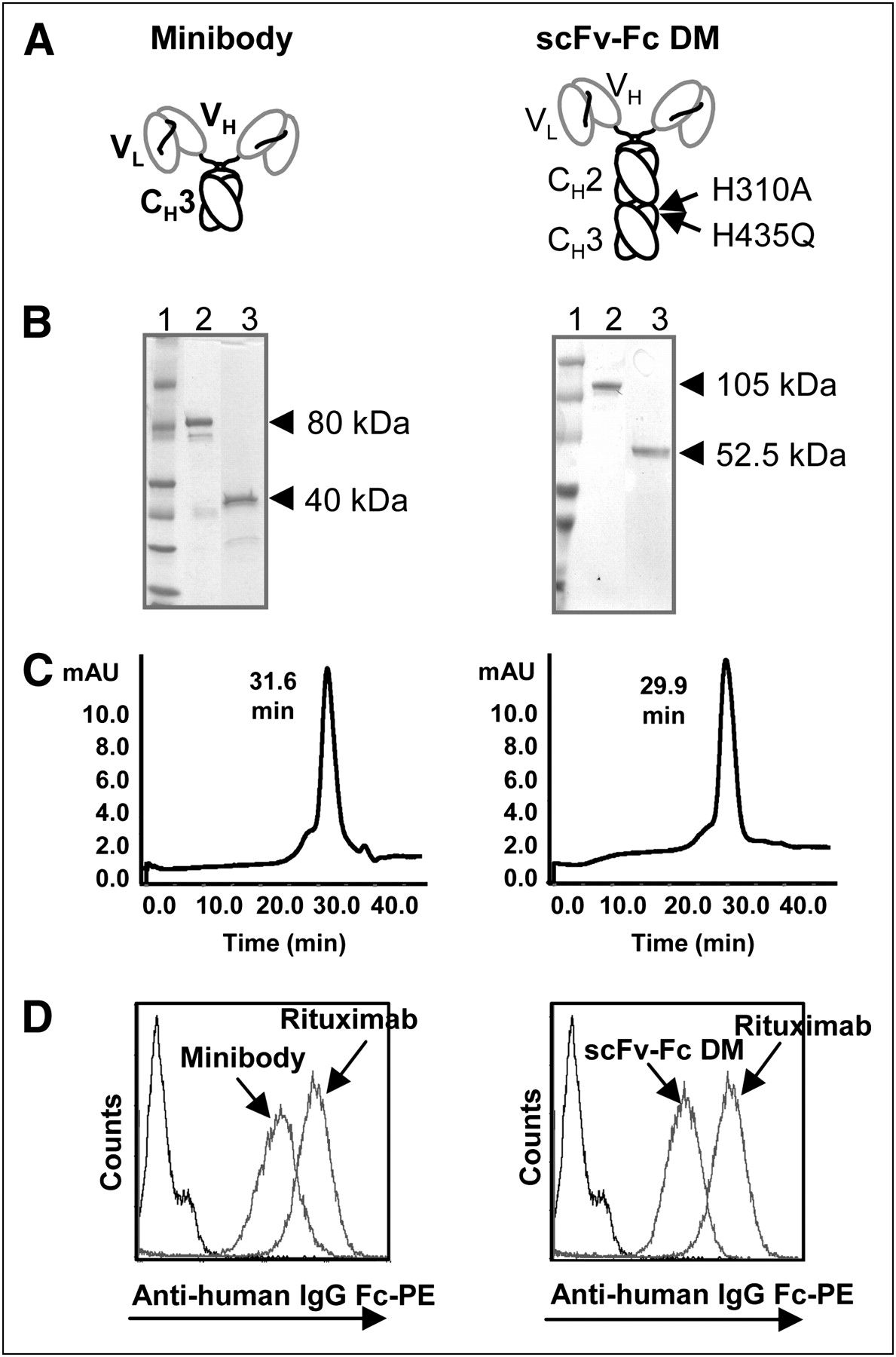
Engineered anti-CD20 antibody fragments (minibody and scFv-Fc DM) (Fig. 1A) were expressed at 4–10 μg/mL in terminal cultures of the mouse myeloma cell line NS0, as determined with an enzyme-linked immunosorbent assay. After affinity chromatography, purified proteins were analyzed by SDS-PAGE (Fig. 1B). Both the minibody and scFv-Fc DM migrated as covalent dimers, consistent with their predicted molecular masses of approximately 80 and 103 kDa, respectively, under nonreducing conditions and as monomers of approximately 40 and 51.5 kDa, respectively, under reducing conditions.

Characterization of purified rituximab minibody and scFv-Fc DM. (A) Schematic presentations of minibody and scFv-Fc DM. Both proteins assemble into covalent bound homodimers through cysteines in hinge region. Two mutations (H310A and H435Q) present in Fc region are indicated. CH = constant heavy; VH = variable heavy; VL = variable light. (B) Coomassie blue staining after SDS-PAGE of purified proteins under nonreducing (lane 2) and reducing (lane 3) conditions. Lane 1 contains molecular marker. (C) Size exclusion chromatography analysis of purified minibody (left) and scFv-Fc DM (right). mAU = milli-absorbance unit. (D) Flow cytometric analysis of CD20 binding by rituximab antibody fragments. Purified proteins were assayed for binding to 38C13-huCD20 cells. Bound protein was detected with PE-conjugated goat antihuman (Fc-specific) antibodies. Rituximab was used as positive control, and secondary antibody alone was used as negative control.
Size Exclusion Chromatography
Size exclusion chromatography verified that the anti-CD20 minibody and scFv-Fc DM were eluted at times corresponding to those of correctly folded dimers with the expected molecular masses (Fig. 1C). As expected, the minibody eluted slightly later than scFv-Fc DM—31.6 versus 29.9 min—consistent with their size difference. The purity of the proteins was determined from the size exclusion chromatography to be greater than 90%.
CD20-Binding Studies
Binding to the target antigen was demonstrated by indirect immunofluorescence cell surface staining of 38C13-huCD20 cells incubated with purified protein by flow cytometry (Fig. 1D). CD20 binding by the minibody and scFv-Fc DM shifted the cells to the right, indicating specific binding. The binding of rituximab to the cells is also shown.
Expression of Target CD20 Antigen by 38C13-huCD20 Lymphoma Cells In Vivo and In Vitro
To evaluate our anti-CD20 immunoPET imaging agents in vivo, we chose to use the 38C13-huCD20 B-cell lymphoma model. This fully syngeneic model allows for physiologic tumor growth in a natural immunocompetent host in which tumor cell adhesion, tumor metastasis, and adaptive immune effector mechanisms are fully functional. As shown in Figure 2A, cultured 38C13-huCD20 cells expressed high levels of human CD20, comparable to those found on human B-cell lymphoma lines commonly used as lymphoma xenografts (32). On subcutaneous injection of 38C13-huCD20 cells into C3H mice, tumors grew rapidly, and immunohistochemical staining of excised tumors demonstrated high levels of human CD20 expression (Fig. 2B). Penetration of intact rituximab into growing tumors was demonstrated by intravenous injection, tumor excision 4 h later, and staining with antibody to human IgG1; prominent staining was observed (data not shown). Together, these results indicated that human CD20 is expressed at levels similar to those seen with typical human B-cell lymphoma lines and that in vivo rituximab can recognize human CD20 expressed in this murine B-cell lymphoma model.

38C13-huCD20 cells express surface human CD20 at levels comparable to human B-cell lymphoma xenograft lines. (A) Flow cytometric analysis of cells stained with PE-labeled anti–human CD20 antibody, showing surface staining of 38C13-huCD20 (dark gray histogram), Daudi (dotted black line), Raji (thick light gray line), Ramos (thick black line), and wild-type, non–CD20-expressing 38C13 (thin black line) cell lines. Controls included unstained 38C13-huCD20 cells (light gray histogram) and PE-labeled isotype control antibody (thin light gray line). (B) Immunohistochemistry for human CD20 expression on 38C13-huCD20 cells in vivo. Day 14–established 38C13-huCD20 tumors grown in wild-type C3H mice were excised, stained with anti–human CD20 antibody, and detected with peroxidase.
In Vivo Characterization of 124I-Labeled Engineered Anti-CD20 Antibody Fragments
Small-Animal PET Imaging with 124I-Labeled Anti-CD20 scFv-Fc DM
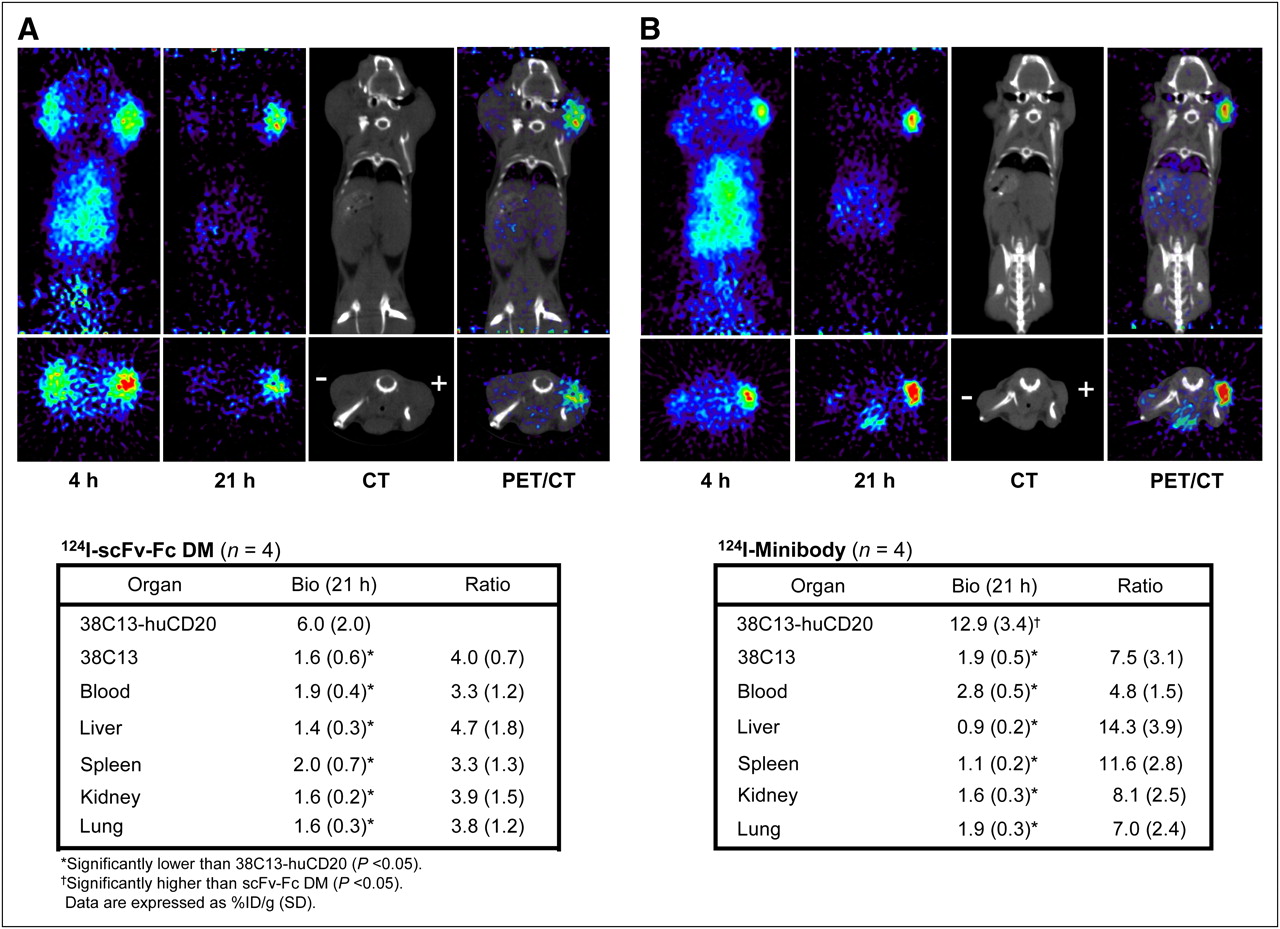
scFv-Fc DM was evaluated in C3H mice bearing 38C13-huCD20 and 38C13 tumors averaging (mean ± SD) 91 ± 92 mg (range, 28–225 mg) and 321 ± 199 mg (range, 93–565 mg) in weight, respectively. These mice were each injected with approximately 3.5 MBq of 124I-scFv-Fc DM (specific activity, 0.066 MBq/μg), and whole-body scans were obtained 4 and 21 h after administration. At 4 h, radioactivity was seen in both tumors and normal organs (Fig. 3A). By 21 h, the nonspecific activity in tumors and organs had cleared, and only CD20-positive tumors showed a strong signal. After the last scan, tumors and organs were harvested, and the %ID/g was calculated and is shown in Figure 3A. The average uptake in CD20-positive tumors was 6.0 ± 2.0 %ID/g, significantly higher than the uptake in control 38C13 (CD20-negative) tumors (1.6 ± 0.6 %ID/g) (P = 0.007) and blood (1.9 ± 0.4 %ID/g) (P = 0.011). Uptake in the liver, kidneys, spleen, and lungs was less than 2 %ID/g and was significantly lower than that in tumors (P < 0.05). In these mice, the ratio of CD20-positive tumors to CD20-negative tumors was 3.9 ± 0.7, and the ratio of CD20-positive tumors to blood ratio was 3.1 ± 1.2. The ratios of CD20-positive tumors to CD20-negative tumors at 4 and 21 h, determined from ROIs, were 1.2 ± 0.2 and 2.4 ± 0.8, respectively, and the ratios of CD-positive tumors to soft tissue were 4.3 ± 1.4 and 9.9 ± 4.6, respectively (Table 1).

Serial small-animal PET images of mice bearing 38C13-huCD20 (+) and wild-type 38C13 (−) tumors at 4 and 21 h after administration of radioiodinated anti-CD20 scFv-Fc DM (A) and minibody (B). Both coronal slices (top row) and transverse slices (bottom row) are shown. PET/CT scan overlay images at 21 h are shown. Biodistribution (Bio) of radioiodinated proteins at time of sacrifice (21 h) and tumor-to-organ ratios are shown in tables accompanying images. Uptake in tissues was measured by ex vivo weighing and counting in γ-counter.
Ratios Derived from PET Images
Small-Animal PET Imaging with124I-Labeled Anti-CD20 Minibody
Tumor targeting of the anti-CD20 minibody was evaluated in mice bearing 38C13-huCD20 and 38C13 tumors averaging 59 ± 32 mg (range, 34–106 mg) and 71 ± 54 mg (range, 10–134 mg), respectively. The mice were each injected with approximately 3.9 MBq of 124I-labeled minibody (specific activity, 0.069 MBq/μg), and whole-body scans were obtained 4 and 21 h after administration. As with scFv-Fc DM, activity in both tumors and normal organs was seen at 4 h, but at 21 h, activity persisted only in CD20-positive tumors (Fig. 3B). At 21 h, mice were sacrificed, tumors were excised, and the %ID/g was calculated (Fig. 2B). The average uptake in CD20-positive tumors was 12.9 ± 3.4 %ID/g, and that in control 38C13 xenografts was significantly lower, at 1.9 ± 0.5 %ID/g (P = 0.003). Also, the liver, kidneys, spleen, and lungs had activities (all <2 %ID/g) that were significantly lower than the activity in CD20-positive tumors (P < 0.05). The activity in the blood (2.8 ± 0.5 %ID/g) was also significantly lower than the activity in CD20-positive tumors (P = 0.004). The ratio of CD20-positive tumors to CD20-negative tumors was 7.0 ± 3.1, and the ratio of CD20-positive tumors to blood was 4.7 ± 1.4. ROIs revealed ratios of CD20-positive tumors to CD20-negative tumors of 2.2 ± 0.8 at 4 h and 4.0 ± 0.7 at 21 h. The ratio of CD20-positive tumors to soft tissue at 4 h was 4.6 ± 0.4; by 21 h, this ratio had increased by more than 4-fold to 17.0 ± 6.7 (Table 1). Thus, both anti-CD20 antibody fragments offered cell surface target-specific imaging in vivo, with the minibody achieving higher-contrast images.
In Vivo Characterization of 64Cu-DOTA-Minibody by Small-Animal PET Imaging
The minibody was also evaluated in the same tumor model after being labeled with a residualizing radiometal to evaluate CD20-positive tumor targeting and normal organ uptake. The minibody was conjugated to DOTA, radiolabeled with 64Cu, and evaluated in mice bearing 38C13-huCD20 tumors (n = 2) and control 38C13 tumors (n = 3) with average weights of 26 ± 20 and 79 ± 38 mg, respectively. The mice were each injected with approximately 3.7 MBq of 64Cu-DOTA-minibody (specific activity, 0.042 MBq/μg). Whole-body images were obtained at 4 and 19 h; after the last scan, mice were euthanized, organs were harvested, and activity was counted to calculate the %ID/g. The average uptake in 38C13-huCD20 tumors was 6.5 ± 3.8 %ID/g, and the uptake in control 38C13 xenografts was 4.7 ± 1.7 %ID/g; these values resulted in a ratio of 1.4, which was 5 times lower than the ratio obtained with the 124I-labeled minibody.
The images obtained at 4 and 19 h showed activity in both types of tumors (Fig. 4). The activities in the tumors, liver, and kidneys, calculated from the biodistributions and ROIs, are shown in Figure 4. ROIs were drawn only on the largest, most clearly imaged CD20-positive tumors in the mouse shown in Figure 4. Levels of uptake in CD20-positive tumors at 4 and 19 h, determined by ROI analyses, were 11.4 and 8.1 %ID/g, respectively; these values corresponded to the activity measured at the time of sacrifice (9.1 %ID/g at 19 h). The ROIs of the CD20-negative tumors (n = 3) resulted in %ID/g values of 4.9 ± 1.1 and 4.2 ± 1.1 at 4 and 19 h, respectively. The resulting ratios of CD20-positive tumors to CD20-negative tumors were 2.3 at 4 h and 1.9 at 19 h. Thus, compared with the results obtained with the 124I-labeled minibody, CD20-positive tumor–specific imaging was less favorable with the 64Cu-DOTA-minibody because of a high background signal in control tumors and normal organs.

Coronal serial small-animal PET images of mouse bearing 38C13-huCD20 (+) and wild-type 38C13 (−) tumors at 4 and 19 h after administration of 64Cu-labeled, DOTA-conjugated anti-CD20 minibody. K = kidney; L = liver. Estimated activities from drawn ROIs in 38C13, liver, spleen, kidney, lung, and soft tissue at 4 and 19 h are shown in table beside image along with activities present in tissues at time of sacrifice (19 h). Uptake in tissues was measured by ex vivo weighing and counting in γ-counter. ROIs for 38C13-huCD20 were drawn from one tumor. Bio = biodistribution.
DISCUSSION
In this work, we sought to develop novel immunoPET imaging agents targeting the CD20 cell surface antigen expressed by most human lymphomas. We described the generation of 2 antibody fragments of different sizes and compared their properties as imaging agents for CD20-expressing tumors. Although both fragments had low immunoreactivities (32%−38%) after radioiodination, they were both able to target the tumors well in vivo. Because the low immunoreactivities could have been attributable to modified tyrosines in the antigen-binding region, site-specific radiolabeling may be a better labeling approach for these fragments. Still, in this study, the minibody exhibited better (2-fold) tumor targeting than scFv-Fc DM at 21 h, producing excellent-quality, high-contrast images. This result was unexpected because iodinated anti-CEA scFv-Fc DM has been shown to have a slightly longer serum half-life (8.0 h) than the iodinated anti-CEA minibody (half-life, 5.3–7.0 h) (13,14,33). This discrepancy may be attributable to the Fc region of the fragments in the 2 systems, namely, IgG1 and IgG4 for anti-CEA and anti-CD20 fragments, respectively. We chose to use the IgG4 Fc region to minimize interactions with other immune cells because IgG1, as opposed to IgG4, permits complement activation and strong effector-mediated cell killing by antibody-mediated cytotoxicity (34). In humans, the mean serum half-life of IgG1 is 36.3 ± 9.2 d, and that of IgG4 is 15.6 ± 4.5 d (35). This difference in serum half-lives was also observed in mice, with residence times of 199.0 ± 9.8 h for IgG1 and 77.3 ± 10.1 h for IgG4 (36). When the IgG4 CH2-CH3 domains were exchanged with the IgG1 CH2-CH3 domains, the serum half-life increased to 281.5 ± 10.1 h; interestingly, this value was higher than that for either parental IgG.
Mutations in the CH2-CH3 domains have identified residues important for high-affinity binding to FcRn (19). The histidine residue at position 435 in the CH3 domain was shown to have a dramatic effect on this interaction for human IgG1, whereas H433 had little effect. However, for human IgG4, H433 has been implicated to be more important than H435 (37). scFv-Fc DM, used in this work, contains 2 mutations in the Fc region, H310A in CH2 and H435A in CH3. The shorter serum half-life and the presence of these mutations in the IgG4 Fc region may explain the lower activity observed in the blood at 21 h relative to that observed with the minibody. Because the minibody contains no mutation in the CH3 domain, it is possible that the minibody has some interaction with FcRn that enables it to stay in the blood slightly longer than the larger fragment, scFv-Fc DM.
The fact that the minibody has a longer residence time in the blood also explains the much higher tumor uptake observed with this fragment at 21 h. The radioactivity measured in tumors with the minibody was about 2-fold higher than that obtained with scFv-Fc DM. Such a difference has not been observed with the anti-CEA minibody and scFv-Fc DM, with reported tumor uptake levels of 12.6, 15.7, and 29.1 %ID/g for the iodinated minibody (12,13,33) and 18.6 %ID/g for iodinated scFv-Fc (38) at 24 h. The anti-CD20 minibody exhibited other superior properties over scFv-Fc DM, such as high ratios of CD20-positive tumors to CD20-negative tumors and soft tissue.
The minibody was also labeled with 64Cu and evaluated for imaging in tumor-bearing mice. However, even at 19 h, this agent showed inferior ratios of CD20-positive tumor uptake to CD20-negative tumor uptake and persistent high-level uptake in normal kidneys, liver, and bowel. Because the molecular mass of the minibody is above the threshold clearance of the kidneys, significant retention of activity in the liver relative to the tumor was not unexpected, as this had been observed previously in other tumor models (15,17). However, this phenomenon could also be attributable to an alteration in the biodistribution properties of the minibody by the DOTA chelator. Such an alteration was observed for an antineuroblastoma F(ab′)2 fragment when it was chelated to 2 analogous copper chelators with different charges (39). Thus, for further development of CD20 immunoPET imaging, we favor the 124I-labeled minibody format.
The human CD20-specific PET agents described here offer new opportunities to image B-cell lymphomas in both preclinical animal models and humans. Our specific imaging of human CD20–positive lymphomas in mice demonstrated that 124I-labeled rituximab minibody fragments can serve as a tool for noninvasive in vivo monitoring of lymphoma growth and metastasis. This imaging technique should allow the assessment of disease spread in mice without the high levels of background signal in the brain, heart, bowel, kidneys, and bladder seen with 18F-FDG PET of lymphomas in mice (40). In the immunocompetent model that we described here, anti-CD20 immunoPET imaging could be used to study responses to therapies depending on an intact adaptive immune system, such as tumor antigen vaccines (41). We recently transduced another murine B-cell lymphoma (A20, BALB/c) with the human CD20 gene for propagation in BALB/c human CD20 transgenic mice (unpublished observations). In clinics, anti-CD20 immunoPET imaging might be used to increase the sensitivity of PET in cases of low tumor burden or in cases of indolent lymphomas, in which the sensitivity of 18F-FDG PET can be as low as 50% (9,10).
CD20-specific imaging might also have use in imaging of the B-cell compartment in other experimental or pathologic states. With further refinements in sensitivity, anti-CD20 immunoPET imaging might be used for the B-cell compartment in human CD20 transgenic mice, either during immune reconstitution after B lymphocyte–depleting therapies (42) or in autoimmune diseases in which collections of autoimmune B cells contribute to pathogenesis (43). Such imaging might increase understanding of B-cell–mediated disease states.
In future studies, it will be important to determine whether pretreatment with rituximab interferes with subsequent anti-CD20 immunoPET imaging of lymphomas via blockade of rituximab-binding sites on CD20. Studies with human CD20 transgenic mice will help clarify this issue. As rituximab levels fall after the cessation of therapy, it is likely that anti-CD20 immunoPET reagents will be able to bind to an increasing number of available sites on the tumor cell surface. Indeed, CD20 immunoPET may be useful for determining whether tumor CD20 sites are saturated after a given dose of rituximab or at specific intervals after rituximab administration.
CONCLUSION
This work demonstrates rapid targeting of CD20-positive lymphomas in vivo by 2 engineered antibody fragments, a minibody and scFv-Fc DM. Both fragments produced high-contrast, target-specific PET images in tumor-bearing mice at 21 h. The better candidate was the minibody, which exhibited superior tumor uptake and, combined with rapid clearance, yielded higher ratios of CD20-positive tumors to CD20-negative tumors and tissues. When the minibody was radiolabeled with 64Cu, lower ratios were obtained because of residual activity in CD20-negative tumors and the liver. Thus, 124I is the preferred radiolabel because of lower background activity in normal tissues, enhancing the overall image.
Acknowledgments
This work was supported by NIH grants P50 CA107399, P50 CA086306, and CA119367 and by the Margaret Early Medical Research Trust. The production of 64Cu at Washington University School of Medicine was supported by NCI grant R24 CA86307. Support was also provided by grants CA33572 (City of Hope Comprehensive Cancer Center), CA16042 (Jonsson Comprehensive Cancer Center), and CI-26-05 (Damon Runyon Cancer Research Foundation). We thank Karl B. Bauer for excellent technical support. We are especially grateful to Dr. David Stout, Waldemar Ladno, and Judy Edwards at the Crump Institute for Molecular Imaging at UCLA for their assistance with the small-animal PET/CT scans. Thanks are also extended to the Jonsson Comprehensive Cancer Center at UCLA and the Center for AIDS Research Flow Cytometry Core Facility at UCLA.
Footnotes
-
Guest Editor: Kenneth T. Cheng, National Institutes of Health
-
COPYRIGHT © 2009 by the Society of Nuclear Medicine, Inc.
References
- Received for publication November 20, 2008.
- Accepted for publication May 28, 2009.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Noninvasive Evaluation of CD20 Expression Using 64Cu-Labeled F(ab')2 Fragments of Obinutuzumab in Lymphoma
- A Novel Engineered Small Protein for Positron Emission Tomography Imaging of Human Programmed Death Ligand-1: Validation in Mouse Models and Human Cancer Tissues
- The Immunoimaging Toolbox
- ImmunoPET of Malignant and Normal B Cells with 89Zr- and 124I-Labeled Obinutuzumab Antibody Fragments Reveals Differential CD20 Internalization In Vivo
- Theranostic Radiolabeled Anti-CD20 sdAb for Targeted Radionuclide Therapy of Non-Hodgkin Lymphoma
- Targeted PET imaging strategy to differentiate malignant from inflamed lymph nodes in diffuse large B-cell lymphoma
- Cerenkov Luminescence Imaging as a Modality to Evaluate Antibody-Based PET Radiotracers
- First-in-Human Imaging with 89Zr-Df-IAB2M Anti-PSMA Minibody in Patients with Metastatic Prostate Cancer: Pharmacokinetics, Biodistribution, Dosimetry, and Lesion Uptake
- Evaluation of Castration-Resistant Prostate Cancer with Androgen Receptor-Axis Imaging
- Use of Fc-Engineered Antibodies as Clearing Agents to Increase Contrast During PET
- Engineered antibody fragments for immuno-PET imaging of endogenous CD8+ T cells in vivo
- Eradication of non-Hodgkin lymphoma through the induction of tumor-specific T-cell immunity by CD20-Flex BiFP
- A Novel Engineered Anti-CD20 Tracer Enables Early Time PET Imaging in a Humanized Transgenic Mouse Model of B-cell Non-Hodgkins Lymphoma
- Advances in Immuno-Positron Emission Tomography: Antibodies for Molecular Imaging in Oncology
- Expression of Antibody Fragments with a Controlled N-Glycosylation Pattern and Induction of Endoplasmic Reticulum-Derived Vesicles in Seeds of Arabidopsis
- Targeted delivery of interferon-alpha via fusion to anti-CD20 results in potent antitumor activity against B-cell lymphoma