


Abstract
Pretargeted radioimmunotherapy (PRIT) with the β-emitting radionuclide 177Lu is an attractive approach to treat carcinoembryonic antigen (CEA)–expressing tumors. The therapeutic efficacy of PRIT might be improved using α-emitting radionuclides such as 213Bi. Herein, we report and compare the tumor-targeting properties and therapeutic efficacy of 213Bi and 177Lu for PRIT of CEA-expressing xenografts, using the bispecific monoclonal antibody TF2 (anti-CEA × anti–histamine-succinyl-glycine [HSG]) and the di-HSG-DOTA peptide IMP288. Methods: The in vitro binding characteristics of 213Bi-IMP288 were compared with those of 177Lu-IMP288. Tumor targeting of 213Bi-IMP288 and 177Lu-IMP288 was studied in mice bearing subcutaneous LS174T tumors that were pretargeted with TF2. Finally, the effect of 213Bi-IMP288 (6, 12, or 17 MBq) and 177Lu-IMP288 (60 MBq) on tumor growth and survival was assessed. Toxicity was determined by monitoring body weight, analyzing blood samples for hematologic and renal toxicity (hemoglobin, leukocytes, platelets, creatinine), and immunohistochemical analysis of the kidneys. Results: The in vitro binding characteristics of 213Bi-IMP288 (dissociation constant, 0.45 ± 0.20 nM) to TF2-pretargeted LS174T cells were similar to those of 177Lu-IMP288 (dissociation constant, 0.53 ± 0.12 nM). In vivo accumulation of 213Bi-IMP288 in LS174T tumors was observed as early as 15 min after injection (9.2 ± 2.0 percentage injected dose [%ID]/g). 213Bi-IMP288 cleared rapidly from the circulation; at 30 min after injection, the blood levels were 0.44 ± 0.28 %ID/g. Uptake in normal tissues was low, except for the kidneys, where uptake was 1.8 ± 1.1 %ID/g at 30 min after injection. The biodistribution of 213Bi-IMP288 was comparable to that of 177Lu-IMP288. Mice treated with a single dose of 213Bi-IMP288 or 177Lu-IMP288 showed significant inhibition of tumor growth. Median survival for the groups treated with phosphate-buffered saline, 6 MBq 213Bi-IMP288, 12 MBq 213Bi-IMP288, and 60 MBq 177Lu-IMP288 was 22, 31, 45, and 42 d, respectively. Mice receiving 17 MBq 213Bi-IMP288 showed significant weight loss, resulting in a median survival of only 24 d. No changes in hemoglobin, platelets, or leukocytes were observed in the treatment groups. However, immunohistochemical analysis of the kidneys of mice treated with 17 or 12 MBq 213Bi-IMP288 showed signs of tubular damage, indicating nephrotoxicity. Conclusion: To our knowledge, this study shows for the first time that PRIT with TF2 and 213Bi-IMP288 is feasible and at least as effective as 177Lu-IMP288. However, at higher doses, kidney toxicity was observed. Future studies are warranted to determine the optimal dosing schedule to improve therapeutic efficacy while reducing renal toxicity.
Targeted radiotherapy with α-emitting radionuclides is a promising strategy for the treatment of cancer. Currently, β-emitting radionuclides such as 177Lu, 90Y, and 131I are most commonly used. However, α-particles present significantly higher energies than β-particles (4–9 MeV vs. 0.1–2.2 MeV), which combined with short path lengths results in high linear energy transfer and a greater probability of generating DNA double-strand breaks on interaction with cell nuclei. This occurs independently of tissue oxygenation, dose rate, and cellular resistance to photon irradiation and chemotherapy (1–5). Therefore, α-particles are highly cytotoxic and promising candidates for targeted radiotherapy.
Colorectal cancer is the third most common type of cancer in both men and women in the Western world (6). Radioimmunotherapy is an attractive new treatment option. Unfortunately, for colorectal cancer, radioimmunotherapy has not been very effective because of the radioresistance of these tumors and the limited radionuclide activity that can be administered safely. Myelotoxicity as a result of continuous radiation exposure of red bone marrow by long-circulating antibodies is the dose-limiting factor (7,8). Pretargeted radioimmunotherapy (PRIT) has been developed to reduce the radiation dose to normal tissues while delivering lethal doses to the tumor. In this approach, a bispecific monoclonal antibody (bsmAb) is administered intravenously, and when it has accumulated in the tumor and cleared from the circulation, a radiolabeled hapten is administered that clears rapidly from the blood but is trapped in the tumor by the anti–hapten-binding arm of the bsmAb (9).
We previously showed that PRIT with the anti-CEA × anti–histamine-succinyl-glycine (HSG) bsmAb TF2, and 177Lu-labeled di-HSG-DOTA peptide IMP288, inhibited the growth of carcinoembryonic antigen (CEA)–positive human colonic tumor xenografts in mice, with limited toxicity (10,11). Moreover, a phase I study in patients with metastatic colorectal cancer showed that this treatment is feasible and safe (12). However, in both the preclinical and the clinical studies, progressive tumor growth was observed after the 177Lu-IMP288 treatment.
To our knowledge, no studies have yet been performed using α-emitting particles for PRIT with TF2 and IMP288. Because of the high cytotoxicity of α-particles, they are potentially more therapeutically effective than 177Lu. Moreover, the extremely rapid tumor-targeting kinetics of IMP288 matches the short half-life of the α-emitter 213Bi (46 min). Therefore, the aim of our study was to compare the tumor-targeting properties and therapeutic efficacy of 213Bi- and 177Lu-IMP288 for PRIT with TF2 in mice bearing CEA-expressing colorectal cancer xenografts.
MATERIALS AND METHODS
Cell Culture and Pretargeting Reagents
The CEA-positive human colorectal cancer cell line LS174T was cultured in RPMI1640 (GIBCO, BRL Life Sciences Technologies), supplemented with 2 mM glutamine (GIBCO) and 10% fetal calf serum (Sigma-Aldrich Chemie BV) at 37°C in a humidified atmosphere with 5% CO2.
The bsmAb TF2 (anti-CEA × anti-HSG), the control bsmAb TF12 (anti-TROP-2 × anti-HSG), and the peptide IMP288 were provided by Immunomedics, Inc., and IBC Pharmaceuticals, Inc. The preparation and binding properties of TF2 and TF12 have been described previously (13–15). IMP288 is a DOTA-conjugated d-Tyr-d-Lys-d-Glu-d-Lys-NH2 tetrapeptide in which both lysine residues are derivatized with an HSG moiety (molecular weight, 1,456 Da). The synthesis and purification were described by McBride et al. (16).
Radiolabeling and Stability
Radiolabeling with 213Bi
IMP288 (0.7–7.0 nmol) was added to 150 μL of 2 M Tris and 50 μL of 20% ascorbic acid. 213Bi was eluted from a 225Ac/213Bi generator (Institute for Transuranium Elements) using 600 μL of 0.1 M HCl and 0.1 M NaI. The eluate was immediately added to the peptide (final pH, 9.0) and incubated for 10 min at 95°C. Labeling efficiency was determined by instant thin-layer chromatography on silica gel chromatography strips (Agilent Technologies) using 0.1 M ammonium acetate and 0.1 M ethylenediaminetetraacetic acid as the mobile phase and by reverse-phase high-performance liquid chromatography on an Agilent 1200 system (Agilent Technologies). A monolithic C18 column (Onyx, 4.6 × 100 mm; Phenomenex) was used at a flow rate of 1 mL/min with the following buffer system: buffer A, 0.1% v/v trifluoroacetic acid in water; buffer B, 0.1% v/v trifluoroacetic acid in acetonitrile; and a gradient of 97% buffer A to 0% buffer A at 5–15 min. The radioactivity of the eluate was monitored using an in-line NaI radiodetector (Raytest GmbH). Elution profiles were analyzed using Gina Star software (version 2.18; Raytest GmbH). For therapy studies, 3.8 μM diethylenetriamine tetraacetic acid was added to a final concentration of 0.14 μM to complex nonincorporated 213Bi. Subsequently, the labeling mixture was trapped on an Oasis HLB Cartridge (30 mg; Waters) and eluted with 500 μL of ethanol. The ethanol was evaporated until less than 50 μL remained, and 213Bi-IMP288 was diluted in phosphate-buffered saline (PBS) containing 0.5% bovine serum albumin. Radiochemical purity exceeded 95% in all experiments.
Radiolabeling with 177Lu
No-carrier-added 177Lu (specific activity, >3,000 GBq/mg) was obtained from Isotope Technologies Garching GmbH). IMP288 was radiolabeled in 0.5 M 2-(N-morpholino)ethanesulfonic acid buffer, pH 5.5, for 15 min at 95°C. After the labeling reaction was complete, 50 mM ethylenediaminetetraacetic acid was added to a final concentration of 5 mM to complex nonincorporated 177Lu. Labeling efficiency was determined as described for the 213Bi conjugates and exceeded 95% in all experiments.
Stability
Stability of 213Bi- and 177Lu-IMP288 was tested in PBS at 37°C. At 30, 60, 90, and 120 min after the start of incubation, radiochemical purity was determined with instant thin-layer chromatography as described above.
In Vitro Studies
Binding and Internalization
Binding and internalization were determined as described previously (17). In short, 200,000–300,000 LS174T cells per well were cultured to confluency in 6-well plates (2–3 d). Subsequently, the cells were preincubated for 1 h in 1 mL of binding buffer (RPMI1640, 0.5% bovine serum albumin) containing 64 nM TF2 or TF12 at 37°C in a humidified atmosphere with 5% CO2. After preincubation, the cells were incubated for 1, 2, and 3 h with 22 nM 177Lu-IMP288 or 213Bi-IMP288. Acidic wash buffer (0.1 M HAc, 0.15 M NaCl, pH 2.8) was used to remove the membrane-bound fraction of the cell-associated 177Lu-IMP288 or 213Bi-IMP288, and the cells were harvested using 0.1 M NaOH. Activity was measured in a shielded well-type γ-counter by measuring the γ-emission of 213Bi (440 keV; Perkin-Elmer). Specific binding and internalization were calculated by subtracting the nonspecific binding and internalization from the total binding and internalization.
Scatchard Analysis
The affinity of 177Lu-IMP288 and 213Bi-IMP288 was determined with Scatchard analysis. Cells were preincubated with TF2 or TF12 as described above. Subsequently, the cells were incubated with increasing concentrations of radiolabeled IMP288 (0.03–100 nM). After 45 min of incubation on ice, the unbound radiolabeled IMP288 was removed and the cell-associated activity was measured in a γ-counter. Dissociation constant and maximum binding were determined using a 1-site specific binding equation in GraphPad Prism.
Animal Studies
Animal studies were performed using female BALB/c nude mice (Janvier) in accordance with the principles established by the revised Dutch Act on Animal Experimentation (1997) and were approved by the institutional Animal Welfare Committee of the Radboud University Nijmegen. At 6–8 wk old, the mice were inoculated subcutaneously with 1 × 106 LS174T cells. The experiments started when tumor size reached approximately 0.1 cm3 (8–10 d after inoculation). Tumor size was determined by caliper measurements in 3 dimensions (radius x, y, and z), using the following formula: 4/3 ⋅ π ⋅ x ⋅ y ⋅ z.
Biodistribution
LS174T tumors were pretargeted by intravenous injection of 5 nmol of TF2 or the nonspecific bsmAb TF12. After 20 h, the mice received an intravenous injection of different doses of 213Bi-IMP288 (group 1, 0.14 nmol and 0.98 MBq; group 2, 0.28 nmol and 1.68 MBq; group 3, 0.56 nmol and 3.12 MBq). One hour after injection, the mice were euthanized and the biodistribution of the radiolabeled peptide was determined ex vivo. Tumor, blood, muscle, lung, spleen, pancreas, intestine, kidney, and liver were dissected, collected, and weighed, and the activity for each was measured in a γ-counter. To calculate the uptake of radiolabeled peptide in each sample as a fraction of the injected dose, aliquots of the injected dose were counted simultaneously.
Subsequently, the pharmacokinetics of 213Bi-IMP288 were determined. Mice with TF2-pretargeted tumors received an intravenous injection of 2.26 MBq 213Bi-IMP288 (0.28 nmol). At 15, 30, 45, and 60 min after injection of 213Bi-IMP288, the mice were euthanized and the biodistribution of the radiolabeled peptide was determined ex vivo.
Finally, the biodistribution of 0.28 nmol of 213Bi-IMP288 was compared with that of 0.28 nmol of 177Lu-IMP288. Mice with TF2-pretargeted tumors were injected intravenously with 13 MBq 213Bi-IMP288 or 10 MBq 177Lu-IMP288. At 60 min after injection of the radiolabeled IMP288, the mice were euthanized and the biodistribution of the radiolabeled peptide was determined ex vivo. The tumors of the 177Lu-IMP288 group were frozen at −80°C for ex vivo autoradiography (tumor weight, 88 ± 72 mg).
Autoradiography
Frozen tumor sections (5 μm) from the mice injected with TF2 and 10 MBq 177Lu-IMP288 (0.28 nmol) were exposed to a Fujifilm BAS cassette 2025 overnight (Fuji Photo Film). Phospholuminescence plates were scanned using a Typhoon FLA 7000 laser scanner (GE Healthcare Life Sciences) at a pixel size of 25 × 25 μm. Images were analyzed with Aida Image Analyzer software (Raytest).
PRIT Study
Six groups of 8 mice each bearing LS174T tumors were pretargeted with 5 nmol of TF2. Twenty hours later, the mice received a single intravenous injection of radiolabeled IMP288. The first 3 groups received 0.28 nmol of 6 MBq 213Bi-IMP288, 12 MBq 213Bi-IMP288, or 17 MBq 213Bi-IMP288. To investigate whether a lower peptide dose would improve the therapeutic efficacy, the fourth group received 0.14 nmol of 12 MBq 213Bi-IMP288. The fifth group received 0.28 nmol of 60 MBq 177Lu-IMP28, which is the maximum tolerable dose as determined previously (11). Mice in the control group received vehicle only (PBS, 0.5% bovine serum albumin).
The general health of the animals was measured by monitoring body weight 3 times per week. Tumor size was measured 3 times per week by measuring tumor diameter in 3 dimensions with a caliper. Mice were removed from the experiment if they had weight loss of more than 20% compared with baseline or more than 15% within 2 d, a tumor size of 2 cm3 or more, or ulcerative tumor growth. Removed animals were inspected for any macroscopic evidence of abnormalities, and the kidneys were harvested, stored in 4% formalin, and embedded in paraffin for further analysis.
Four separate groups of non–tumor-bearing mice (4 per group) received 0.28 nmol of 6 MBq 213Bi-IMP288, 12 MBq 213Bi-IMP288, 17 MBq 213Bi-IMP288, or 60 MBq 177Lu-IMP88. Blood samples were collected from these mice before treatment and at weeks 2, 4, 6, and 8 after treatment to measure hematologic toxicity (hemoglobin, leukocytes, thrombocytes). Renal toxicity was analyzed by measuring plasma creatinine levels before therapy and at weeks 1, 3, and 5 after treatment. Furthermore, kidney sections were stained with hematoxylin–eosin and periodic acid–Schiff reagent. Renal damage was microscopically graded from 0 (no damage) to 4 (severe damage) by an experienced pathologist, according to Table 1 (18).
Criteria for Assessment of Renal Damage
Statistical Analyses
Statistical analyses were performed using PASW Statistics, version 18.0, and GraphPad Prism, version 5.03, for Windows (Microsoft). Continuous data were described as mean and SD, unless stated otherwise. Comparisons were performed using nonparametric Kruskal–Wallis and Mann–Whitney U testing. Survival was described as median, and survival curves were compared with the log-rank test. A P value below 0.05 was considered statistically significant.
RESULTS
Radiolabeling and Stability
IMP288 was labeled with more than 95% efficiency at maximum specific activities of 320 and 214 MBq/nmol for 213Bi and 177Lu, respectively. After 2 h in PBS at 37°C, no significant loss of the radionuclide was observed. Radiochemical purity was 99.96% and 99.75% for 213Bi-IMP288 and 177Lu-IMP288, respectively.
In Vitro
177Lu-IMP288 and 213Bi-IMP288 showed similar binding to LS174T cells pretreated with TF2, and only a small fraction of the cell-associated activity (<20%) was internalized (Table 2). The internalization rate did not differ significantly between the two tracers. Furthermore, both tracers showed high affinity for binding to TF2 on LS174T cells (dissociation constant, 0.45 ± 0.20 nM and 0.53 ± 0.13 nM for 213Bi- and 177Lu-IMP288, respectively) (Fig. 1).
Comparison of In Vitro Characteristics of 213Bi-IMP288 and 177Lu-IMP288
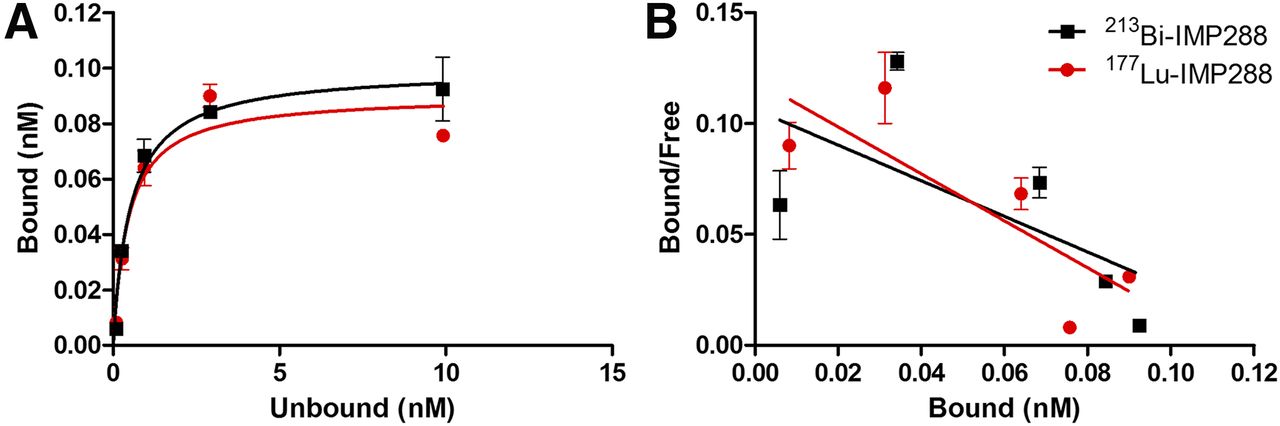
Binding plot (A) and Scatchard plot (B) of 177Lu-IMP288 and 213Bi-IMP288 to TF2-pretargeted LS174T cells.
Biodistribution
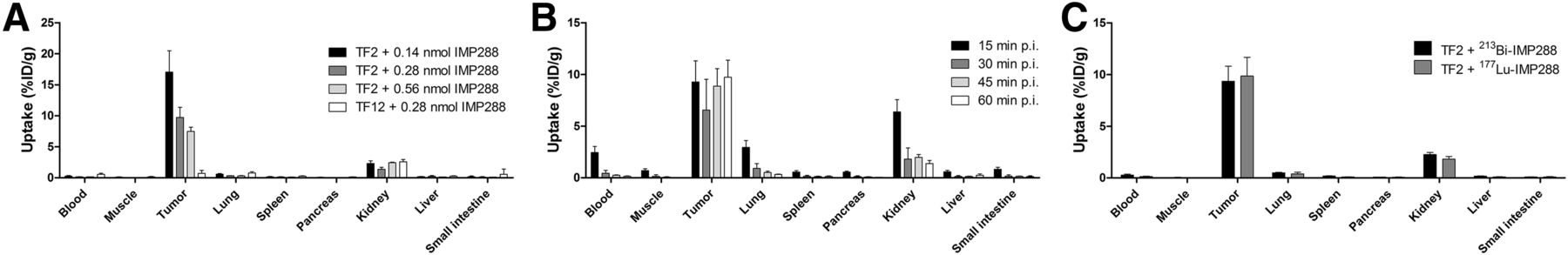
Tumor uptake of 213Bi-IMP288 was the highest in mice receiving 0.14 nmol of 213Bi-IMP288 (17.1 ± 3.4 percentage injected dose [%ID]/g), followed by the groups receiving 0.28 nmol (9.7 ± 1.6 %ID/g) and 0.56 nmol (7.5 ± 0.7 %ID/g, P = 0.005; Fig. 2A). Uptake in LS174T tumors pretargeted with the control bsmAb TF12 was significantly lower (0.7 ± 0.5 %ID/g, P = 0.008). Uptake of 213Bi-IMP288 in healthy tissue was low, except for the kidneys (1.4 ± 0.3 %ID/g in the 0.28-nmol group). Subsequently, the biodistribution of 0.28 nmol 213Bi-IMP288 was determined at several time points after injection. Tumor uptake remained stable between 15 min and 60 min after injection (9.2 ± 2.0 %ID/g, 6.6 ± 3.0 %ID/g, 8.9 ± 1.7 %ID/g, and 9.7 ± 1.6 %ID/g, at 15, 30, 45, and 60 min after injection, respectively) (Fig. 2B). The radiolabeled peptide cleared rapidly from the circulation; 30 min after injection, the 213Bi-IMP288 concentration in the blood was 0.44 ± 0.28 %ID/g. Kidney uptake at 30 min after injection was 1.8 ± 1.1 %ID/g. Overall, the uptake of 213Bi-IMP288 in tumor and normal tissue was similar to that of 177Lu-IMP288 (Fig. 2C).

Biodistribution of radiolabeled IMP288. (A) 213Bi-IMP288 biodistribution in TF2- or TF12-pretargeted LS174T tumor–bearing mice at 60 min after injection. (B) Biodistribution of 213Bi-IMP288 (0.28 nmol) in mice bearing TF2-pretargeted LS174T xenografts at 15, 30, 45, and 60 min after injection. (C) Biodistribution of 213Bi-IMP288 (0.28 nmol) or 177Lu-IMP288 (0.28 nmol) in mice bearing TF2-pretargeted LS174T xenografts at 60 min after injection. p.i. = after injection.
Autoradiography
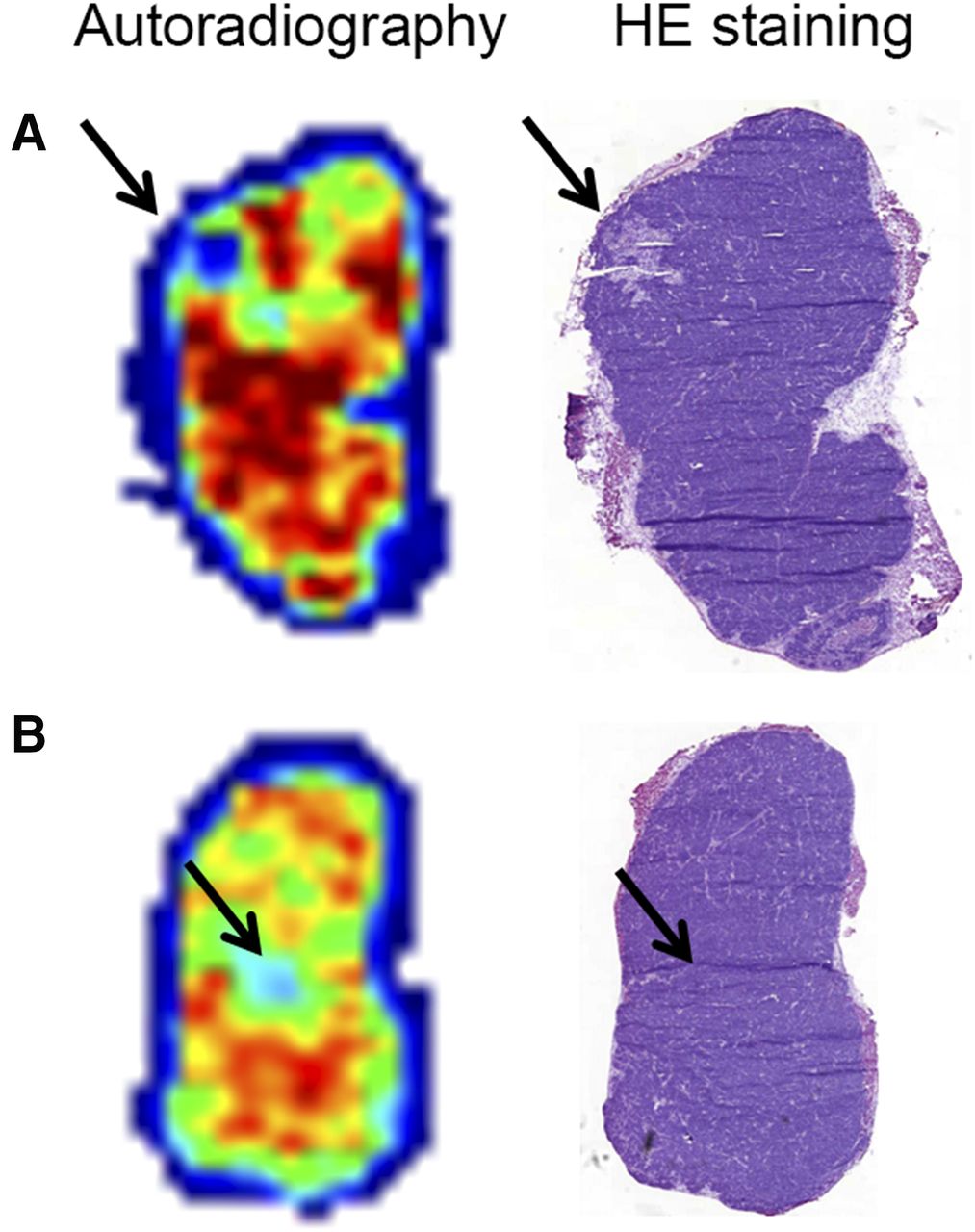
Autoradiographic analysis of tumor sections showed that 177Lu-MP288 was distributed heterogeneously within the tumor. Hematoxylin–eosin staining of the same tumor sections showed low uptake of radiolabeled IMP288 both in areas of necrosis and in areas of viable tumor (Fig. 3).

Autoradiography and hematoxylin–eosin (HE) staining of LS174T tumor xenografts of mice injected with TF2 and 177Lu-IMP288. Autoradiography reveals heterogeneous uptake of 177Lu-IMP288. Areas with low uptake were found in both necrotic (A) and viable (B) tumor tissue (arrow).
PRIT Study
Body Weight of Non–Tumor-Bearing Animals After Radionuclide-IMP288 Treatment
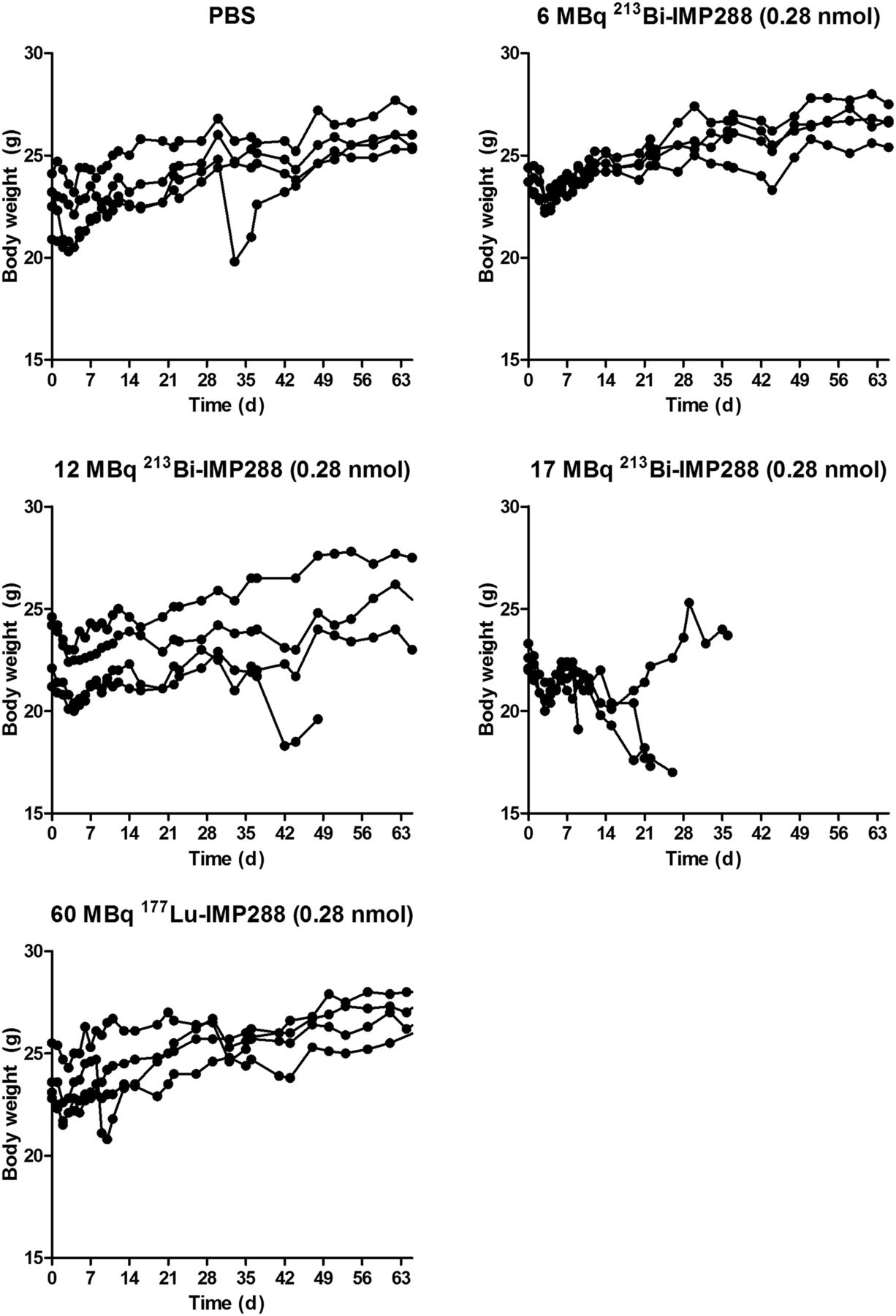
Mice treated with 6 MBq 213Bi-IMP288 or 60 MBq 177Lu-IMP288 showed almost no effect on body weight compared with control animals (Fig. 4). Mice administered 12 MBq 213Bi-IMP288 showed an initial drop in body weight, which slowly recovered. Treatment with 17 MBq 213Bi-IMP288 significantly reduced body weight during the first weeks, resulting in removal of all animals from the experiment within 10–39 d after treatment.

Body weight of non–tumor-bearing mice treated with TF2 and different activity doses of 213Bi-IMP288 or 177Lu-IMP288.
Hematologic and Renal Toxicity of Non–Tumor-Bearing Animals After Radionuclide-IMP288 Treatment
None of the treatment groups showed significant changes in hemoglobin, leukocytes, thrombocytes, or creatinine levels (Supplemental Fig. 1; supplemental materials are available at http://jnm.snmjournals.org). Immunohistochemical analysis of the kidneys treated with PBS, 60 MBq 177Lu-IMP288, or 6 MBq 213Bi-IMP288 showed normal morphology or only mild abnormalities (grade 0–1). Kidney damage in mice treated with 12 or 17 MBq 213Bi-IMP288 varied from grades 1 to 3, as indicated by the observed tubular dilation, flat tubule epithelium, and inflammatory infiltrate in the glomeruli (Fig. 5).

Representative kidney slices stained with periodic acid–Schiff from mice treated with 213Bi-IMP288 or 177Lu-IMP288. Arrows indicate tubular damage in mice treated with 12 or 17 MBq 213Bi-IMP288.
Tumor Growth and Survival of Tumor-Bearing Mice After Radionuclide-IMP288 Treatment
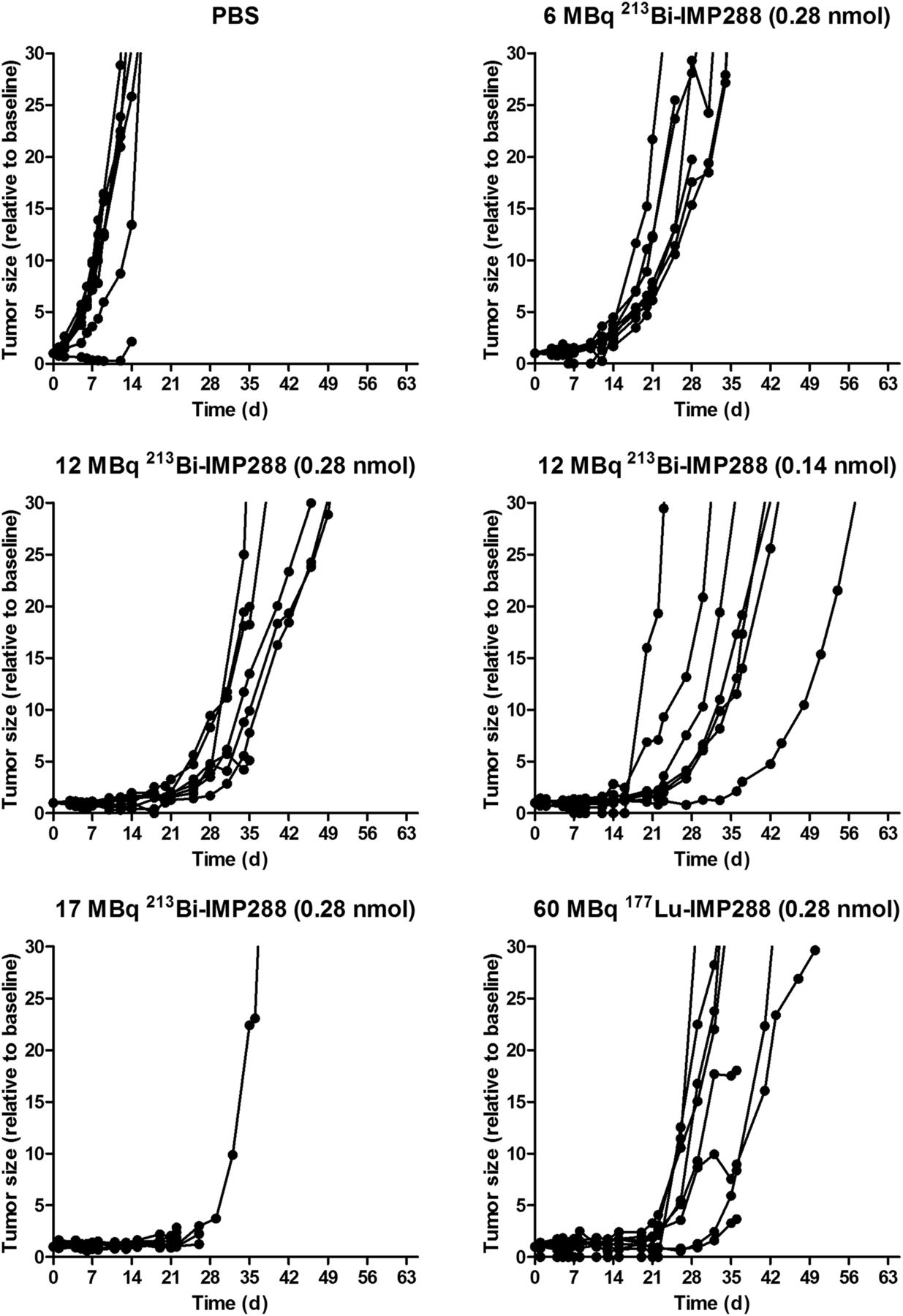
Tumor growth was significantly inhibited by 213Bi-IMP288 treatment (Fig. 6). Mice treated with 17 MBq 213Bi-IMP288 showed a significant decrease in body weight (≥20% compared with baseline) and had to be removed from the experiment within 23–41 d after therapy. The delay in tumor growth did not differ between mice that received 0.28 or 0.11 nmol of 12 MBq 213Bi-IMP288. Tumor growth inhibition in these groups was similar to that of mice receiving 60 MBq 177Lu-IMP288 (0.28 nmol). Mean tumor doubling time for PBS, 6 MBq 213Bi-IMP288, 12 MBq 213Bi-IMP288 (0.28 nmol), 12 MBq 213Bi-IMP288 (0.14 nmol), and 60 MBq 177Lu-IMP288 was 3.2 ± 0.6, 6.5 ± 1.2, 9.6 ± 2.7, 10.7 ± 7.8, and 10.6 ± 9.2 d, respectively. The overall survival significantly increased for mice treated with 6 MBq 213Bi-IMP288 (P = 0.037), 12 MBq 213Bi-IMP288 (0.28 nmol, P = 0.003), 12 MBq 213Bi-IMP288 (0.14 nmol, P = 0.001), and 60 MBq 177Lu-IMP288 (P < 0.001) as compared with the PBS group. Overall survival of the mice that received 17 MBq 213Bi-IMP288 did not increase significantly. Kaplan–Meier curves are presented in Figure 7. Median survival for the PBS, 6 MBq 213Bi-IMP288, 12 MBq 213Bi-IMP288 (0.28 nmol), 12 MBq 213Bi-IMP288 (0.14 nmol), and 60 MBq 177Lu-IMP288 groups was 22, 31, 45, 45, and 42 d, respectively. Median survival for the 17 MBq 213Bi-IMP288 group was 24 d. Supplemental Table 1 summarizes the criteria by which animals were removed from the experiment (weight loss or tumor growth).
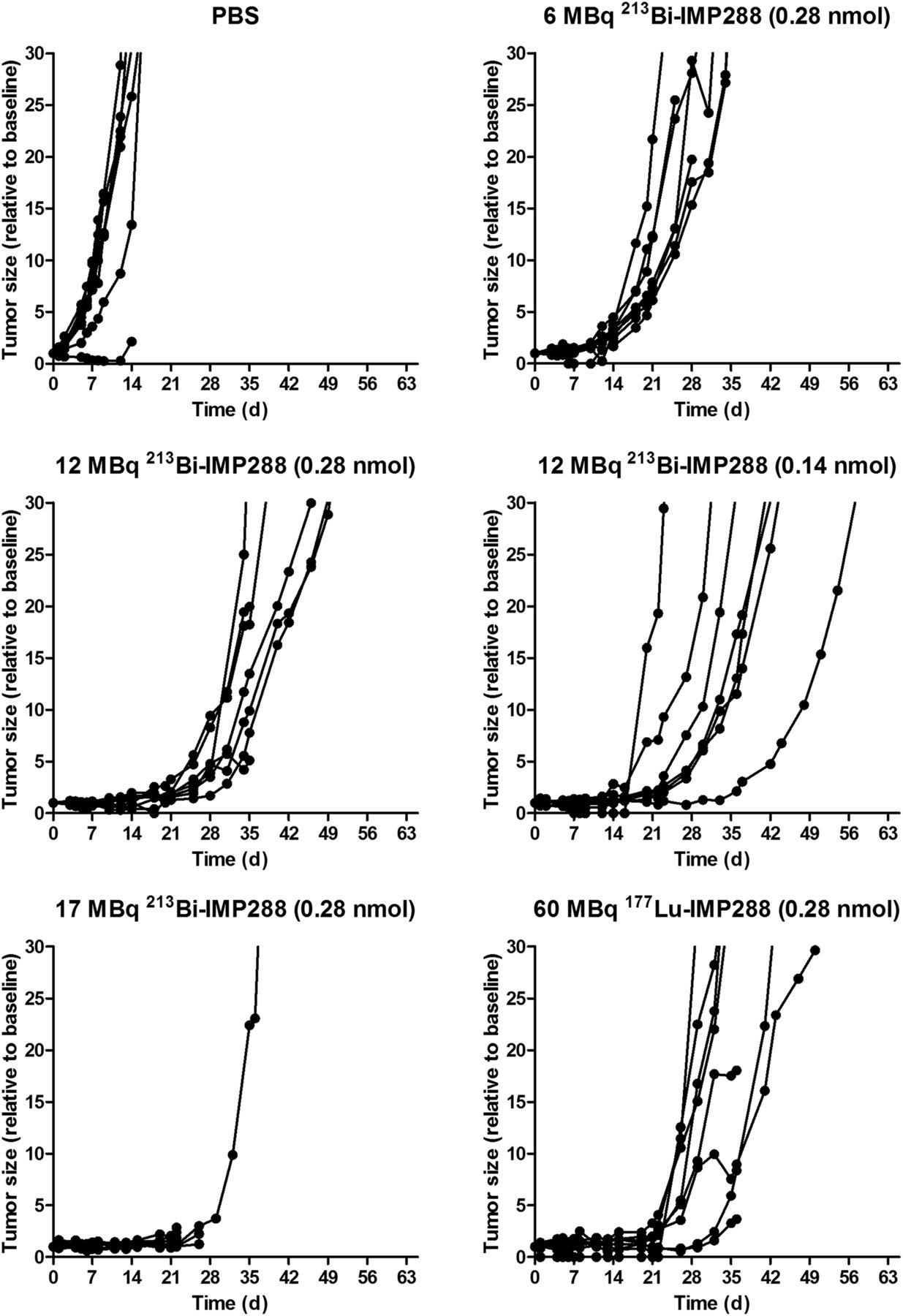
Tumor size of mice treated with TF2 and different activity doses of 213Bi-IMP288 or 177Lu-IMP288.
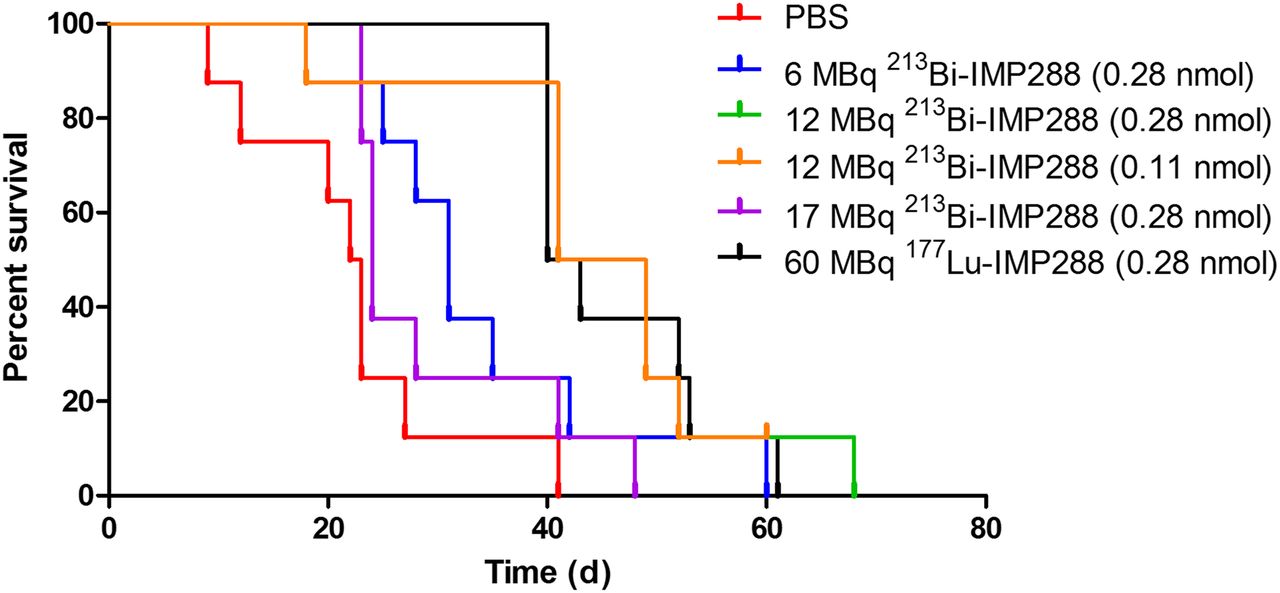
Survival curves of mice treated with TF2 and different activity doses of 213Bi-IMP288 or 177Lu-IMP288.
DISCUSSION
Pretargeting with the bsmAb TF2 and radiolabeled IMP288 allows rapid and specific targeting of CEA-expressing tumors. Therapy with 177Lu-IMP288 has been shown to be safe and feasible (10,11,19,20). However, tumors eventually show progressive growth. Because of their high linear energy transfer, α-emitting radionuclides deliver a significantly higher dose to tumors than β-emitting radionuclides and may improve the therapeutic efficacy of PRIT. To our knowledge, ours is the first study showing the potential of PRIT with TF2 and IMP288 using the α-emitting radionuclide 213Bi.
213Bi-IMP288 has high binding affinity toward TF2-pretargeted LS174T cells and showed efficient targeting to TF2-pretargeted xenografts, similar to that of 177Lu-IMP288. In vitro, internalization rates were low for both tracers, which is expected since CEA is a slowly internalizing antigen (21). In vivo, tumor uptake was observed as early as 15 min after injection and the peptide cleared rapidly from normal tissue via the kidneys. Therefore, the tumor-targeting kinetics of the peptide match the short half-life of 213Bi (45.6 min).
A single dose of TF2 and 213Bi-IMP288 significantly delayed tumor growth and prolonged the survival of tumor-bearing mice, as is in line with the literature on pretargeted α-therapy with antibody–streptavidin constructs and 213Bi-labeled biotin (22–24). The therapeutic efficacy of 213Bi-IMP288 was as least as effective as a single injection of 177Lu-IMP288 at the previously reported maximum tolerated dose. This finding is contrary to other preclinical studies, which have shown that α-emitting radionuclides are more effective than β-emitting radionuclides (23,25,26). For example, Pagel et al. have shown that pretargeted α-therapy with an anti-CD45 antibody–streptavidin complex and 29.6 MBq 213Bi labeling was more effective than 4.1 MBq 90Y-labeled biotin (23). The differences between our results and the literature can be attributed to several factors, such as the type of α- and β-emitter used, the dosing schedule, the radiosensitivity of the tumor model, and the tumor size at the start of therapy. Furthermore, 177Lu and 213Bi have also been shown to have similar therapeutic effectiveness, such as for radioimmunotherapy in a gastric cancer model using the mutant E-cadherin targeting antibody d9mAb (27).
Recent clinical studies have shown that targeted α-therapy is effective in patients who have been treated inefficiently with β-emitting radionuclides. For example, 225Ac-PSMA-617 and 213Bi-DOTATOC can overcome radioresistance to targeted radionuclide therapy with β-emitting radionuclides in patients with neuroendocrine tumors and prostate cancer, respectively (28,29). The therapeutic efficacy of PRIT with 213Bi-IMP288 may be further improved using a fractionated dosing schedule, as has been shown for 177Lu-IMP288 and other 213Bi-labeled targeting agents (10,26,30,31). Furthermore, because of the long retention of radiolabeled IMP288 in the tumor, α-emitting radionuclides with a longer half-life, such as 225Ac (10 d), may be more effective (10,19).
The markedly higher specific activity that can be attained with 213Bi-IMP288 than with 177Lu-IMP288, together with the lower activity doses required to achieve a therapeutic effect, allow for a significant reduction in the administered peptide dose. This is a potential advantage since a lower peptide dose results in higher IMP288 tumor uptake, which may enhance the therapeutic efficacy. However, this increased therapeutic efficacy was not observed in our study. Autoradiography showed heterogeneous uptake of radiolabeled IMP288 in the tumor. Given that the range of α-particles is only 40–100 μm (a few cell diameters), tumor cells in areas of low 213Bi-IMP288 uptake are likely to receive sublethal radiation doses by the α-particles. In contrast, the β-particles emitted by 177Lu have a longer range (1.5 mm), which results in a more homogeneous irradiation of the tumor bed, allowing for effective cell killing in areas with low IMP288 uptake due to a crossfire effect. Pretargeted α-therapy might be more effective for smaller and more homogeneous tumor lesions; for example, in the LS174T intraperitoneal tumor model or in more differentiated CEA-positive xenograft models such as SW1222 (11,32).
213Bi-IMP288 at 12 or 17 MBq caused nephrotoxicity in a subgroup of mice. The retention of radiolabeled IMP288 in the kidney is low, whereas the peptide is cleared rapidly from the circulation. Within 30 min, almost all IMP288 has been filtered by the glomeruli and has passed the proximal and distal tubuli. Because 213Bi has a short half-life of only 45.6 min, within the first 30 min a high dose is delivered to the kidneys. Several other studies have also observed renal toxicity in mice after the intravenous administration of targeted α-therapy (22,26,33,34). Potentially, renal toxicity might be reduced using dose fractionation or α-emitting radionuclides with a longer half-life (26,30,31). More important, renal toxicity profiles have not been reported in clinical studies using targeted α-therapy (28,29).
CONCLUSION
This study showed, for the first time to our knowledge, that PRIT with 213Bi-IMP288 is feasible and at least as effective as 177Lu-IMP288. However, because kidney toxicity was observed at higher doses, the optimal dosing schedule needs to be determined in future studies. Furthermore, α-emitting radionuclides with a longer half-life might improve therapeutic efficacy and reduce renal toxicity.
DISCLOSURE
This work was supported by the National Science Foundation (DGE-1256259), the National Institutes of Health (NIBIB/NCI 1R01CA169365 and T32-GM008349), and the Dutch Cancer Society. David M. Goldenberg is, and William J. McBride was, an employee of Immunomedics, Inc., and IBC Pharmaceuticals, Inc., which have patented this technology and reagents. These two authors are also inventors on these patents. David M. Goldenberg has stocks in Immunomedics. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Birgit Blechert, Cathelijne Frielink, Bianca Lemmers-van de Weem, Henk Arnts, Iris Lamers-Elemans, and Kitty Lemmens-Hermans for excellent technical assistance.
Footnotes
Published online Feb. 23, 2017.
- © 2017 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication November 14, 2016.
- Accepted for publication January 31, 2017.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Theranostic GPA33-Pretargeted Radioimmunotherapy of Human Colorectal Carcinoma with a Bivalent 177Lu-Labeled Radiohapten
- Efficacy of HER2-Targeted Intraperitoneal 225Ac {alpha}-Pretargeted Radioimmunotherapy for Small-Volume Ovarian Peritoneal Carcinomatosis
- Profound immunomodulatory effects of 225Ac-NM600 drive enhanced anti-tumor response in prostate cancer
- Pretargeting: A Path Forward for Radioimmunotherapy
- Mathematical Modeling of Preclinical Alpha-Emitter Radiopharmaceutical Therapy