Article Text

Abstract
Background and aims—The liver is frequently involved in amyloidosis but the significance of hepatic amyloid has not been systematically studied. We have previously developed scintigraphy with 123I serum amyloid P component (123I-SAP) to identify and monitor amyloid deposits quantitatively in vivo and we report here our findings in hepatic amyloidosis.
Methods—Between 1988 and 1995, 805 patients with clinically suspected or biopsy proven systemic amyloidosis were evaluated. One hundred and thirty eight patients had AA amyloidosis, 180 had AL amyloidosis, 99 had hereditary amyloid syndromes, and 67 had dialysis related (β2 microglobulin) amyloid. One hundred and ninety two patients with amyloidosis were followed for six months to eight years.
Results—Hepatic amyloid was found in 98/180 (54%) AL and 25/138 (18%) AA patients but in only 1/53 patients with familial transthyretin amyloid polyneuropathy and in none with dialysis related amyloidosis. There was complete concordance between hepatic SAP scintigraphy and the presence or absence of parenchymal amyloid deposits on liver histology. Amyloidosis was never confined to the liver. Mortality was rarely due to hepatic failure, although hepatic involvement with AA amyloid carried a poor prognosis. Successful therapy to reduce the supply of amyloid fibril protein precursors was followed by substantial regression of all types of amyloid.
Conclusions—SAP scintigraphy is a specific and sensitive method for detecting and monitoring hepatic amyloid. Liver involvement is always associated with major amyloid in other organ systems and carries a poor prognosis in AA type. Appropriate therapy may substantially improve prognosis in many patients.
- systemic amyloidosis
- serum amyloid P component
Statistics from Altmetric.com
Amyloidosis is a disorder of protein metabolism in which autologous proteins are deposited extracellularly in a characteristic abnormal fibrillar form. Amyloid can be focal, localised, or systemic in distribution and the deposits may be hereditary or acquired. They are composed mainly of amyloid fibrils which can be derived from many different proteins and are associated with a wide spectrum of disease. The non-fibrillar glycoprotein, serum amyloid P component (SAP), a normal plasma protein, is present in all amyloid deposits.1
Until recently, the presence of amyloid could only be confirmed by histology, and the finding of apple green birefringence when tissues stained with Congo red are viewed microscopically through crossed polarising filters remains the diagnostic gold standard. However, tissue biopsy can provide only limited information on the extent and distribution of amyloid generally and none on its rate of accumulation and/or regression. The introduction of radiolabelled SAP as a specific nuclear medicine tracer for amyloid has therefore been a significant advance, providing a quantitative whole body survey of amyloid deposits. The technique has been extensively validated.2-4
Amyloid can be deposited in the liver in most forms of systemic amyloidosis, as shown in many previous retrospective studies, mainly based on postmortem findings. Liver biopsy causes haemorrhage in approximately 5% of amyloid patients5-7 and is best avoided unless there are compelling indications. In systemic amyloidosis there are always widespread vascular deposits, and sometimes also interstitial amyloid, which may enable the diagnosis to be made on biopsy of clinically unaffected tissue, for example subcutaneous fat or rectal mucosa. Diffuse vascular wall amyloid without parenchymal involvement is frequently seen in the liver,8 and inclusion of such cases may have confounded earlier studies of the natural history of hepatic amyloid.9 ,10 The only substantial clinical series reported in the past 20 years in which hepatic histology for amyloid was obtained during life was confined to AL amyloidosis and details of liver histology were not provided.5 In a more recent series by the same group, hepatic involvement was diagnosed on clinical grounds alone in 11 of 19 patients.11
Iodine-123 labelled SAP scintigraphy has already provided a number of new insights into the natural history of the various amyloid disorders and their response to treatment. For example, the rate of amyloid accumulation varies greatly between patients and between different organs in the same patient and most encouragingly, amyloid deposits regress substantially in many cases when the supply of the fibril precursor protein is reduced.12 We report here the findings of 123I-SAP scintigraphy in 484 patients with systemic amyloidosis with respect to the diagnosis, evolution, and significance of amyloid deposits in the liver.
Patients and methods
PATIENT CHARACTERISTICS
Between 1988 and 1995, 805 patients with clinically suspected or histologically proven amyloidosis were referred to the Immunological Medicine Unit at the Hammersmith Hospital for clinical evaluation and123I-SAP scintigraphy. Although many patients were diagnosed in our unit, or seen soon after diagnosis, some were only referred after a considerable delay. Physical examination and full haematological and biochemical profiles were performed in all patients together with other investigations at the discretion of the examining doctor. All cases were reviewed by a senior physician (PNH). Amyloidosis, as the presumptive or suspected diagnosis made by clinicians elsewhere, was excluded in 274 patients, including seven in whom previous biopsy specimens had been reported as positive. Forty seven patients had localised amyloid syndromes. Patients with systemic amyloidosis included 138 with AA amyloid, 180 with AL type, 99 with hereditary amyloid syndromes, and 67 patients with dialysis related amyloid.
There were 62 females and 76 males with AA amyloidosis. Median age at diagnosis was 30 years (range 8–87 years) and the median duration of underlying illness at diagnosis was 12 years (range 1–33 years). Ninety four patients were referred following histological diagnosis of amyloid at other centres (median time from diagnosis to referral 10 months, range one month to 15 years). Forty four patients (32%) were diagnosed by SAP scintigraphy alone. Seven had already undergone rectal biopsy in which amyloid was not shown but histology of subsequent renal biopsy in two of these confirmed the diagnosis. Histological confirmation was obtained in nine others (seven by renal biopsy and two by rectal biopsy). The most common diseases associated with AA amyloidosis were juvenile rheumatoid arthritis and rheumatoid arthritis (table 1).
Underlying conditions in patients with systemic AA amyloidosis
There were 83 female and 97 male patients with systemic AL amyloidosis whose median age at diagnosis was 59 years (range 28–87 years). One hundred and sixty five patients had biopsy proven amyloid at referral (median time from diagnosis to referral four months, range one month to five years). Fifteen further patients (8%) were diagnosed by SAP scintigraphy alone. Three of these had already undergone rectal biopsy but amyloid had not been shown and the diagnosis was later confirmed histologically at endomyocardial biopsy in one further patient. Table 2 presents characteristics of the underlying monoclonal gammopathy.
Underlying conditions in patients with systemic AL amyloidosis
Patients with hereditary systemic amyloid syndromes included 33 with the transthyretin Val30Met variant and 21 patients with 13 other point mutations in the transthyretin gene. The variant amyloidogenic protein in the remaining patients was apolipoprotein AI in 30, fibrinogen α chain in seven, cystatin C in three, lysozyme in four, and gelsolin in one (table 3).
SAP scintigraphy in hereditary amyloidosis
Fifty four of the patients with dialysis related amyloid had received haemodialysis for a median of 14 years (range 4–26 years); the 13 other patients had received only continuous ambulatory peritoneal dialysis (median 6.5, range 5–17 years).
HISTOLOGY
Histological evidence of amyloid in the liver was obtained either by needle biopsy or at postmortem examination. Formalin fixed paraffin wax embedded tissue sections were stained with alkaline alcoholic Congo red13 and examined under polarised light for characteristic apple green birefringence. Immunohistochemistry was performed using standard peroxidase antiperoxidase techniques with monospecific polyclonal antisera to AA protein,14 κ and λ immunoglobulin light chains, transthyretin, lysozyme (Dakopatts, Glostrup, Denmark) and apolipoprotein AI (Medix Biotech antisera, California, USA). In each case positive staining was controlled using primary antisera that had been absorbed with an excess of the appropriate specific antigen.
123I-SAP SCINTIGRAPHY
Preparation and radiolabelling of SAP
Serum amyloid P component was isolated from the plasma of a single donor accredited by the British National Blood Transfusion Service and iodinated with 123I to a specific activity of 3000 MBq/mg as previously described.2 ,4
Scanning protocol
Following thyroid blockade with potassium iodide,123I-SAP (3 MBq/kg) was administered by intravenous bolus injection. There were no adverse effects. After 24 hours, anterior and posterior whole body images and appropriate regional views were obtained with a General Electric Starcam gamma camera (IGE, Slough, UK) or an MS2 gamma camera (Siemens, Erlangen, Germany) using a medium energy parallel hole collimator. All scintigraphs were assessed by a single assessor (PNH).
Follow up scintigraphy was performed six monthly in AL patients and annually in patients with AA amyloid and hereditary amyloid syndromes. Eighty five AA patients were studied prospectively for eight to 100 months (median 43 months) and 62 AL patients were studied for six to 96 months (median 20 months).
All data were prospectively entered into a computerised clinical database (Microsoft Access).
Results
CORRELATION BETWEEN LIVER HISTOLOGY AND 123I-SAP SCINTIGRAPHY
Liver biopsy was performed successfully in 38 patients and tissue was subsequently obtained at postmortem examination in a further 21 cases. Biopsy was complicated by significant haemorrhage in two patients. SAP scintigraphy of the liver was positive in every case with parenchymal or stromal amyloid on liver histology and was negative in all cases where only diffuse vascular deposits were shown (table4).
Correlation between hepatic amyloid shown by liver histology and SAP scintigraphy
PREVALENCE OF HEPATIC AMYLOIDOSIS
The proportion of patients with hepatic amyloid identified by SAP scintigraphy varied greatly between different types of amyloidosis. At first evaluation in our unit, liver deposits were found in 98/180 (54%) patients with AL amyloid but in only 25/138 (18%) with AA disease. In neither type was there any significant difference between patients with amyloid in the liver and those without with respect to age at presentation or duration of underlying disease.
Familial amyloid polyneuropathy is caused by deposition of amyloid derived from genetically variant transthyretin, a plasma protein produced only in the liver and choroid plexus. However, among the 53 patients studied, the only one with hepatic amyloid was an individual with a unique familial amyloid polyneuropathy phenotype associated with the previously unreported Leu12Pro TTR variant. Table 3 shows the frequency of liver involvement in other hereditary amyloid syndromes. Hepatic amyloid was not found in any of the 67 patients with dialysis related (β2 microglobulin) amyloidosis.
FINDINGS IN THE 25 PATIENTS WITH HEPATIC AA AMYLOIDOSIS AT PRESENTATION
Extrahepatic amyloid deposition
Splenic deposits were present in all AA patients when first seen in our unit. Renal deposits were seen in 15 patients but the kidneys were obscured in eight by intense signal arising from the adjacent spleen and liver. The kidneys were not visualised in two patients who already had end stage renal failure. Adrenal deposits were identified in eight patients.
Laboratory findings
Proteinuria exceeding 5 g/day was present in 10/14 (71%) patients in whom complete urine collections were obtained at presentation to our unit. Proteinuria was found in a similar proportion of patients without hepatic amyloid. The median serum alkaline phosphatase activity (ALP) in patients with hepatic amyloid was 141 IU/l (range 72–1350 IU/l; normal less than 130 IU/l) at first review in our unit and was significantly higher (p<0.001) than in the 113 unaffected patients (median 92, range 11–466 IU/l) (fig 1), although there was much overlap. Serum bilirubin and aspartate aminotransferase (AST) were normal in all patients with hepatic amyloid except one, an intravenous drug user with a raised AST. This patient had chronic active hepatitis, associated with hepatitis B infection.
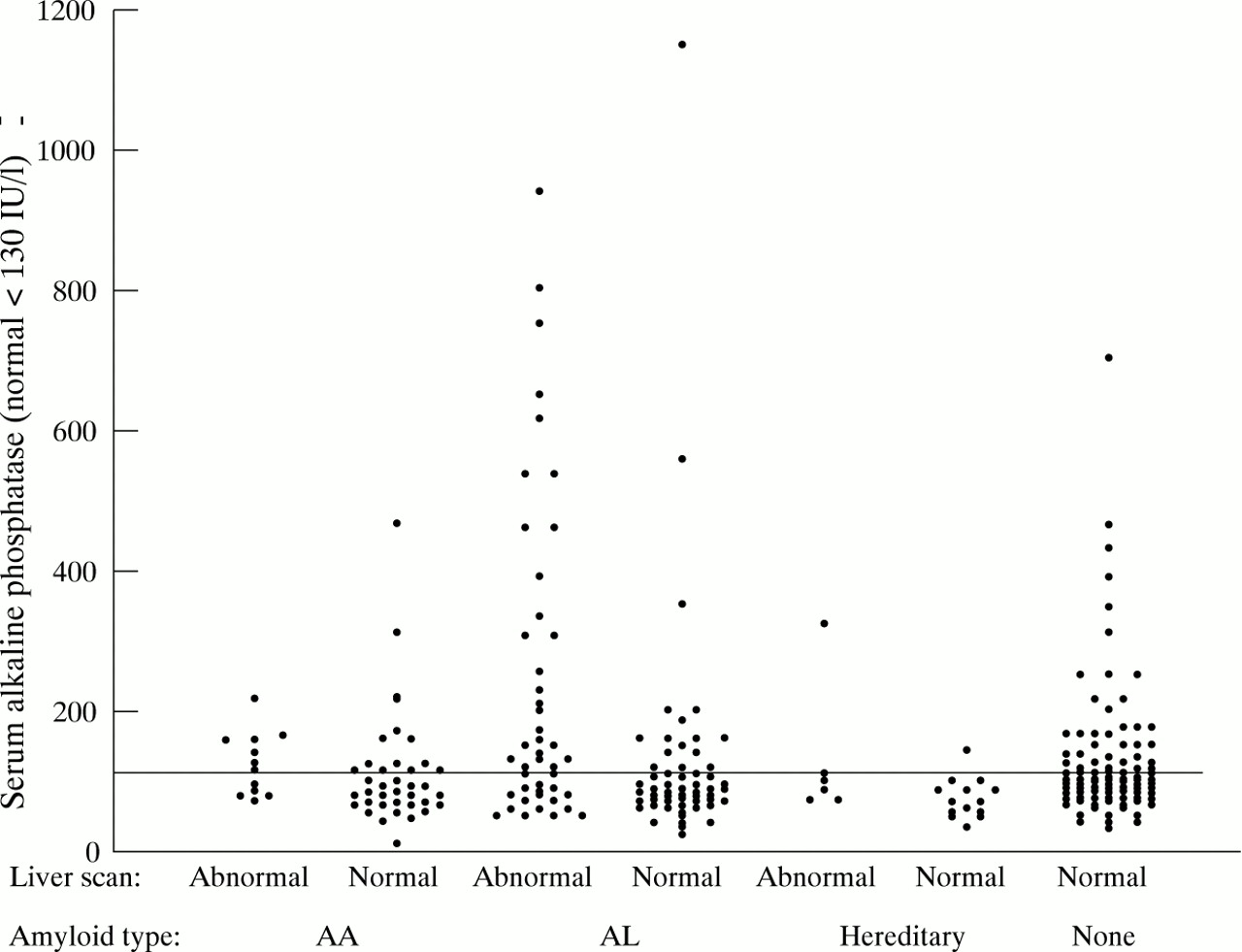
Serum alkaline phosphatase in 691 patients who underwent liver scintigraphy with 123I-SAP showing poor predictive value of elevated serum alkaline phosphatase for liver amyloidosis.
FOLLOW UP STUDIES IN 85 PATIENTS WITH AA AMYLOIDOSIS
All patients were thoroughly investigated to determine the disorder that had been complicated by reactive AA amyloidosis, and were treated vigorously to control the primary disease. The acute phase response was monitored initially by serum C reactive protein levels and since 1992 by monthly estimation of serum amyloid A protein, providing direct information on the rate of production of the amyloid fibril precursor.14 Follow up scans were performed in eight patients who had liver involvement at baseline. In six patients serum amyloid A protein concentrations were reduced to normal and this was associated with substantial regression of amyloid in the liver and elsewhere. Nevertheless, two of these patients, who were already on haemodialysis, died after 2.5 and 5.8 years respectively. Amyloid deposits initially regressed in the remaining two patients but stabilised or reaccumulated later. The first had had Crohn’s disease for 15 years prior to the diagnosis of AA amyloid in 1989. He underwent renal transplantation for amyloid associated renal failure in 1990. The acute phase response was incompletely controlled and although the hepatic amyloid regressed during the first two years of follow up, it had reaccumulated at four years. No further change was seen at five years and he remains in good health. The second patient developed juvenile rheumatoid arthritis at 10 years of age and was treated with steroids. AA amyloidosis was diagnosed 15 years later, in 1988. He received a renal transplant in 1989 and by 1994 some regression of liver amyloid was seen. However, more recently, the acute phase response has been only partially controlled, and six years after transplantation some amyloid could be identified in the grafted kidney, although the hepatic deposits have been stable.
Five patients who, despite treatment, continued to have substantial activity of the underlying inflammatory disease, first developed liver amyloid during follow up (fig 2). One of these cases developed end stage renal failure, was established on haemodialysis, but died after five years.

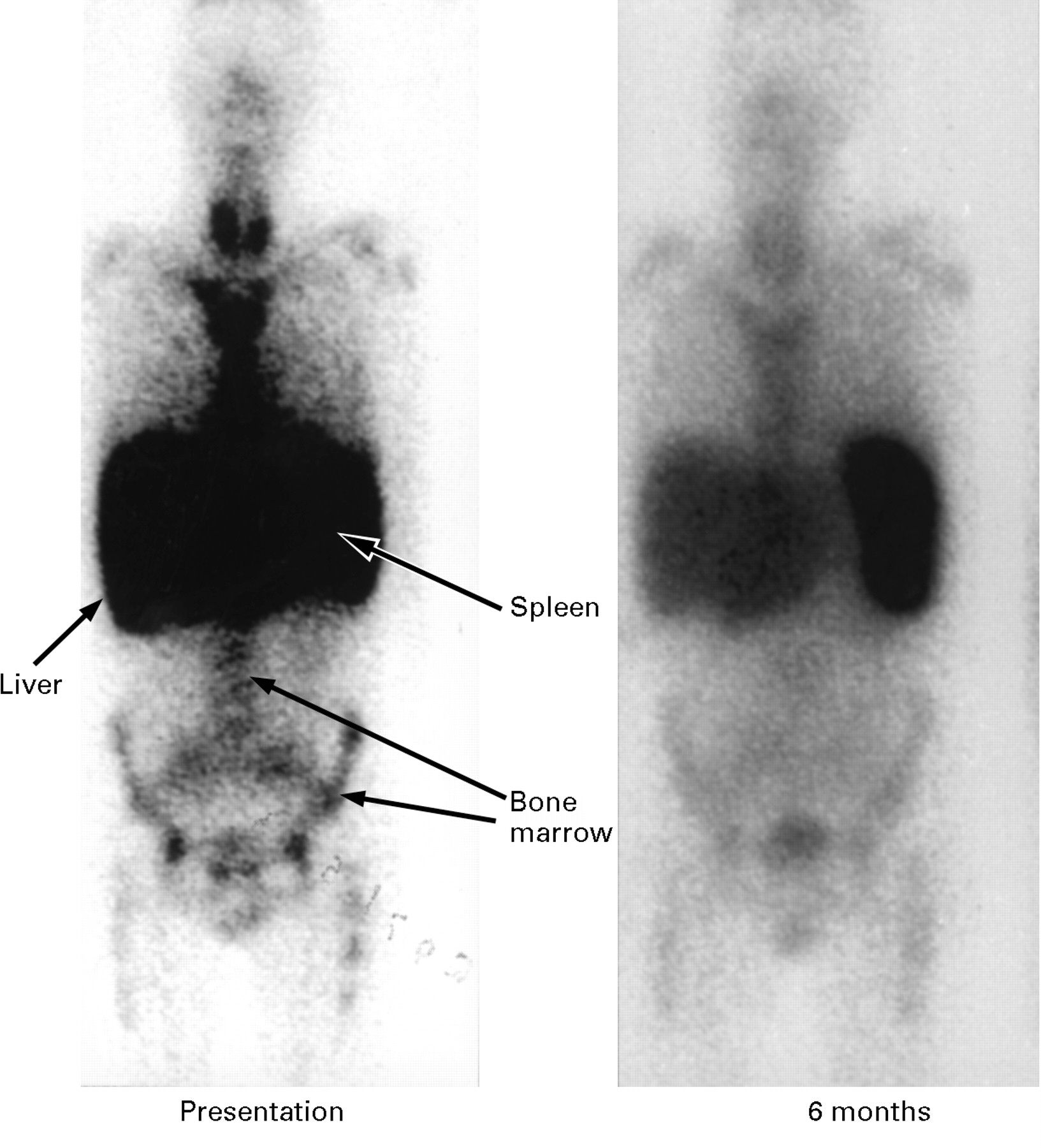
Serial posterior abdominal 123I-SAP scans of a patient with AA amyloidosis complicating rheumatoid arthritis. The scan at presentation (A) shows uptake of the tracer into amyloid deposits in the spleen, kidneys, and adrenal glands. The patient did not respond to anti-inflammatory treatment and the follow up scan two years later (B) shows additional localisation of tracer into the liver; reduction of the renal signal is due to end stage renal failure.
Survival
Hepatic amyloid deposition in systemic AA amyloidosis carries a poor prognosis, with the predicted five year survival falling to 43% in cases with liver involvement from 72% in cases without (fig 3). Most patients died from complications of renal amyloid and none died of hepatic failure. Interestingly, in patients with underlying diseases in which AA amyloidosis is a well recognised complication, liver involvement was found in 17/124 (13.8%), compared with 7/14 patients (50%) with diseases with which amyloid is rarely associated (χ2=0.0007) (table 1). There had been considerable delay in the diagnosis of amyloid in these latter cases because the index of suspicion was low. These data suggest that hepatic amyloid deposition is associated with the presence of extensive systemic amyloid and occurs late in the evolution of the condition.
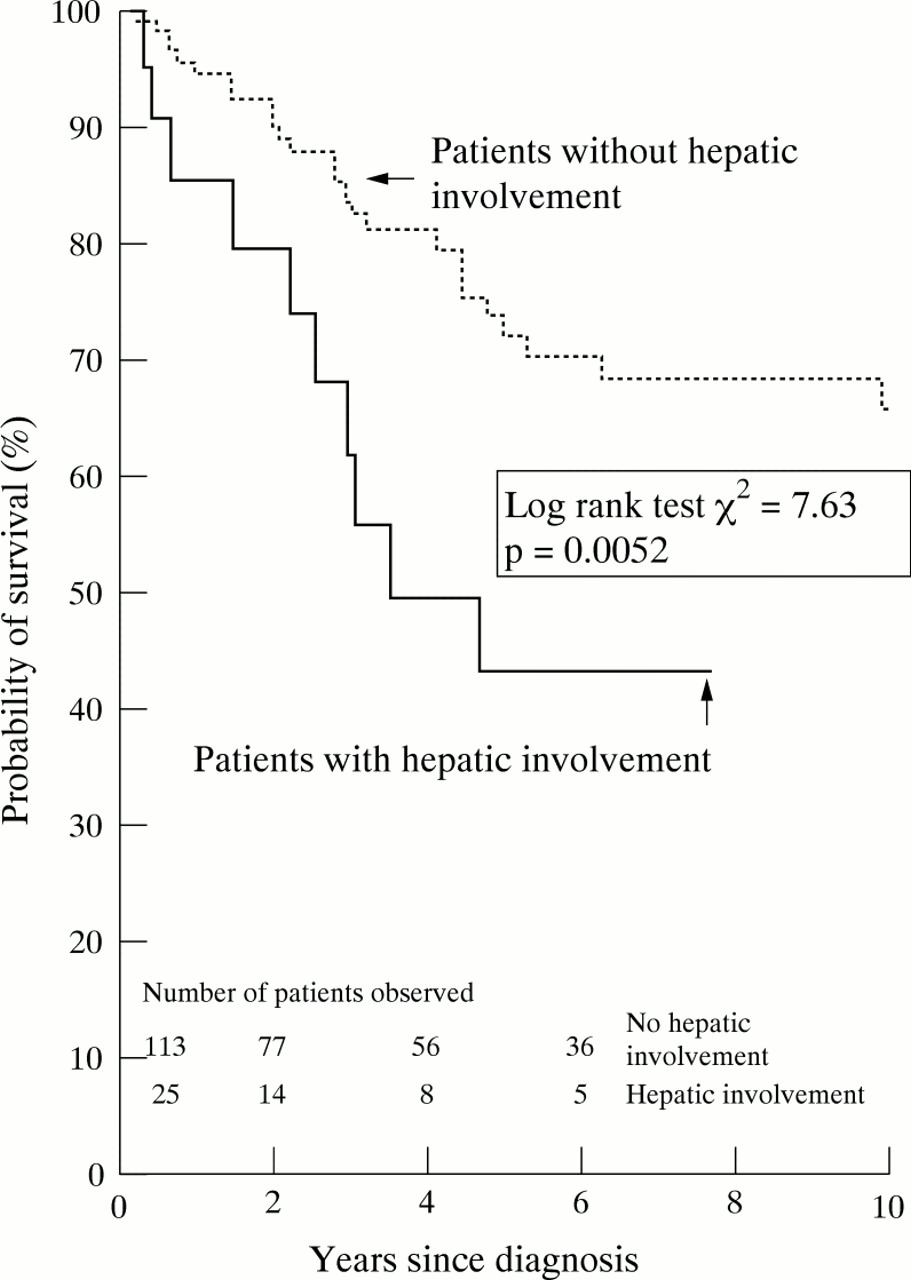
Kaplan-Meier estimate of survival in patients with systemic AA amyloidosis with and without hepatic involvement on123I-SAP scintigraphy.
FINDINGS IN THE 98 PATIENTS WITH HEPATIC AL AMYLOIDOSIS AT PRESENTATION
Extrahepatic amyloid deposition
Splenic amyloid deposition was present in all but one patient. Renal deposits were seen in 52 patients but the kidneys could not be evaluated in 22 patients because of the intensity of the neighbouring hepatic and splenic signals. In one further patient, who was in end stage renal failure, the kidneys were not visualised, as is often the case because of reduced renal perfusion in this situation. Bone marrow uptake of tracer occurred in 48 patients and was completely specific for AL amyloid.
Laboratory findings
Proteinuria in excess of 0.2 g/day was present in 59/72 patients (82%) at first review in our unit and 33/72 (46%) had proteinuria in excess of 5 g/day. Proteinuria (more than 0.2 g/day) was present in 37/64 patients (58%) without hepatic amyloid. Mean proteinuria was significantly higher in patients with hepatic amyloid than in those without (5.65 versus 2.64 g/day, p=0.0016).
The median serum ALP activity in patients with hepatic amyloid was 150 IU/l (range 50–1497 IU/l; normal less than 130 IU/l) at presentation to us and was significantly lower (84 IU/l, range 23–1146 IU/l, p<0.0001) in the 82 patients without liver deposits (fig 1). As in AA amyloidosis, there was a wide overlap of ALP values between patients with and without liver involvement. Median AST was 30 IU/l (range 11–420; normal less than 40 IU/l) in 77 patients with hepatic amyloid and 28 IU/l (range 9–96) in 86 patients without. Elevation of AST was almost invariably accompanied by raised serum ALP activities. Serum bilirubin concentrations exceeded twice the upper limit of normal in only five patients in this whole series, and all had AL amyloid. Although none had biliary obstruction on ultrasound examination, their mean survival was only four months and none survived beyond nine months after diagnosis, except one patient who received a liver transplant (see below).
A prolonged thrombin time was present in 23/62 patients (37%), and also in 10/49 patients (20%) without hepatic involvement. There was prolongation of the prothrombin time in 11/64 patients (17%) and of the activated partial thromboplastin time in 7/64 AL patients (11%) but neither was specific for hepatic amyloidosis. All patients with prolonged prothrombin time or activated partial thromboplastin time, regardless of hepatic amyloid infiltration, had major splenic amyloidosis.
FOLLOW UP STUDIES IN 62 PATIENTS WITH AL AMYLOIDOSIS
Sixty two patients, of whom 32 had liver involvement at first presentation to us, were scanned serially at six monthly intervals. Median follow up was 20 months (range 6–98 months). Two patients developed hepatic deposits after six months follow up, and a further patient who presented with amyloid localised to the bronchial tree and was not treated with cytotoxic drugs developed systemic amyloid with liver involvement after three years. One patient with hepatic amyloid at presentation received no cytotoxic therapy and his amyloid progressed. Among 17 subjects who received low dose oral chemotherapy consisting of monthly melphalan and prednisolone for up to two years,15 progression of liver amyloid was seen in five patients (29%), the deposits were stable in 11 others (65%), and regression of liver deposits was seen in only one patient, and not until three years after treatment was started. Fourteen patients received more intensive cytotoxic regimens (most commonly vincristine, adriamycin, and dexamethasone). Amyloid progressed in two of these who were followed for 12 and 30 months respectively and remained unchanged in six others who were followed for a mean of 26 months (range 9–72 months). However, regression of amyloid was seen in six patients (43%), within as little as six months of commencing treatment (fig 4). In addition, many treated patients reported improvement in well being even when the amyloid load did not alter.
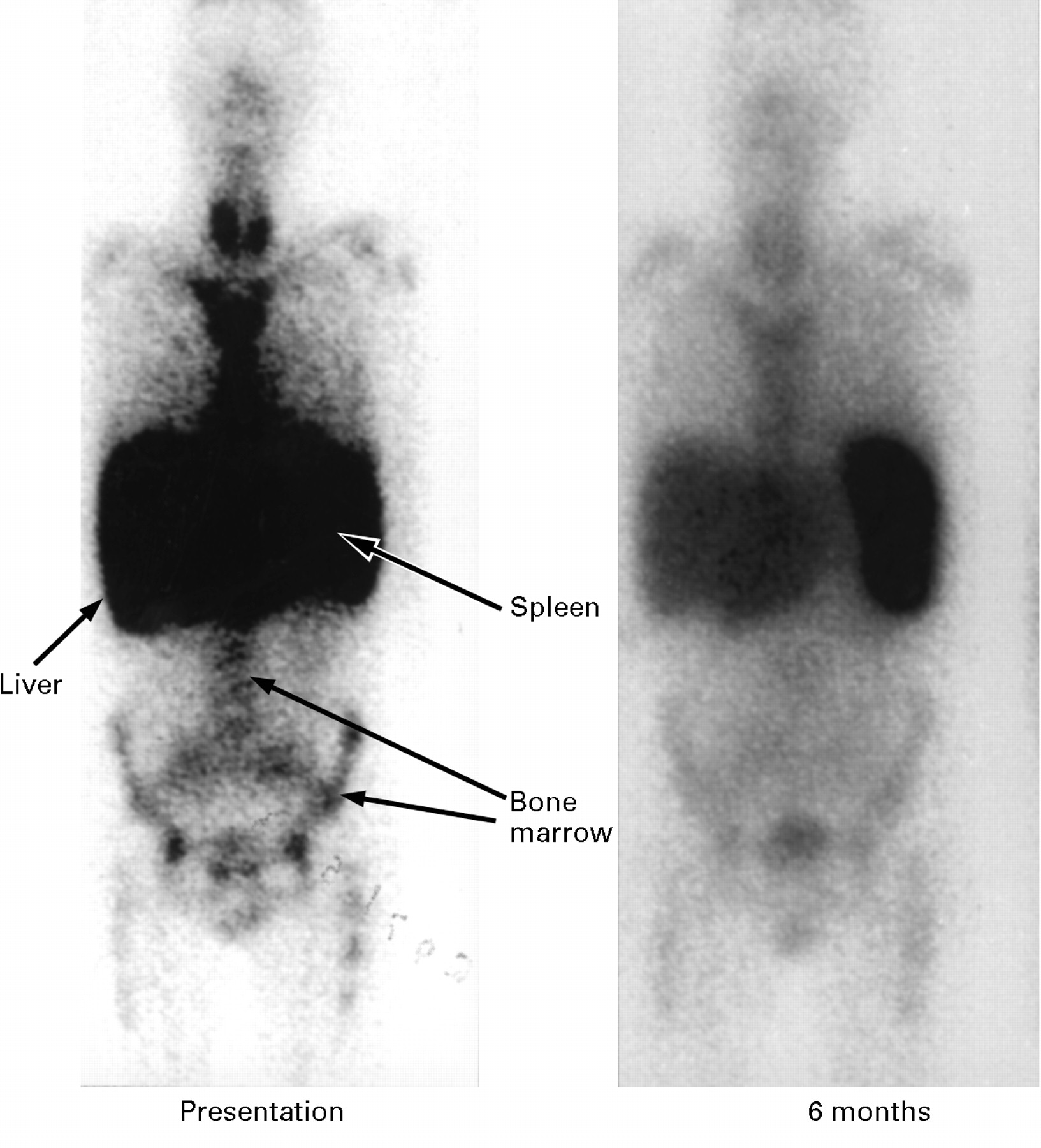
Serial anterior whole body 123I-SAP scans in a patient with AL amyloidosis who presented with hepatomegaly and liver dysfunction. The initial scan (A) shows massive uptake of tracer into amyloid deposits in the liver, spleen, and bone marrow. The follow up scan six months later (B), following intensive chemotherapy, shows much less uptake of labelled SAP into the liver and bone marrow indicating substantial regression of the amyloid in these organs. Liver function had also returned to near normal levels.
Survival
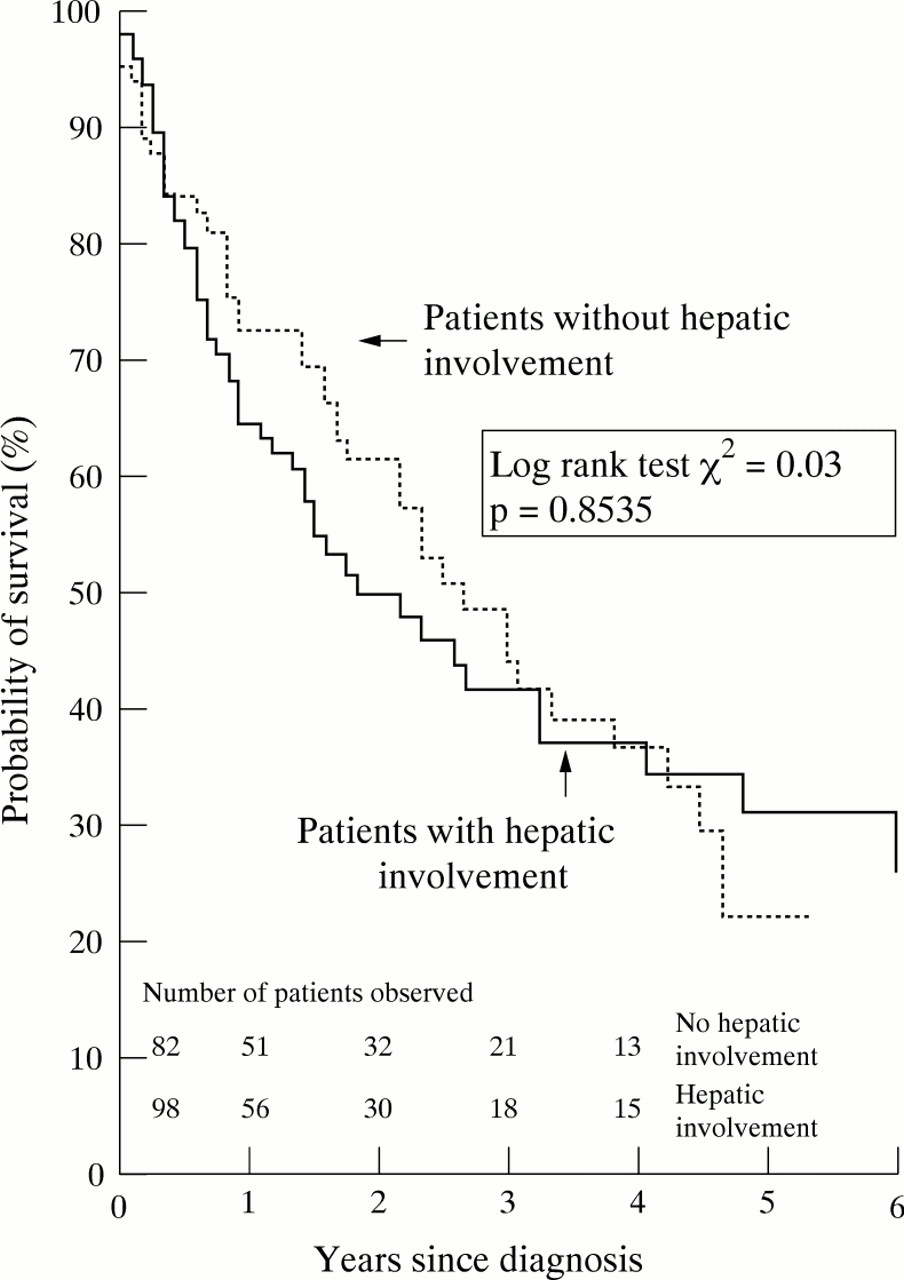
Hepatic involvement is more common in AL than in AA amyloidosis, and does not significantly influence prognosis (log rank test χ2=0.03, p=0.8535). In our patients the estimated time of survival from diagnosis was 26 months (fig 5). The only laboratory test that was prognostic was an elevated bilirubin concentration. However, treatment aimed at reducing the supply of monoclonal immunoglobulin light chains resulted in improved mean survival and preliminary analysis indicates that the prognosis may be notably better in those treated with regimens more intensive than melphalan and prednisolone alone.16
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Kaplan-Meier estimate of survival in patients with systemic AL amyloidosis with and without hepatic involvement on123I-SAP scintigraphy.
LIVER TRANSPLANTATION FOR SYSTEMIC AMYLOIDOSIS
Although massive hepatic amyloidosis was common in our series, clinically significant liver failure was very rare. Four patients with AL amyloid died of hepatic failure although one of these also had cirrhosis associated with hepatitis C. One patient underwent orthotopic liver transplantation for liver failure due to amyloid and remained alive at 30 months. Emergency liver transplantation was performed in one other patient, a 15 year old boy who presented with uncontrollable bleeding from spontaneous hepatic rupture. The liver contained amyloid composed of the Asp67His lysozyme variant and he is a heterozygous carrier of the corresponding gene mutation. Interestingly, both his father and grandfather also died of spontaneous liver rupture due to the same condition.17 This patient remains well 36 months postoperatively with normal hepatic function and with no evidence as yet of recurrent liver amyloid.
Orthotopic liver transplantation has been used most widely for amyloid as a form of “surgical gene therapy” which is potentially curative for familial amyloid polyneuropathy associated with variants of transthyretin. It has now been performed in over 150 patients worldwide. We have performed serial SAP scintigraphy in eight patients with familial amyloid polyneuropathy who have received liver transplants and have shown regression of renal and splenic amyloid deposits in seven, within three years of transplantation. This has been associated with stabilisation of peripheral neuropathy and substantial improvement in autonomic and gastrointestinal function and general well being in most patients.
Discussion
The clinical significance of hepatic amyloidosis has hitherto been obscure because the only available method for studying amyloid deposits was histological examination of biopsied tissue. Needle biopsy is invasive and yields an extremely small sample of the whole organ. It provides only limited information about amyloid in the liver and none on deposits elsewhere. Previous histological studies have therefore not been able to quantify the total amount of hepatic amyloid, or prospectively monitor its evolution and response to treatment.5 ,9 ,10
Scintigraphy with radiolabelled SAP provides a method for non-invasively showing amyloid in vivo, based on the reversible but high affinity binding of SAP to all types of amyloid fibrils. Deposition of SAP occurs in proportion to the quantity of amyloid that is present and SAP does not accumulate in tissues that do not contain amyloid.2 Scintigraphic localisation of labelled SAP to amyloid is thus a highly specific phenomenon in which the tracer equilibrates freely with SAP in the plasma and the much greater amount concentrated within the amyloid deposits. This unique dynamic behaviour enables labelled SAP to be used prospectively for quantitative monitoring of amyloid during all phases of its deposition, steady state, and mobilisation.18 We have not encountered any adverse effects of SAP scintigraphy and the radiation dosage is small, being less than a typical barium series.
Our present results confirm that SAP scintigraphy is both specific and sensitive for detecting hepatic amyloid deposits which, in many cases, were clinically silent. The diffuse vascular amyloid that occurs throughout the body in most patients with systemic amyloid, and is often observed histologically in liver biopsy specimens,8 ,19 is not visualised by the technique. On the other hand, when stromal and parenchymal liver amyloid is present, massive uptake of labelled SAP can be detected long before the organ becomes enlarged or there is any functional disturbance. The present study is thus the first in which deposition of amyloid in the liver has been systematically compared with functional effects in vivo, and it convincingly supports our previous observations that amyloidosis is a dynamic process which frequently regresses when supply of the fibril precursor protein is reduced.20 Although SAP scintigraphy is currently available at only a few centres, our recent success in radiolabelling SAP with technetium-99m21 and in producing fully functional recombinant human SAP should facilitate general availability of the technique in due course.
The present series confirms that liver amyloid deposits are much more common in AL than in AA amyloidosis, and shows directly in vivo that hepatic involvement is extremely rare in familial amyloid polyneuropathy, even though the fibril precursor protein, transthyretin, is produced almost exclusively in the liver. We also did not detect liver amyloid in dialysis related amyloidosis. Amyloid deposits, derived from β2 microglobulin, are almost universally present in the large joints and carpal tunnel of patients after five years or more of long term haemodialysis for end stage renal failure.1 Although systemic and visceral deposition have been reported as a late event in this type of amyloid, none of the patients in our series had liver involvement even after more than 20 years’ haemodialysis.
All previously reported patients with hepatic amyloid who were thoroughly investigated also had systemic deposits,22 and in the present series it was usually associated with major amyloid deposits in the spleen and kidneys, even in those individuals who presented with liver dysfunction. Serum alkaline phosphatase activities are commonly increased in both AA and AL amyloidosis, but this can be a manifestation of the acute phase response and was not specific for the presence of liver deposits in our patients. Indeed, many patients with major hepatic amyloid deposits have no biochemical derangement of liver function. In their study of 80 patients with hepatic AL amyloid, Gertz and Kyle5 concluded that hepatomegaly out of proportion to liver enzyme abnormalities, proteinuria, the presence of a serum paraprotein, and hyposplenism were all diagnostic clues. The findings here indicate that these features were suggestive of AL amyloidosis but were not specific for liver involvement. On the other hand, bone marrow amyloid seen on SAP scintigraphy was 100% specific for AL amyloid and its sensitivity for diagnosis compares well with biopsy or postmortem liver histology.8-10 ,23 The only laboratory test of prognostic significance in our series of hepatic AL amyloid cases was elevation of serum bilirubin above 34 μmol/l, which occurred in only 5% of the patients.
Active inflammatory disease causing persistently high serum concentrations of serum amyloid A protein is associated with progressive AA amyloidosis,24 whereas occasional cases of clinical improvement suggesting amyloid regression have been reported in patients in whom the underlying disease has remitted.25Here we show that there was net accumulation of AA amyloid in the liver and elsewhere in most patients whose underlying inflammatory disorder remained active, and that parenchymal liver infiltration was a late sign with a poor prognosis. Abnormalities of liver function tests were common but non-specific, and proteinuria was almost universal whether hepatic amyloid was present or not. In the absence of any direct treatment that promotes mobilisation of amyloid, a vital objective in management of AA amyloidosis is to reduce the synthesis of serum amyloid A protein, the precursor of the AA amyloid fibril protein, using pharmacological and, if appropriate, sometimes also surgical treatment, for example in Castleman’s disease.20Frequent estimation of the plasma serum amyloid A protein is highly desirable.14 Substantial and fairly rapid regression of amyloid occurs in about one half of patients whose underlying acute phase response is controlled.12 However the rate of amyloid regression is highly variable, and there is a poor correlation between the quantity of amyloid and the degree of resulting organ dysfunction.
Similarly, in AL amyloidosis, chemotherapy regimens that reduce monoclonal immunoglobulin light chain production have been used with some success.15 Our preliminary studies suggest that more intensive regimens give better response rates,16 and in the present series, regression of hepatic amyloid was seen earlier and more frequently in patients treated aggressively.
Hepatic failure is an unusual cause of death in patients with amyloidosis of the liver. It is most commonly due to intrahepatic cholestasis26 although obstruction of the bile ducts by amyloid deposits has also been reported.27 Only four of our patients with AL disease died of hepatic insufficiency, and none of the AA patients died. The only patient described in the literature with AA amyloidosis and hepatic failure had a primary hepatocellular carcinoma,28 a condition known to cause an acute phase response.29 In AL amyloidosis, projected median survival with intensive cytotoxic chemotherapy is more than four years16 and liver transplantation may be a useful supportive measure in patients who develop hepatic failure before they have responded to treatment. Spontaneous hepatic rupture is a very rare complication of amyloidosis and has only been reported in six previous cases.30-32
In conclusion, hepatic amyloid deposition is a dynamic and reversible process that is always associated with amyloidosis in other tissues. Although it is common in patients with systemic amyloidosis, severe clinical effects are unusual and liver failure rarely occurs. The outcome can be improved by treatment that reduces production of the respective fibril precursor proteins. Supportive liver transplantation may be appropriate in selected cases.
Acknowledgments
This work was partly supported by MRC Programme Grant G7900510 to MBP and PNH. LBL is supported by an MRC Research Training Fellowship.