

Abstract
Radiolabeled compounds used for brain imaging with PET must readily cross the blood–brain barrier (BBB) to reach their target. Efflux transporters at the BBB—P-glycoprotein (P-gp) and the breast cancer resistance protein (BCRP)—could limit their uptake by the brain. Methods: We developed and validated an in vitro model using MDCKII cells transfected with human multidrug resistance (MDR1) or BCRP genes and assessed the transport of selected PET ligands by the concentration equilibrium technique. The tested compounds included befloxatone, (R,S)-CGP-12177, clorgyline, R-(−)-deprenyl, diprenorphine, DPA-714, fallypride, flumazenil, 2-fluoro-A-85380, LBT-999, loperamide, p-MPPF, PE2I, Pittsburgh compound B (PIB), (R,S)-PK11195, raclopride, R-(+)-verapamil, and WAY-100635. The assays were performed using the nonradioactive form of each compound (ultraviolet high-performance liquid chromatography analysis) and, when available, the 18F-labeled analogs (γ-counting). Results: Befloxatone appeared to be transported solely by BCRP. Loperamide, verapamil, and diprenorphine were the only P-gp substrates. Other ligands were transported by neither P-gp nor BCRP. Conclusion: The present method can readily be used to screen new-compound transport by P-gp or BCRP, even before any radiolabeling. Compounds that were previously thought to be transported by P-gp in rodents, such as p-MPPF, WAY-100635, and flumazenil, cannot be considered substrates of human P-gp. The impact of BCRP and P-gp at the BBB on the transport of befloxatone and diprenorphine in vivo remains to be evaluated with PET.
- P-glycoprotein
- breast cancer resistance protein
- blood–brain barrier
- positron emission tomography
- radiotracers
- ABC proteins
All drugs that act on the central nervous system must cross the blood–brain barrier (BBB) to reach the brain parenchyma. It is now well established that there are several carrier-mediated systems that interact with many xenobiotics at the BBB (1). These BBB transporters can affect the rate and extent of central nervous system drug distribution and influence their pharmacodynamics. Similarly, the way in which central nervous system radiotracers interact with BBB transporters may produce differences in their distribution between species or even individuals.
The adenosine triphosphate–binding cassette (ABC) efflux transporters—P-glycoprotein (P-gp: ABCB1, multidrug resistance [MDR1]) and breast cancer resistance protein (BCRP: ABCG2)—are now considered to influence the distribution of many structurally unrelated substrates within the central nervous system (1). Some genetic variants of the ABCB1 and ABCG2 genes are believed to influence the pharmacokinetics of their substrates, as reported for the opiates methadone (2) and fentanyl (3), which are putative P-gp substrates, whose distributions may be influenced by P-gp activity (4). Regional variations in the function of P-gp at the BBB of individuals with neurologic disorders such as epilepsia, Parkinson disease, and Alzheimer disease (1,5) may occur. These variations may account for some variability in brain imaging: several PET ligands have been identified as P-gp substrates, and their crossing of the BBB is therefore controlled by an active efflux kinetic process (6). Some of these drugs, such as R-(+)-verapamil, loperamide, and its nonopioid derivative N-desmethyl-loperamide, have been specifically used in PET studies to determine the physiologic impact of P-gp (5).
The U.S. Food and Drug Administration has now made it mandatory that the P-gp substrate status of licensed drugs be evaluated (7). Variations in the transport of a drug and potential drug–drug interactions at the BBB can significantly influence the distribution of a drug. Most studies on the carrier-mediated transport of radiotracers across the BBB are performed on rodents using chemical inhibition or physical disruption of the P-gp or Bcrp transporter genes (8). However, some differences in the spectra of ABC substrates of the human and rodent isoforms of P-gp indicate that models of human ABCs should be used (9,10). A few studies have been done on humans, but they require expensive, time-consuming experimental protocols using cyclosporine A or newer-generation P-gp inhibitors (11,12). These technical issues readily explain why the interactions between clinically useful PET radiotracers and human P-gp are poorly documented. BCRP was discovered more recently, and its impact at the human BBB remains to be assessed (13).
The present study characterized the transport by human P-gp and human BCRP of several ligands that are routinely radiolabeled for PET. These compounds were tested using both their nonradioactive form and, when available, their 18F-radiolabeled analogs in a concentration equilibrium assay using Madin–Darby canine kidney II (MDCKII) cells transfected with the hMDR1 or hBCRP genes.
MATERIALS AND METHODS
Chemicals
The chemical structures of tested compounds are presented in Figure 1. Befloxatone, R-(−)-deprenyl, and (R,S)-PK11195 ((R,S)-N-methyl-N-(1-methylpropyl)-1-(2-chlorophenyl)isoquinoline-3-carboxamide) were gifts from Sanofi-Aventis, (R,S)-CGP-12177 (4-[3-[(1,1-dimethylethyl)amino]2-hydroxypropoxy]-1,3-dihydro-2H-benzimidazol-2-one hydrochloride) was from Ciba, flumazenil from Roche, and PSC833 (valspodar) from Novartis. Clorgyline, fumitremorgin C, loperamide, p-MPPF (4-(2′-methoxyphenyl)-1-[2′-[N-(2″-pyridinyl)-p-flourobenzamido]ethyl]piperazine), prazosin, raclopride, R-(+)-verapamil, and WAY-100635 (N-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-N-(2-pyridyl)cyclohexanecarboxamide) were all purchased from Sigma-Aldrich; diprenorphine from Reckitt and Colman; and fallypride (N-([(2S)-1-(2-propenyl)-2-pyrrolidinyl]methyl)-5-(3-fluoropropyl)-2,3-dimethoxybenzamide) and Pittsburgh compound B (PIB) from ABX GmbH. DPA-714 (N,N-diethyl-2-(2-(4-(2-fluoroethoxy)phenyl)-5,7-dimethylpyrazolo[1,5-α]pyrimidin-3-yl)acetamide) (14), 2-fluoro-A-85380 (2-fluoro-3-[2(S)-2-azetidinylmethoxy]pyridine) (15), LBT-999 (8-((E)-4-fluoro-but-2-enyl)-3β-p-tolyl-8-aza-bicyclo[3.2.1]octane-2β-carboxylic acid methyl ester) (16), and PE2I ((E)-N-(3-iodoprop-2-enyl)-2β-carbomethoxy-3β-(4′-methylphenyl)nortropane) (17) were synthesized in-house according to published methods.

Chemical structure of PET ligands tested on concentration equilibrium assay using MDCKII-MDR1 and -BCRP cells.
Radiochemicals
3H-prazosin and 3H-raclopride were purchased from Perkin-Elmer. Over 99% radiochemically pure 18F-labeled DPA-714, fallypride, 2-fluoro-A-85380, and LBT-999 were prepared from cyclotron-produced 18F-fluoride (Cyclone-18/9 cyclotron; IBA) on the basis of already published standard conditions (18F-DPA-714 (14), 18F-fallypride (18), 2-18F-fluoro-A-85380 (15), and 18F-LBT-999 (16)) using a commercially available TRACERLab FX-FN synthesizer (GE Healthcare). All preparations included a final high-performance liquid chromatography (HPLC) purification (performed on semipreparative X-Terra RP18 [Waters], SunFire C-18 [Waters], ZorbaxSB [Hewlett Packard], and Symmetry C18 [Waters] HPLC columns, respectively) and a formulation for intravenous injection (using a SepPakPlus C-18 cartridge [Waters]–based removal of the HPLC solvents). Quality controls were performed on an aliquot of the ready-to-inject preparations, in compliance with our in-house quality control and quality assurance specifications.
Cell Culture
MDCKII-hMDR1 and MDCKII-human BCRP cells were generous gifts from Alfred Schinkel (National Cancer Institute, The Netherlands). They were grown in Dulbecco modified Eagle medium (Invitrogen) containing 10% heat-inactivated fetal bovine serum, glutamine, a 100 U/mL concentration of penicillin, and a 100 μg/mL concentration of streptomycin (Invitrogen) at 37°C in a humidified atmosphere of 95% air/5% CO2 and passaged twice a week. The presence of transporters was assessed by Western blotting of whole-cell lysates (19).
Bidirectional Transport Study
Bidirectional transport was studied as previously described (19,20). Briefly, cells were seeded (3 × 105 cells/cm2) on 12-well Corning Costar Transwell polycarbonate membranes (ATGC) and grown to confluence (3 d). Preliminary checks, including Lucifer yellow permeation (marker of the paracellular route) and transepithelial resistance measurement, indicated that these culture conditions were optimal for producing a confluent monolayer (19). Transepithelial resistance was determined using a Millicell ERS ohm-meter (Millipore) before and after incubation to ensure monolayer integrity. Monolayers with a transepithelial resistance of less than 200 Ω·cm2 were discarded. On the day of the experiment, the culture medium was replaced by Hanks balanced salt solution buffer containing CaCl2 and MgCl2 supplemented with 1 mM pyruvate and 10 mM N-2-hydroxyethylpiperazine-N-2-ethanesulfonic acid (Invitrogen) in both the apical compartment (compartment A, 0.5 mL) and the basolateral compartment (compartment B, 1.5 mL), with or without a transport inhibitor, and preincubated for 1 h. P-gp was inhibited with 5 μM PSC833, and BCRP with 10 μM fumitremorgin C. Permeation in both directions, apical to basolateral (A to B) and basolateral to apical (B to A), was measured. Transport medium containing the compound to be tested, with or without specific inhibitor, was then placed in the donor compartment. p-MPPF (10 μM) was used to ensure that the concentration in the opposite compartment was above the limit of quantification of the analytic procedure. Monolayer efflux was stopped after 90 min. The concentrations of tested compound in the receiver compartment were determined. The apparent permeability coefficient Papp (nm·s−1) is determined from the amount of compound cumulating in the receiver compartment over time (dQr/dt; μmol·s−1) according to the following equation:
Concentration Equilibrium Assay
The concentration equilibrium assay was performed according to previous studies (21) using the same cell culture and seeding conditions as the bidirectional transport assay. The preincubation buffer was removed by aspiration and replaced in both compartments by the same buffer containing the test compound (5 μM), with or without inhibitor. The dual P-gp and BCRP substrate 3H-prazosin (∼3.7 kBq/mL) was added to the solution as an internal positive control. The 18F-radiolabeled analogs of some of the screened compounds were also tested at about 11 kBq/mL when they were available. Plates were then placed at 37°C for 3 h under a humidified atmosphere and gentle shaking to allow polarized P-gp– or BCRP-mediated transport through the cell monolayer. The concentrations in the apical and basolateral compartments were then determined at the end of incubation.
Analytic and β- or γ-Counting Procedures
Nonradioactive compounds were assayed by ultraviolet HPLC analysis; the radioactivities of their β− or β+ radiolabeled analogs were counted. The analytic methods used are summarized in Table 1.
Analytic Methods to Determine Concentration of Test Compounds in Incubation Buffer
A Cobra Quantum D5003 counter (Perkin-Elmer) was used for γ-counting, and tritium radioactivity was measured in a Tri-Carb 2250CA analyzer (Packard) and Ultima-Gold scintillation cocktail (Perkin-Elmer) 2 d later, to allow β+ decay when 18F-radiolabeled compounds were tested.
For ultraviolet HPLC, samples of the initial incubation solution, apical and basolateral compartments, were centrifuged and the supernatant injected directly onto the Alliance chromatographic system, which consisted of a Symmetry C-18 column, 4.6 × 150 mm (5 μm), coupled to a 2996 photodiode array detector (Waters). The mobile phase flow rate was set at 1 mL·min−1. The separation and detection parameters of each compound are described in Table 1.
Condition A: gradient elution from T0 min (A/B = 90/10) to T5 min (A/B = 10/90) − eluent: A = 99.95/0.05 water/formic acid (v/v); B = 99.95/0.05 acetonitrile/formic acid (v/v).
Condition B: isocratic elution – eluent: 89.95/10/0.05 water/acetonitrile/formic acid (v/v/v).
Condition C: gradient elution from T0 min (A/B = 90/10) to T10 min (A/B = 10/90) − eluent: A = 99.95/0.05 water/formic acid (v/v); B = 99.95/0.05 acetonitrile/formic acid (v/v).
Condition D: isocratic elution – eluent: 64.97/34.98/0.05 acetonitrile/water/formic acid (v/v/v).
Concentration was calculated on the basis of a calibration curve (0–20 μM) performed for each compound, plotting ultraviolet HPLC peak area as a function of the concentration. Final concentrations in the concentration equilibrium assay are expressed as a percentage of the initial concentration C0 determined experimentally. Polarized transport was identified when the concentration in the apical compartment at the end of incubation was significantly greater than that in the basolateral compartment using unpaired 2-tailed Student t tests (n = 3–6 for each experiment). The threshold of significance was set at a P value of less than 0.05. Any polarized transport had to be abolished by P-gp or BCRP inhibitors to ensure its specificity.
RESULTS
Bidirectional transport was assessed to determine the permeability of p-MPPF through MDCKII-MDR1 cells: Papp (A to B) = 313 ± 15 nm·s−1 and Papp (B to A) = 248 ± 11 nm·s−1 in the absence of inhibitor (efflux ratio = ∼0.8). The p-MPPF efflux ratio was not reduced by the P-gp inhibitor PSC833, in contrast to that of the typical substrate prazosin (efflux ratio = 2.1 and 1.0, without and with inhibitor, respectively).
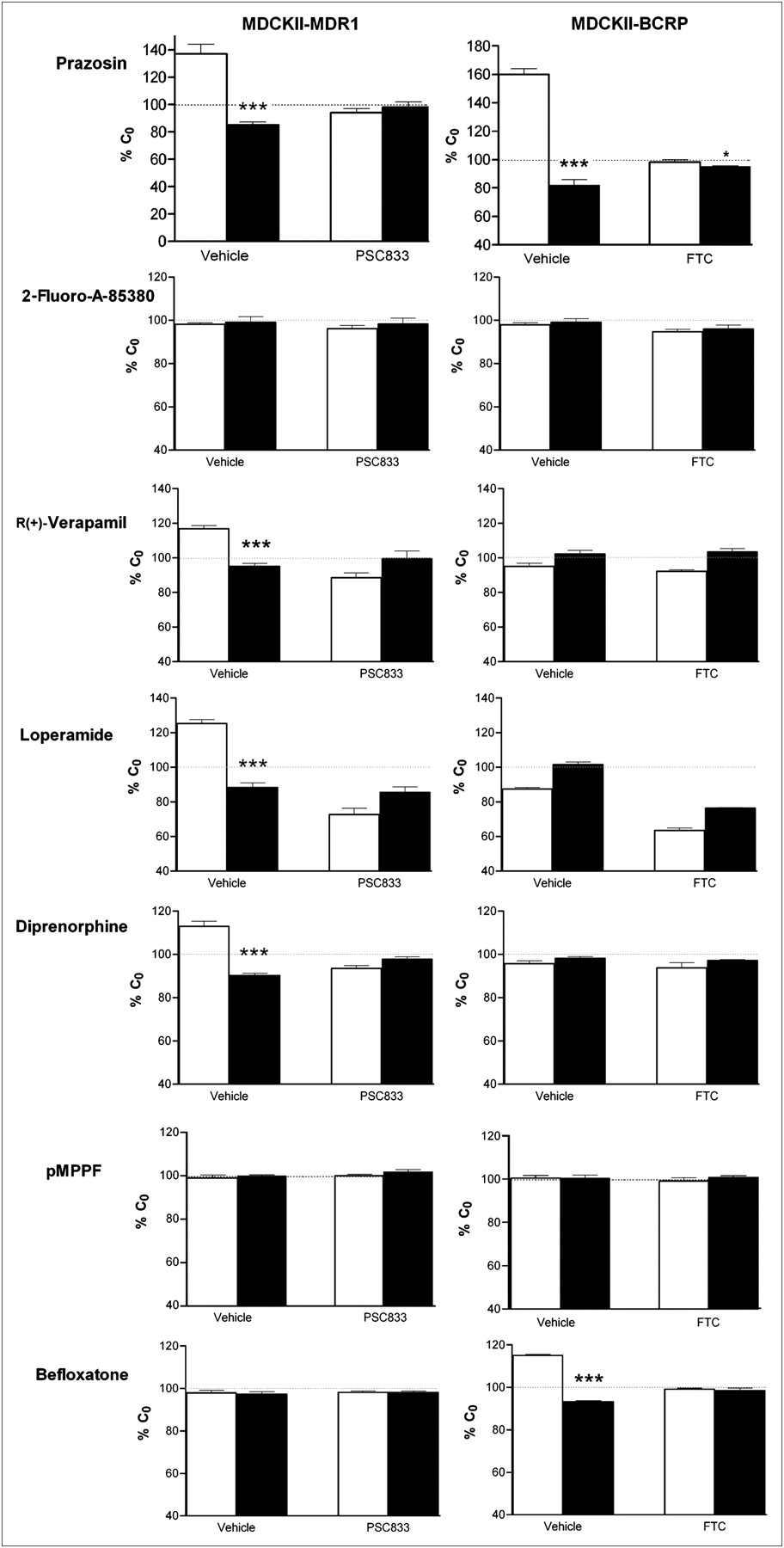
The functionality of the concentration equilibrium assay was checked by the time-dependent transport of prazosin through the MDR1 and BCRP cell monolayers, indicated by an increased concentration in the apical compartment and a simultaneous decrease in the basolateral compartment, which was significant after 120 min of incubation (Fig. 2), as previously reported (21). We therefore used an incubation time of 180 min (Figs. 2 and 3). The polarity of prazosin transport remained slightly significant in BCRP cells in the presence of the BCRP inhibitor fumitremorgin C, suggesting the presence of basal endogenous canine P-gp transport activity, as shown for an MDCKII clone (22).

Percentage ± SD of initial concentration (C0) vs. time of dual P-gp and BCRP substrate prazosin in apical compartment (compartment A) and basolateral compartment (compartment B) using concentration equilibrium assay performed on MDCKII cells transfected with human MDR1 (top) and BCRP (bottom) without (closed symbols) or with (open symbols) transport inhibitor. *P < 0.05. ***P < 0.001.

{kind=link}
{kind=link}
{kind=link}
Percentage ± SD of initial concentration (C0) obtained in apical compartment (compartment A, white columns) and basolateral compartment (compartment B, black columns) at end of incubation after concentration equilibrium assay performed using MDCKII-MDR1 (left) and BCRP cells (right) with or without inhibitor. *P < 0.05. ***P < 0.001.
There was no significant P-gp– or BCRP-mediated transport of CGP-12177, clorgyline, deprenyl, DPA-714, fallypride, flumazenil, 2-fluoro-A-85380, LBT-999, p-MPPF, PE2I, PIB, PK11195, raclopride, or WAY-100635 (Table 2). Diprenorphine, loperamide, and R-(+)-verapamil were confirmed as P-gp substrates. The concentrations of R-(+)-verapamil and loperamide were higher in compartment B than in compartment A at the end of incubation in the absence of functional P-gp, suggesting the presence of another transport system that carries them in the opposite direction to the P-gp efflux. There was significant polarized transport of befloxatone through cells bearing BCRP (but not P-gp). This transport was completely inhibited by fumitremorgin C. None of the other compounds tested were BCRP substrates (Fig. 3).
Transport of PET Compounds by Human P-gp and BCRP Using In Vitro Concentration Equilibrium Assay with MDCKII Cells Transfected with MDR1 and BCRP Genes
DISCUSSION
We tested several PET compounds to determine their transport by P-gp and BCRP using a sensitive, human-specific in vitro assay (Fig. 1; Table 2). Some of our findings on the role of P-gp in the transport of some PET radioligands differ from those previously published, possibly because the previous in vivo PET experiments were performed on rodents. We also suggest that the newly described BCRP is involved in the transport of some compounds.
In vitro bidirectional transport studies across epithelial cells transfected with the human MDR1 gene are widely used for nonclinical drug studies (7). The Food and Drug Administration considers these techniques to be a reference tool for testing the transport of new chemical entities by P-gp, as species-to-species extrapolation of the interactions of compounds with P-gp is hazardous in vivo (10). The correlation between the interaction of many compounds in vitro with human MDR1 and mouse Mdr1a is poor (9,23), and recently published data suggest that there are differences between rodents and humans in the transporter profiles of the brain capillary endothelial cells forming the BBB (24). Thus, a specific human model is needed to clarify the P-gp substrate status of the drugs, so as to prevent any drug–drug interaction that could occur in clinical practice and variability in the brain distribution due to variations in transporter expression. However, bidirectional transport studies may fail to identify poor P-gp substrates, especially drugs that rapidly permeate biologic membranes (20), because the cell monolayer is exposed to a concentration gradient: molecular diffusion along this gradient should be greater than the transporter-mediated efflux. This was recently illustrated when conventional in vitro models failed to identify some antiepileptic drugs as P-gp substrates (21). In the concentration equilibrium assay, the drug is initially added at identical concentration to both sides of a polarized P-gp– or BCRP-overexpressing cell monolayer instead of being applied to either the apical or the basolateral side for studying bidirectional transport. This method allows active transport to be evaluated independently of the passive diffusion component and was shown to be more sensitive in demonstrating any carrier-mediated transport of highly permeable compounds (21). These findings justify the use of the concentration equilibrium assay to study the carrier-mediated transport of drugs with significant BBB permeability. This in vitro model also provides reliable specificity toward ABC transporter efflux. Polarized transport observed through selectively transfected MDR1 and BCRP cell lines can be characterized by measuring the effect of selective reversal agents. In this work, the nonimmunosuppressive cyclosporine derivative PSC833 was used as a P-gp inhibitor because of its negligible effect on human BCRP at 5 μM (25). In the same way, the fungal toxin fumitremorgin C (10 μM) was chosen for its specific inhibitory effect on human BCRP (26).
The P-gp– and BCRP-mediated transport of SPECT radiopharmaceuticals such as 99mTc-sestamibi, 99mTc-tetrofosmin, 99mTc-hexamethyl-propyleneamine oxime, and 123I-metaiodobenzylguanidine has been studied in vitro (27,28). However, most studies on the P-gp–mediated transport of compounds used for PET have been performed in vivo on rodents. An extensive study on rats tested 8 PET ligands; the enhanced brain uptake of 11C-flumazenil and 11C-donepezil after P-gp inhibition with cyclosporine A suggested that there was P-gp transport for these compounds, unlike 11C-PK11195 (29). Similarly, three 5-hydroxytryptamine receptor 1A radioligands—11C-(R)-(−)-RWAY (11C-2,3,4,5,6,7-hexahydro-1{4-[1[4-(2-methoxyphenyl)-piperazinyl]]-2-phenylbutyry}-1H-azepine) (30), 18F-MPPF (31), and 11C-WAY100635 (32)—were identified as P-gp substrates in rodents. However, the results obtained in monkeys were different; 11C-(R)-(−)-RWAY was not found to be a P-gp substrate after inhibition with tariquidar (33). Studies with the P-gp inhibitor DCPQ ((2R)-anti-5-{3-[4-(10,11-dichloromethanodibenzo-suber-5-yl)piperazin-1-yl]-2-hydroxypropoxy}quinoline trihydrochloride) showed that P-gp had no effect on the brain distribution of the cerebral cannabinoid receptor radioligand 11C-MePPEP in monkeys (34). However, the results of these in vivo inhibition studies must be interpreted carefully because the P-gp structure has multiple binding sites that have a significant impact on drug transport properties (35). Thus, P-gp should not be considered a transporter with a single site of broad substrate specificity that could simply be “switched off” by modulators. Interactions between inhibitor and substrate molecules involve complex allosteric interactions at distinct binding sites (36). Consequently, a lack of increase in brain uptake obtained with a P-gp modulator does not necessarily mean that there is no P-gp–mediated transport.
Some newly developed central nervous system radiotracers designed for histamine H3 receptor mapping were recently tested in an in vitro model of human P-gp to ensure that they did not interact with P-gp at a preclinical stage (37). However, there is a real need to determine how the compounds used for PET are transported by human P-gp and BCRP (6). Some of the compounds we have tested have been evaluated in conventional bidirectional transport studies. These studies showed that verapamil is a poorer P-gp substrate (efflux ratio of 1.7) than loperamide (efflux ratio of 9.9) (20,38). They also revealed that deprenyl and flumazenil are not P-gp substrates. Our concentration equilibrium assay studies confirmed these previous results, showing that deprenyl and flumazenil are not transported by human P-gp or BCRP. These data differ from those obtained in vivo in rats by Ishiwata (29), suggesting that P-gp influences the brain uptake of flumazenil. This difference could be due to interspecies variations in the P-gp–mediated transport of flumazenil. p-MPPF and WAY-100635 are also considered to be P-gp substrates (increased uptake after P-gp inhibition) in rodents. However, to our knowledge, the transport of p-MPPF by human P-gp has never been evaluated either in vitro or in vivo. Our results obtained using the conventional bidirectional assay and the more sensitive concentration equilibrium assay suggest that p-MPPF is not transported by human P-gp. Likewise, WAY-100635 is not influenced by human P-gp in vitro.
The respective transport of verapamil enantiomers has already been described and has shown no evidence of stereoselectivity (39). However, 11C-(R)-verapamil, which is less metabolized and has a lower affinity for the calcium channel, is usually preferred over the racemate for P-gp activity imaging. In the same way, some screened compounds are preferentially—for PET purposes—used as enantiomers instead of racemate (i.e., (R)-PK11195 and (S)-CGP-12177). Racemate solutions were tested in this screening for these 2 compounds. If an active transport has been found, a further investigation would have been undertaken to evidence a putative enantioselectivity, as described for (R)- and (S)-methadone (19). However, our results show that none of the PK11195 or CGP-12177 enantiomers were transported by P-gp or BCRP, which gives reliable information for whenever the racemic mixture or an enantiomer is used for PET.
Diprenorphine was found as a P-gp substrate with the concentration equilibrium assay, whereas conventional bidirectional studies failed to reveal this transport using Caco-2 cells (40). The polarized transport of diprenorphine through Caco-2 cells (efflux ratio, ∼2) was attributed to a transporter other than P-gp by Hassan et al. (40), because it was not inhibited by the dual P-gp/BCRP inhibitor GF120918 (N-(4-[2-(1,2,3,4-tetrahydro-6,7-dimethoxy-2-isoquinolinyl)ethyl]-phenyl)-9,10-dihydro-5-methoxy-9-oxo-4-acridine carboxamide) and verapamil. However, we found no polarized transport of diprenorphine in BCRP cells, in contrast to MDR1 cells, where it is inhibited by PSC833, suggesting a reliable P-gp transport specificity (Fig. 3).
Our findings raise the question of whether bias is introduced in vivo by the ABC protein–mediated efflux of PET tracers when used on humans (6). Recent publications suggest that Alzheimer disease could be linked to a loss of P-gp function at the BBB (1,5). 11C-PIB is a useful PET radiopharmaceutical for imaging Aβ deposits in Alzheimer disease. We find that PIB is not transported by P-gp or BCRP. Thus, the cerebral accumulation of PIB is probably not influenced by any gradual change in P-gp–mediated efflux. The consequence of inhibiting P-gp on the 11C-diprenorphine distribution volume remains to be evaluated in vivo. Hume et al. suggested that this could be a factor influencing variations in the brain uptake of 11C-diprenorphine (41). (R,S)-PK11195 has been found to be a potent inhibitor of the human P-gp and BCRP on multidrug resistant cells in vitro (42). However, (R,S)-PK11195 is not transported by P-gp in vitro (42) or in vivo (29), as confirmed by the present in vitro study.
We find that the monoamine oxidase A antagonist befloxatone is specifically transported by human BCRP and not by P-gp. Therefore, radiolabeled 11C-befloxatone could be a candidate for estimating the specific involvement of BCRP at the BBB. Further studies should focus on the brain distribution of befloxatone in the presence of a BCRP inhibitor in vivo. The bias introduced by the BCRP-mediated transport on the brain kinetic parameters of 11C-befloxatone could be evaluated in baboons and humans using previously described quantification methods (43).
CONCLUSION
We have used a specific, sensitive in vitro model to show that several PET compounds, including p-MPPF, WAY-100635, and flumazenil, are not transported by human P-gp or BCRP. Some of these results do not agree with data obtained in rodents, showing the importance of using specific human in vitro models. The impact of BCRP and P-gp on the kinetics of their respective substrates 11C-befloxatone and 11C-diprenorphine remains to be established in vivo to evaluate the influence of this active transport on the quantification performed in preclinical and clinical protocols. The method described here can readily be used to screen new-compound transport by P-gp or BCRP, even before any radiolabeling. The method can also be useful for identifying new radiotracer candidates designed to interact specifically with these 2 major transporters.
Acknowledgments
This work was supported by a grant from the CEA (Commissariat à l’énergie atomique et aux énergies alternatives) and the AP-HP (Assistance Publique–Hôpitaux de Paris). The English text was edited by Dr. Owen Parkes.
- © 2011 by Society of Nuclear Medicine
REFERENCES
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.
- 12.
- 13.
- 14.
- 15.
- 16.
- 17.
- 18.
- 19.
- 20.
- 21.
- 22.
- 23.
- 24.
- 25.
- 26.
- 27.
- 28.
- 29.
- 30.
- 31.
- 32.
- 33.
- 34.
- 35.
- 36.
- 37.
- 38.
- 39.
- 40.
- 41.
- 42.
- 43.
- Received for publication May 28, 2010.
- Accepted for publication August 6, 2010.