

Abstract
Pretargeted radioimmunotherapy (PRIT) with bispecific antibodies in combination with a radiolabeled peptide reduces the radiation dose to normal tissues, especially the bone marrow. In this study, the optimization, therapeutic efficacy, and toxicity of PRIT of colon cancer with a 177Lu-labeled peptide was determined in mice with carcinoembryonic antigen (CEA)-expressing human tumors. Methods: To obtain the optimal therapeutic efficacy, several strategies were evaluated to increase the total amount of radioactivity targeted to subcutaneous LS174T colon cancer tumors in BALB/c nude mice. First, the maximum amount of bispecific anti-CEA and antihapten antibody TF2 and the peptide IMP288 that could be targeted was determined. Second, the tumor targeting of repeated administrations of radiolabeled IMP288 was investigated. Mice received 1 TF2 injection, followed by multiple IMP288 injections (3-h interval) or multiple cycles, with each IMP288 administration preceded by a new TF2 injection (72-h interval). PRIT was administered at maximum doses of TF2 and 177Lu-labeled IMP288 in groups of 9 mice with subcutaneous LS174T tumors. Mice received 1, 2, or 3 successive cycles of treatment (26 MBq/mouse/cycle) or carrier only. The primary endpoint was survival; secondary endpoints were tumor growth, body weight, bone marrow, and renal toxicity. Results: The highest amount of radioactivity delivered to a subcutaneous colon tumor was achieved by the administration of 5.0 nmol of TF2 and 0.28 nmol of IMP288 in 3 successive cycles, with each IMP288 preceded by a new TF2 injection (72-h interval). PRIT effectively delayed tumor growth and prolonged survival significantly. Higher activity doses, administered in successive cycles, correlated with longer survival: the median survival of untreated mice was 13 d (range, 6–20 d), whereas that of mice treated with 1, 2, or 3 cycles of PRIT was 24 (range, 24–31 d), 45 (range, 38 ≥ 130 d), and 65 (range, 48 ≥ 130 d) days, respectively. Toxicity was limited: no significant changes in mean body weight were measured. Minimal changes in leukocyte counts were measured at 2 and 3 wk after injection, with full recovery within 7 wk after treatment. Platelet counts were unaffected. Serum creatinine levels were not increased significantly; thus, there was no indication of acute renal toxicity. Conclusion: This study indicates that PRIT in mice is an effective treatment modality against colon cancer, with limited toxicity.
- radioimmunotherapy
- pretargeting
- bispecific antibody
- colonic cancer
- mice
The survival of patients with colorectal cancer depends mainly on the development of distant metastases. In patients with metastasized disease, chemotherapy with capecitabine, oxaliplatin, and irinotecan used in combination protocols prolongs median overall survival from 8.0 to 11.7 mo (1). The addition of bevacizumab to those chemotherapy schedules increases median overall and progression-free survival by 3.7–4.7 mo (2), resulting in an overall survival of more than 20 mo in patients with metastatic disease (3).
Novel systemic strategies are needed for patients who become refractory or intolerant to these treatments. Radioimmunotherapy, the selective targeting of tumor-associated antigens expressed on the tumor cells with radiolabeled antibodies, is an attractive candidate. This concept has been investigated extensively and has proven to be effective in patients with non-Hodgkin lymphoma (4–7), leading to the approval of 2 radiolabeled anti-CD20 monoclonal antibody preparations: 90Y-ibritumomab tiuxetan (Zevalin; Bayer Schering Pharma AG) and 131I-tositumomab (Bexxar; GlaxoSmithKline). Although radioimmunotherapy has not been as successful in solid tumors, presumably because these tumors are less radiosensitive, an 131I-labeled anti-CEACAM5 IgG given to colorectal cancer patients as an adjuvant therapy after the resection of hepatic metastases improved survival, compared with a contemporaneous group of patients (8–9). The activity dose in radioimmunotherapy is limited by myelotoxicity as a result of the continuous radiation exposure of the red marrow to the slow-clearing antibody. Pretargeting techniques were developed to circumvent this problem (10). In pretargeting, an unlabeled bifunctional reagent, with affinity for the tumor and a small radiolabeled molecule, is administered to prelocalize in the tumor (11–13). In this approach, a bispecific monoclonal antibody (bsMAb) is administered intravenously and given time to accumulate in the tumor and clear from the circulation. Then, a radiolabeled hapten peptide is given that clears rapidly from the blood and body but is trapped in the tumor by the anti–hapten-binding arm of the bsMAb, greatly reducing the radiation dose to normal tissues, especially the bone marrow. The retention of the radiolabeled hapten peptide in the tumor is enhanced when 2 haptens are included in the hapten peptide (14).
In our pretargeting system, peptides are substituted with the hapten histamine-succinyl-glycine (HSG), creating a flexible pretargeting system, because these HSG-substituted peptides can be conjugated with various chelating moieties (diethylenetriaminepentaacetic acid, 1,4,7-tri-azacyclononane-N,N′,N″-triacetic acid (NOTA), DOTA, N3S chelates, etc.). Thus, the peptide can be radiolabeled stably with a variety of radionuclides, such as 111In and 99mTc for SPECT (13); 18F and 68Ga for PET (15–18); or 131I, 90Y, and 177Lu for pretargeted radioimmunotherapy (PRIT) (19).
Until recently, the bsmAbs used in pretargeting were produced either by chemical conjugation of Fab fragments or via the quadroma technology. A new method, the dock-and-lock (DNL) technology, has been described previously (20–21). Significant therapeutic responses with DNL constructs to target B-cell lymphoma and pancreatic cancer have been reported in animals with subcutaneous xenografts, using a 90Y-labeled di-HSG peptide (22–23). The DOTA-di-HSG peptide can also be stably labeled with 177Lu. Herein, we report the optimization, therapeutic efficacy, and toxicity of an anti–carcinoembryonic antigen (CEA) DNL bsmAb construct in combination with a 177Lu-labeled di-HSG peptide in mice bearing CEA-expressing tumors.
MATERIALS AND METHODS
Pretargeting Reagents TF2 and IMP288
The trivalent anti-CEACAM5 and anti-HSG bsmAb construct TF2 (molecular weight, 157 kDa) and IMP288 peptide (molecular weight, 1,456 Da) were provided by IBC Pharmaceuticals, Inc., and Immunomedics, Inc., respectively. The preparation and binding properties of TF2 have previously been described (21,24–26). Size-exclusion chromatography showed that TF2 could bind more than 90% of the added 111In-IMP288 peptide.
IMP288 was synthesized and purified as described by McBride et al. (16).
In some studies, 125I-TF2 (0.4 MBq) was coinjected with unlabeled TF2 to confirm tumor accretion. IMP288 was labeled with either 111In for biodistribution studies or with 177Lu for radioimmunotherapy. Mice received TF2 and IMP288 intravenously in 0.2–0.3 mL of phosphate-buffered saline (PBS) and 0.5% bovine serum albumin.
TF2 was radioiodinated with a trace amount of 125I (Perkin Elmer) by the IODO-GEN (Pierce) method, and 125I-labeled TF2 was purified as described previously (27).
IMP288 was labeled with 111In (Covidien) at a specific activity of 32 MBq/nmol and with 177Lu (IDB Holland BV) at a specific activity of 86 MBq/nmol, and radiolabeling was performed as described previously (18).
The radiochemical purity of the radiolabeled TF2 and IMP288 preparations was determined using instant thin-layer chromatography on silica-gel strips (Pall Life Sciences). The percentage of radiolabeled TF2 was determined using 0.1 M citrate buffer, pH 6.0, as the mobile phase. To determine the percentage of radiolabeled IMP288, 0.1 M NH4Ac:0.1 M ethylenediaminetetraacetic acid was used as the mobile phase.
Furthermore, 111In-IMP288 and 177Lu-IMP288 were analyzed by reversed-phase high-performance liquid chromatography as described previously (18).
The radiochemical purity of the 125I-TF2 and 111In- and 177Lu-IMP288 preparations always exceeded 95%.
Animal Experiments
The experiments were performed in male nude BALB/c mice (age, 6–8 wk; weight, 20–25 g). Mice were acclimated to laboratory conditions for at least 1 wk before experimental use and were housed under nonsterile standard conditions in filter-topped cages with free access to animal chow and water. All studies were approved by the institutional Animal Welfare Committee of the Radboud University Nijmegen Medical Centre and conducted in accordance with their guidelines (revised Dutch Act on Animal Experimentation, 1997).
Tumor xenografts were induced by subcutaneous inoculation of 0.2 mL of a suspension of 1 × 106 LS174T cells, a CEA-expressing human colon carcinoma cell line (American Type Culture Collection). TF2 and IMP288 were injected intravenously in 0.2–0.3 mL, with an interval of 16 h, because this period was shown to be sufficient to clear TF2 from the circulation, to allow for IMP288 tumor targeting without significant complexation in the circulation.
Biodistribution Studies
In this study, the maximum amount of IMP288 that could be captured by LS174T tumors (0.02–0.2 g) was assessed. First, the optimal TF2 dose that could bind the maximum amount of IMP288 in the tumor was determined. Groups of 5 tumor-bearing nude mice were injected intravenously with 1.3, 2.5, 5.0, or 10.0 nmol of TF2 (∼0.2 to 1.57 mg), labeled with a trace amount of 125I (0.4 MBq), and 16 h later, 0.10 nmol of IMP288 was given. One hour after injection of IMP288, all mice were euthanized by CO2/O2 asphyxiation. Blood was obtained by cardiac puncture, and tumor and organs of interest were dissected, weighed, and counted in a γ-counter with standards prepared from the injected products, using appropriate energy windows for the radionuclide used. The percentage injected dose per gram of tissue (%ID/g) was calculated.
Subsequently, the maximum amount of IMP288 that could be captured under these pretargeting conditions was assessed. Therefore, groups of 5 mice received 5.0 nmol of TF2 intravenously, and 16 h later, 0.035, 0.070, 0.140, 0.280, or 0.410 nmol of IMP288 (50, 100, 200, 400, or 600 ng, respectively) was given, labeled with a trace amount of 111In (0.4 MBq). One hour later, all mice were euthanized for determination of tumor uptake and organ distribution.
In a third experiment, the accumulation and retention in the tumor of the maximum TF2 and IMP288 doses were investigated. Tumor-bearing nude mice received 5.0 nmol of 125I-TF2 (0.4 MBq) intravenously and 16 h later, 0.28 nmol of 111In-IMP288 (0.4 MBq). Groups of 5 mice were euthanized at 1, 6, 24, and 48 h for determination of tumor uptake and organ distribution.
The radioactivity of 177Lu that could be delivered was limited by the maximum peptide mass, because the maximum specific activity of 177Lu-IMP288 was 90 MBq/nmol. A single dose of 25 MBq of IMP288 (0.28 nmol), which was the maximum IMP288 dose that was specifically targeted to the tumor, was well below the maximum-tolerated radioactivity level. Therefore, our treatment strategy included the use of repeated treatment cycles. Biodistribution studies were performed to assess 2 different approaches. The first strategy was to administer multiple injections of the radiolabeled peptide after a single injection of the bsMAb. The tumor uptake was measured in 3 groups of 5 mice that received one 5.0-nmol dose of TF2, and then 16 h later, 1, 2, or 3 injections of 0.28 nmol of IMP288 were given, with each IMP288 injection separated by 3 h. In the multiply dosed groups, only the last dose of IMP288 was labeled with 111In.
The second strategy investigated whether subsequent injections of IMP288 could be targeted more efficiently by preceding each IMP288 with a new TF2 injection. Three groups of 5 mice received 1, 2, or 3 cycles of 5.0 nmol of TF2, combined 16 h later with 0.28 nmol of IMP288, with each cycle separated by 72 h. In the multiply dosed groups, the last dose of TF2 was labeled with 125I and the last dose of IMP288 was labeled with 111In. One hour after the injection of the 111In-labeled peptide, mice were euthanized and dissection and analyses were performed as described above.
PRIT Studies
The therapeutic efficacy of PRIT with TF2 and 177Lu-IMP288 was determined by random assignment of 9 mice with subcutaneous LS174T xenografts per group. Therapy studies were initiated at 10 d after tumor inoculation, when the median tumor size was 40 mm3 (range, 12–120 mm3). They received 1, 2, or 3 treatment cycles of TF2 (5.0 nmol) and 177Lu-IMP288 (26 MBq/0.28 nmol per cycle) or PBS and 0.5% bovine serum albumin, with each cycle separated by 3 d, because this regime resulted in the highest absolute amount of radiolabeled peptide in the tumors.
The primary endpoint was overall survival. The secondary endpoint was toxicity, with special attention to myelosuppression and acute renal toxicity; toxicity was evaluated by monitoring the general condition of the animals, body weight, blood cell counts, and creatinine levels. Animals were observed daily by independent, experienced biotechnicians who measured body weight and tumor size twice weekly. Standardized humane endpoints used to euthanize animals were failure to eat or drink and loss of 15% or more of body weight in 1 or 2 d or loss of 20% or more of their starting weight, or, in the case of tumor progression, a tumor size exceeding 1.0 cm3 or excessive ulceration of the tumor. Tumor size was measured in 3 dimensions (length, width, and height) with a caliper, and tumor volume was calculated, assuming tumors were ellipsoid, using the formula tumor volume (mm3) = 4/3π × (length/2) × (width/2) × (height/2).
Blood samples of 0.1 mL were collected via submandibular bleeding before therapy for baseline full-blood counts and serum creatinine and weekly starting at 2 wk after therapy. White blood cell counts and platelet counts were analyzed by the ADVIA 120 Hematology System (Siemens Medical Solutions Diagnostics). Serum creatinine levels were analyzed by Aeroset (Abbott Diagnostics). Timing of the white blood cell and platelet count measurement was scheduled to determine the nadir, at 2 and 3 wk after the first injection of 177Lu-IMP288, and bone marrow recovery at 7 wk after injection.
Statistic Analysis
All values are mean ± SD. Statistical analysis was performed using a nonparametric, 2-tailed Mann–Whitney test and Kruskal–Wallis test using SPSS software (version 16.0; SPSS Inc.). Survival curves were compared using the log-rank test. The level of significance was set at a P value of less than 0.05.
RESULTS
Determination of Maximum Peptide Dose
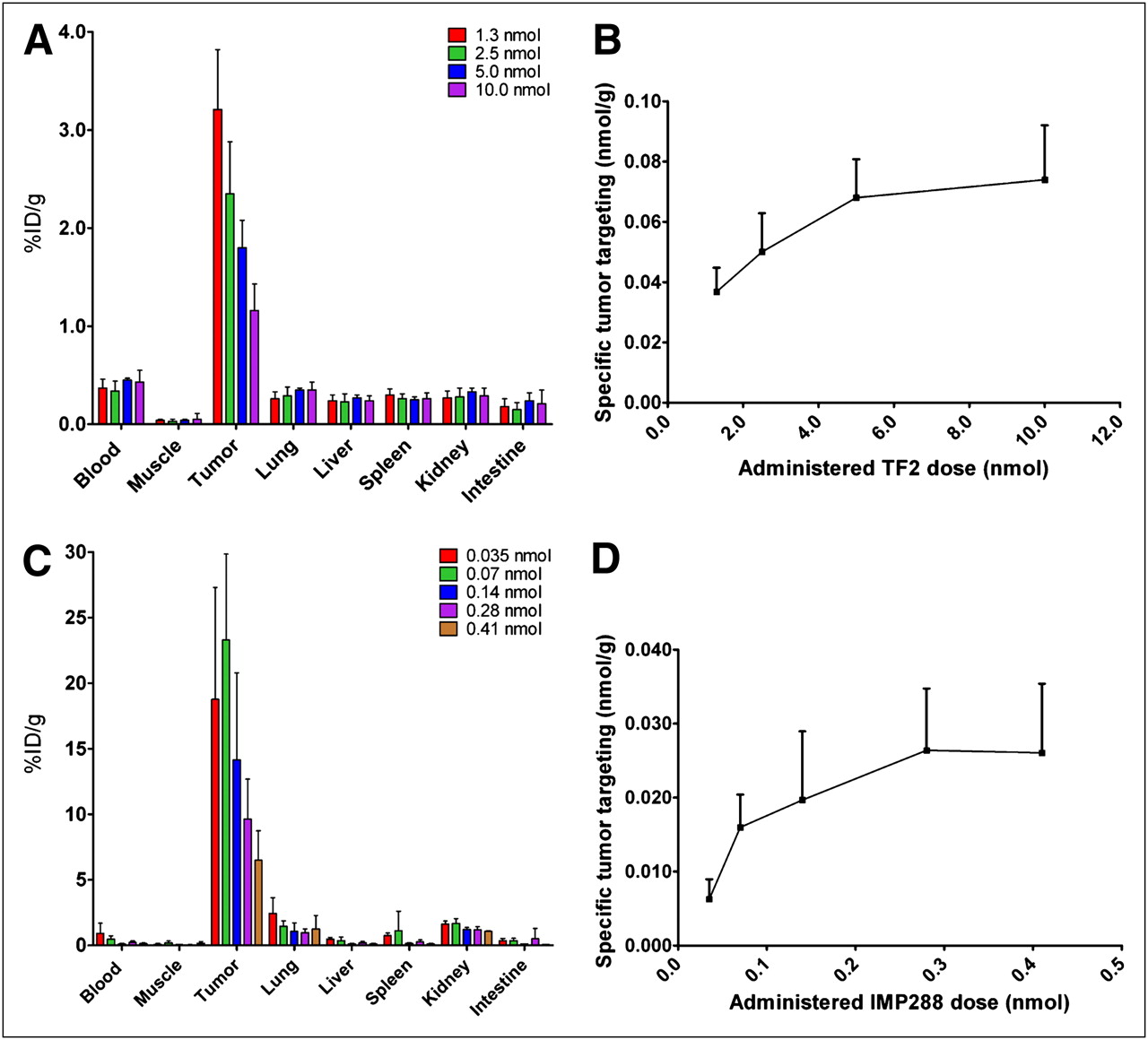
The maximum amount of TF2 and IMP288 that could be targeted specifically to LS174T tumors was determined. As shown in Figure 1A, the percentage uptake of TF2 in the tumor decreased from 3.21 ± 0.61 %ID/g at the 1.0-nmol dose to 1.16 ± 0.27 %ID/g at 10.0-nmol dose. Figure 1B illustrates the absolute amount of TF2 that accreted specifically in the tumor (subtracting the blood concentration), showing that the total amount in the tumor did not increase at administered doses greater than 5.0 nmol (i.e., antigen-saturating dose). Therefore, the 5.0-nmol dose was selected for further PRIT experiments.
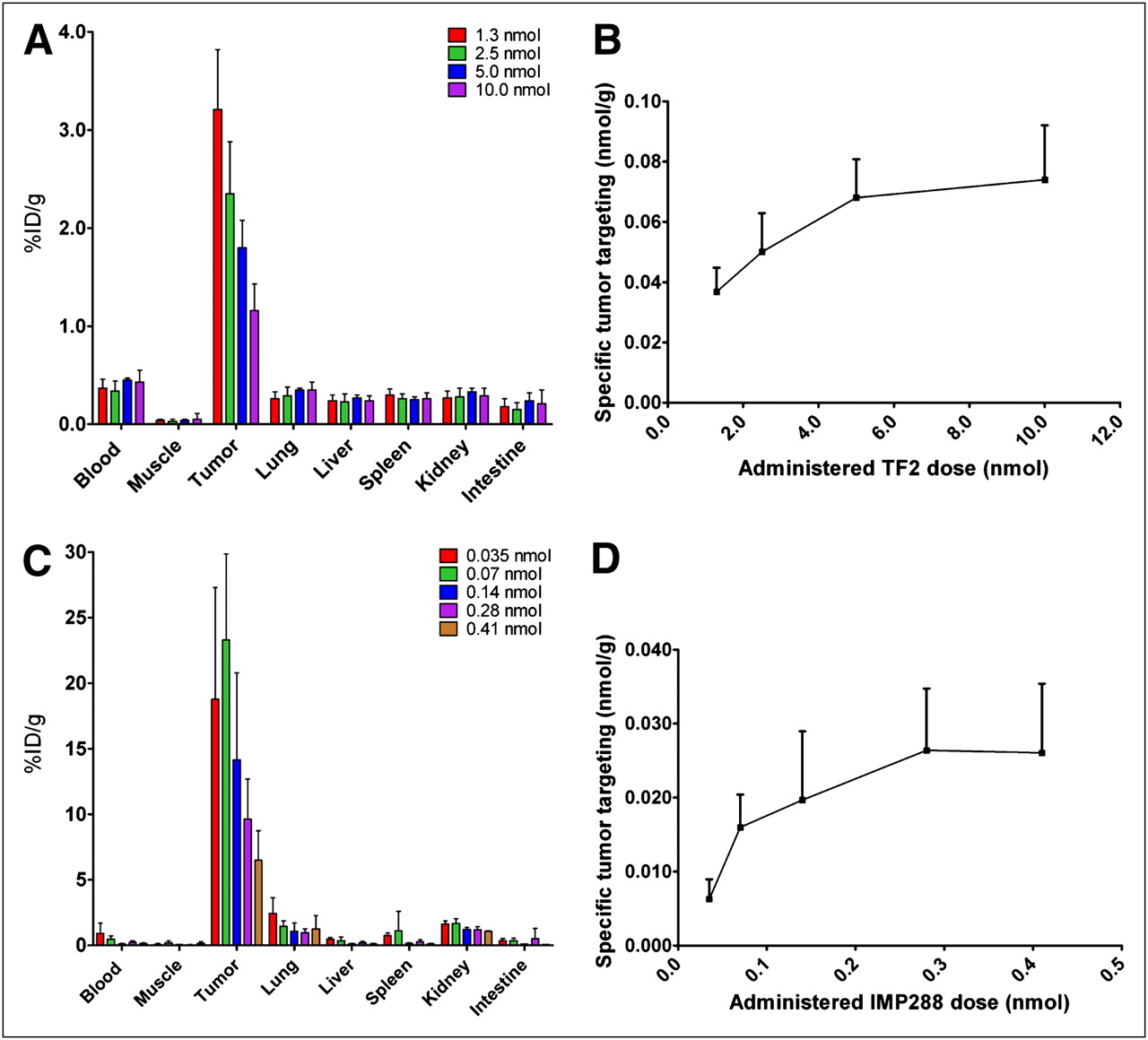
(A and B) Biodistribution and specific tumor targeting of escalating bsmAb doses (1.0–10.0 nmol of 125I-TF2, 0.4 MBq) in BALB/c nude mice with 0.02- to 0.2-g subcutaneous LS174T tumors. bsmAb was followed by injection of 0.10 nmol of IMP288 16 h later, and 1 h after that injection, mice were euthanized. (C and D) Biodistribution and specific tumor targeting of escalating peptide doses (0.035–0.41 nmol of 111In-IMP288, 0.4 MBq), injected 16 h after pretargeting with TF2 (5.0 nmol). Specific tumor targeting of 125I-TF2 and 111In-IMP288 was calculated as nmol per gram of tumor, corrected for blood concentration. Values represent mean ± SD (n = 5).
The maximum amount of IMP288 that could be captured in the tumor with this amount of TF2 was determined. At 1 h after injection, 111In-IMP288 accumulated effectively in the tumor at all peptide doses, with low uptake in all normal tissues. The highest tumor uptake in terms of %ID/g was obtained at the 0.035 and 0.070 nmol of IMP288 doses (18.8 ± 8.1 %ID/g and 23.3 ± 6.6 %ID/g, respectively, P = 0.33; Mann–Whitney exact test, 2-tailed), with decreasing uptake at higher IMP288 doses (14.2 ± 6.6 %ID/g, 9.6 ± 3.0 %ID/g, and 6.5 ± 2.2 %ID/g at 0.140, 0.280, or 0.410 nmol, respectively) (Fig. 1C). Most important, the absolute amount of peptide targeted to the tumors did not increase at injected peptide doses higher than 0.28 nmol of IMP288 (Fig. 1D). Therefore, this dose was selected for PRIT, because it would result in the highest amount of radioactivity in the tumor. For example, although 0.07 nmol of 177Lu-IMP288 would allow 23 %ID/g uptake, based on the specific activity of 90 MBq/nmol for 177Lu-IMP288, only 6.3 MBq of 177Lu activity could be administered, delivering 1.45 MBq to the tumor. However, with 0.28 nmol, even though the percentage uptake is lower (∼10 %ID/g), 25.2 MBq could be administered, delivering 2.5 MBq to the tumor.
Blood and Tumor Retention
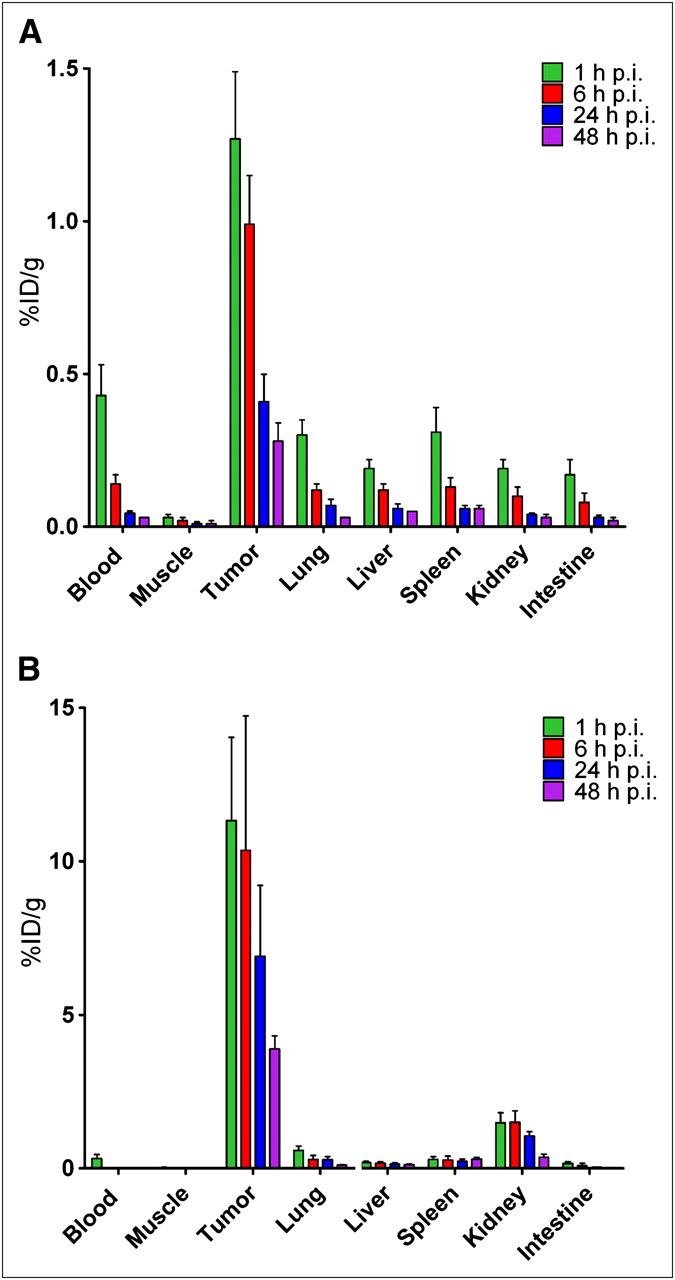
Tumor uptake and tissue distribution were examined over a 48-h period after an 111In-IMP288 injection using 5.0 nmol of TF2 and 0.28 nmol of IMP288 (Fig. 2). TF2 cleared rapidly from the blood and normal tissues, with less than 0.1% in the blood at 17 h after injection (1 h after 111In-IMP288), but with tumor uptake averaging 1.3 ± 0.22 %ID/g (Fig. 2A). At this same time, 111In-IMP288 tumor uptake was 11.3 ± 2.7 %ID/g, with the kidneys having the highest uptake among normal tissues, averaging 1.5 ± 0.3 %ID/g at this 1-h interval (Fig. 2B). Tumor uptake of TF2 and IMP288 gradually decreased over time at a somewhat similar rate. The tumor-to-blood ratio exceeded 1,400:1 at all times.

Accumulation and retention of 125I-TF2 (A) and 111In-IMP288 (B) in BALB/c nude mice with subcutaneous LS174T tumors. TF2 (5.0 nmol) and IMP288 (0.28 nmol) were injected, and animals underwent necropsy at 1, 6, 24, and 48 h after 111In-IMP288 injection. Values represent mean ± SD (n = 5). p.i. = after injection.
Successive Cycles of Therapy
The data from the previous study indicated that the maximum radioactivity dose of 177Lu-IMP288 that could be administered with 0.28 nmol of IMP288 was far below the expected maximum tolerated dose (MTD). Therefore, we evaluated the efficiency of successive doses of IMP288 (i.e., administering 1 dose of 5 nmol of TF2 and 16 h later 3-hourly 1, 2, or 3 111In-IMP288 (0.28 nmol) injections or 1, 2, or 3 cycles of 5 nmol of TF2, followed 16 h later by 0.28 nmol of 111In-IMP288 every 3 d).
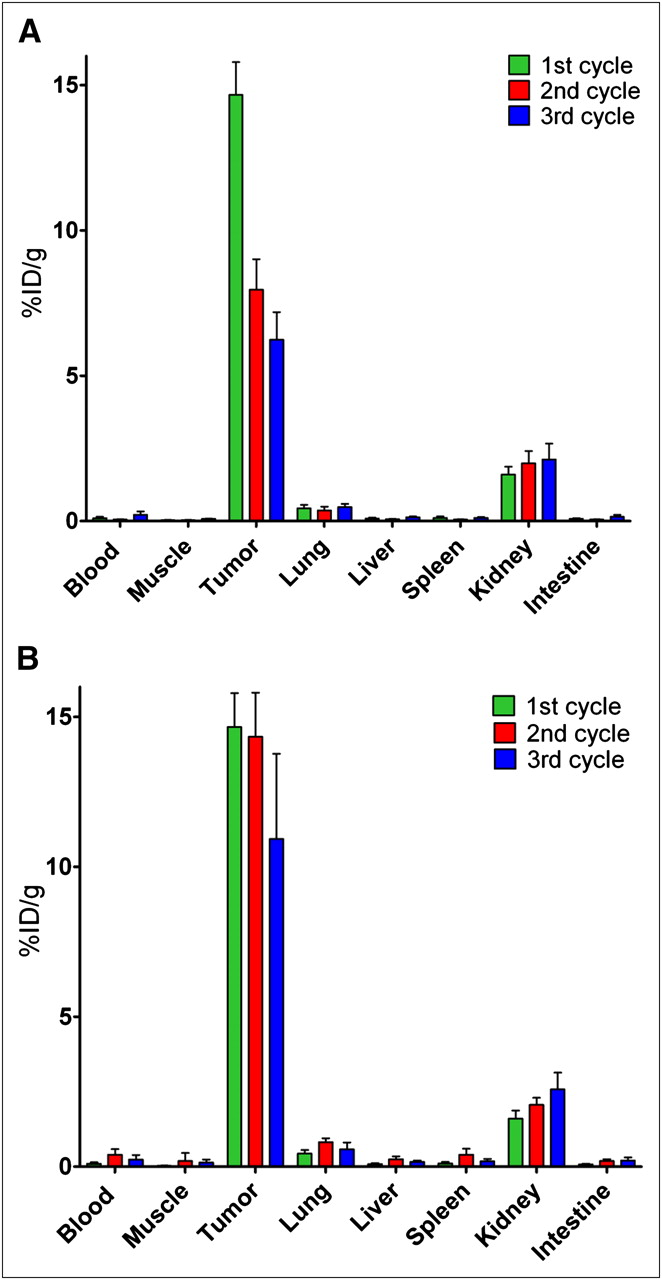
The successive administrations of 111In-IMP288 after 1 injection of TF2 resulted in significantly lower uptake of the second and third injections of peptide than of the first injection (8.0 ± 1.1 %ID/g and 6.2 ± 1.0 %ID/g vs. 14.3 ± 1.5 %ID/g, respectively; Mann–Whitney exact test, 2-tailed, P < 0.05) (Fig. 3A).
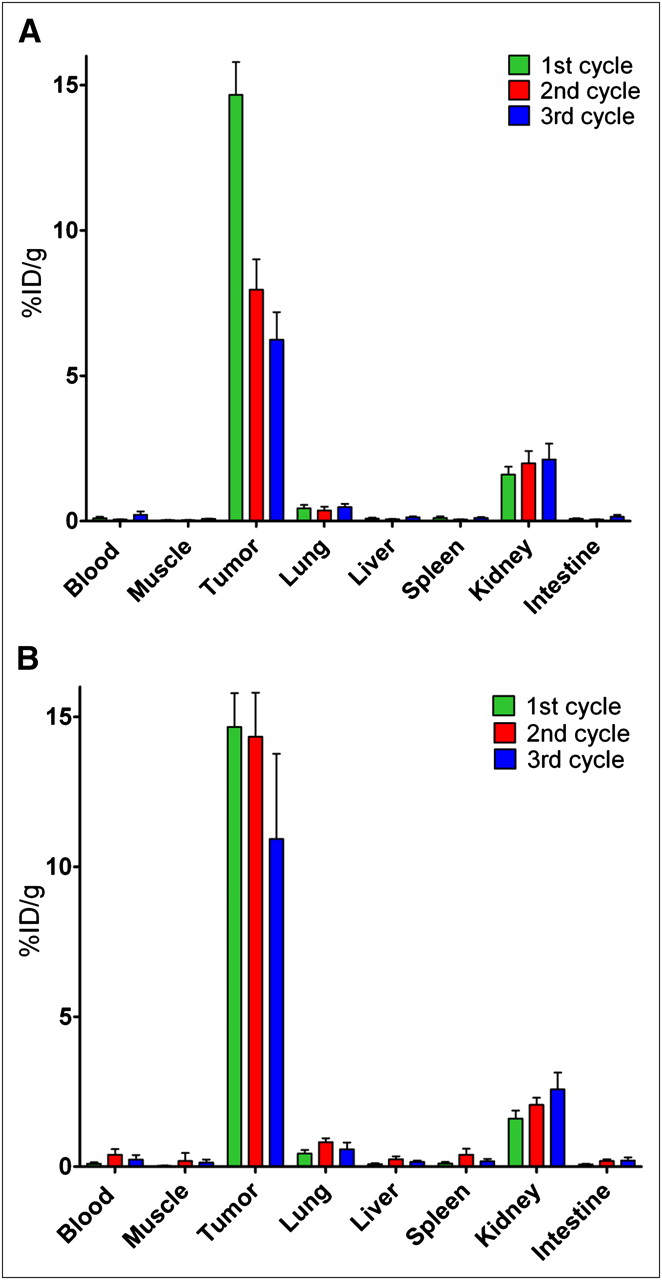
Biodistribution of successive 3-hourly administrations of 111In-IMP288 (A), and of successive 3-daily cycles of TF2 combined with 111In-IMP288 (B). (A) Three groups of 5 mice received 1 injection of 5.0 nmol of TF2 and 16 h later 1, 2, or 3 injections of 0.28 nmol of IMP288, with each IMP288 injection separated by 3 h. (B) Three groups of 5 mice received 1, 2, or 3 cycles of 5.0 nmol of TF2 and 0.28 nmol of IMP288, with each cycle separated by 72 h. In each group, last (or only) dose of IMP288 was labeled with 111In. One hour after IMP288 injection of labeled peptide, all mice were euthanized. Values represent mean ± SD (n = 5).
The administration of an extra second and third dose of TF2 before each successive dose of IMP288, with a 3-d interval, resulted in similar tumor accretion of the second and third injections of bsmAb to that seen with the first injection (2.6 ± 0.4 %ID/g, 2.0 ± 0.4 %ID/g, and 2.3 ± 0.1 %ID/g for the first, second, and third doses, respectively). Most important, in this regime the first and second doses of 111In-IMP288 had similar uptake (14.7 ± 1.1 %ID/g and 14.3 ± 1.5 %ID/g, respectively, P = 0.91, Mann–Whitney exact test, 2-tailed), but by the third cycle, the 111In-IMP288 uptake was slightly lower than with the first and second injections (10.9 ± 2.8 %ID/g, P = 0.018, Mann–Whitney exact test, 2-tailed) (Fig. 3B). Renal uptake also increased slightly with each progressive dose, but tumor-to-kidney ratios were always favorable (9.3 ± 1.3, 7.0 ± 1.0, and 4.3 ± 1.1 after the first, second, and third cycles, respectively). Thus, this latter approach was selected for therapeutic evaluation.
PRIT
PRIT in the treated group of mice, compared with the control mice, effectively delayed tumor growth (Fig. 4). Tumors in all untreated mice rapidly increased, whereas in the treated mice, the tumors did not start growing until days 14, 24, and 41 after 1, 2, and 3 cycles of PRIT, respectively.
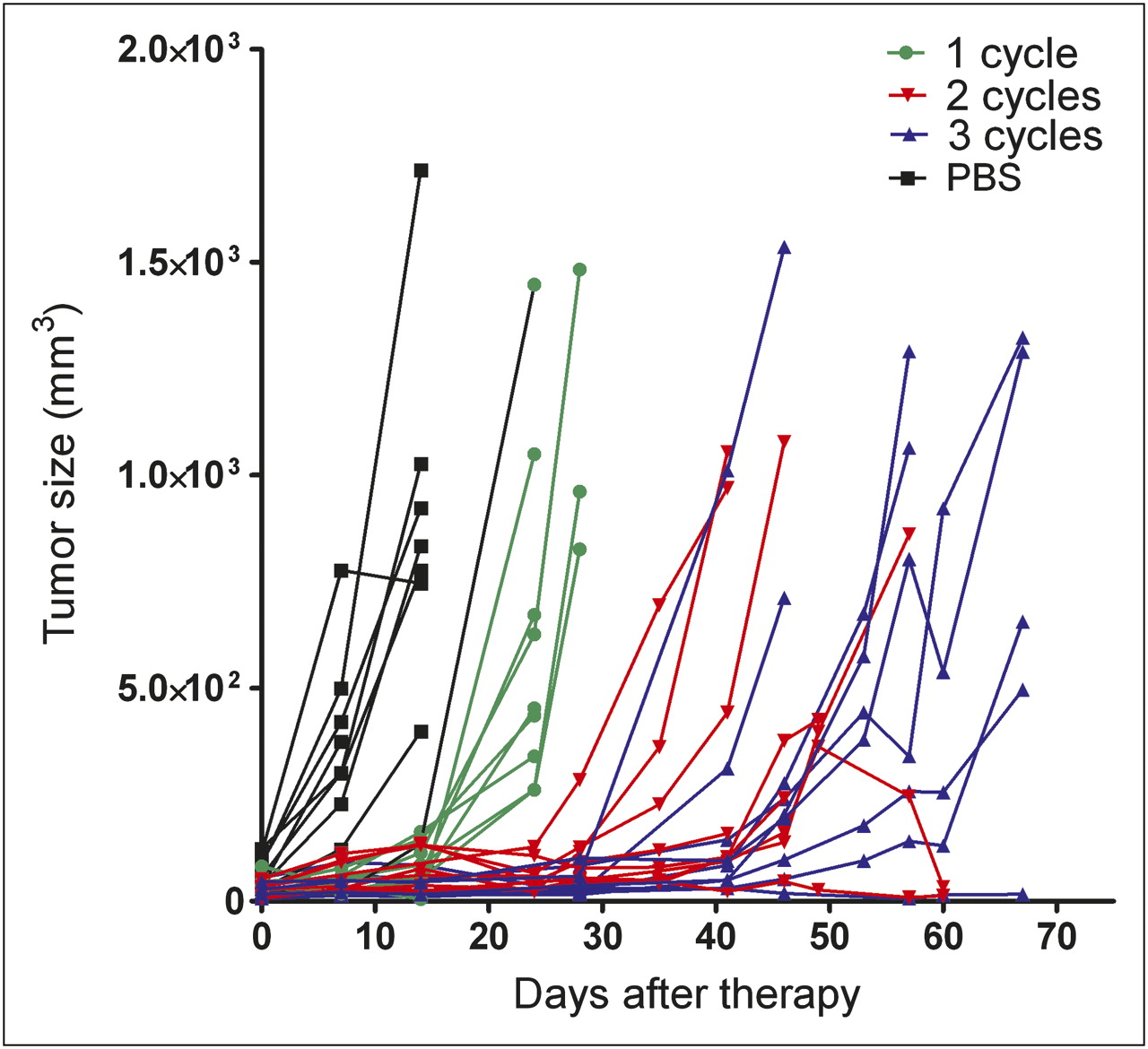
Tumor growth in mice that received 1, 2, or 3 cycles of TF2 (5.0 nmol) and 177Lu-IMP288 (26 MBq/0.28 nmol per cycle) or PBS. Size of tumors of individual mice is depicted.
The survival of the animals in the 4 groups of mice is depicted in Figure 5. PRIT prolonged survival. The median survival of untreated mice was 13 d (range, 6–20 d), whereas the median survival of mice treated with 1, 2, and 3 cycles of PRIT was 24 (range, 24–31 d), 45 (range, 38 ≥ 130), and 65 (range, 48 ≥ 130) days, respectively, with significant differences between all pairs of survival curves (P < 0.001, log-rank test), except for 2 cycles of PRIT, compared with 3 cycles (P = 0.22, log-rank test).
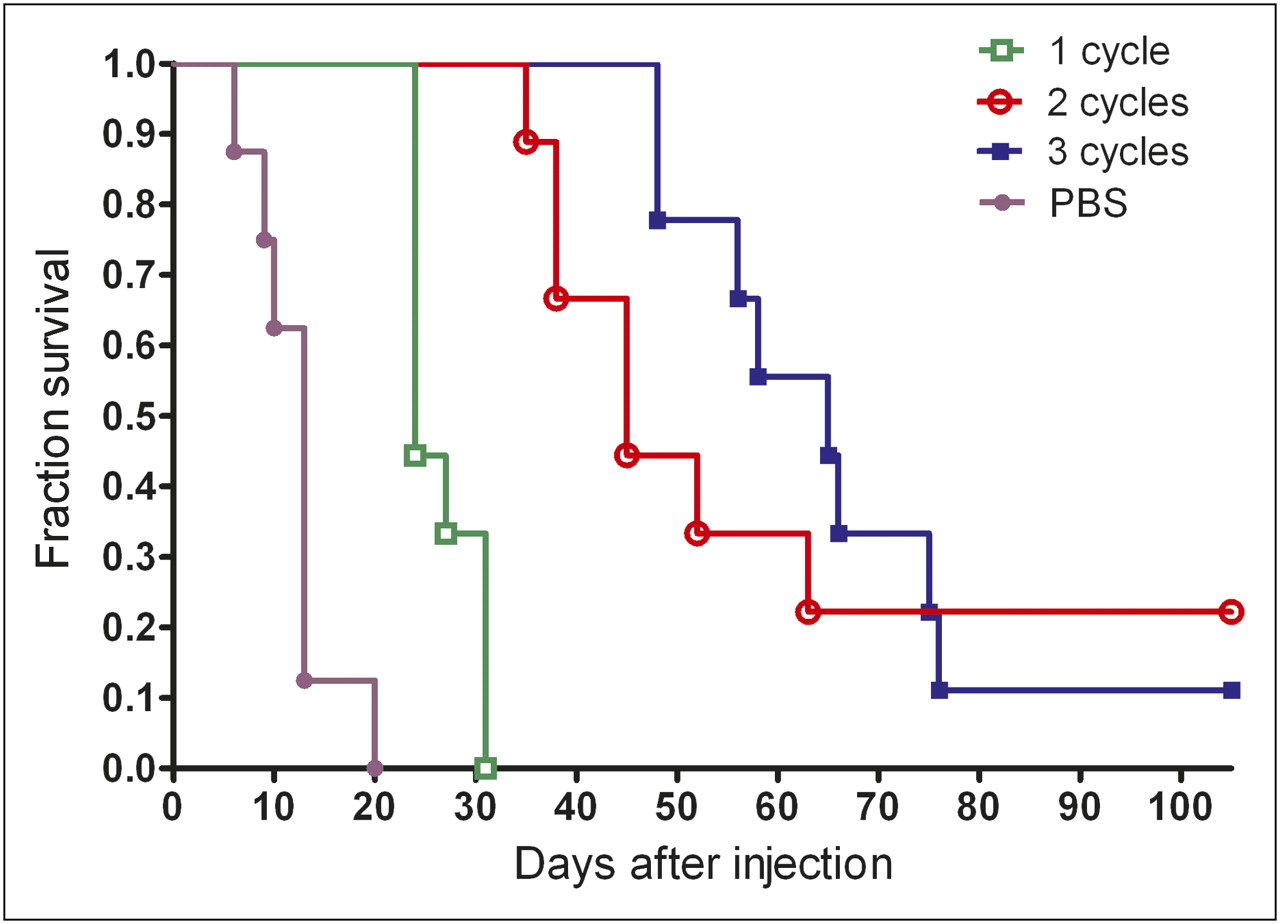
Survival of animals in groups of 9 mice that were treated with 1, 2, or 3 treatment cycles of TF2 (5.0 nmol) and 177Lu-IMP288 (26 MBq/0.28 nmol per cycle) or PBS.
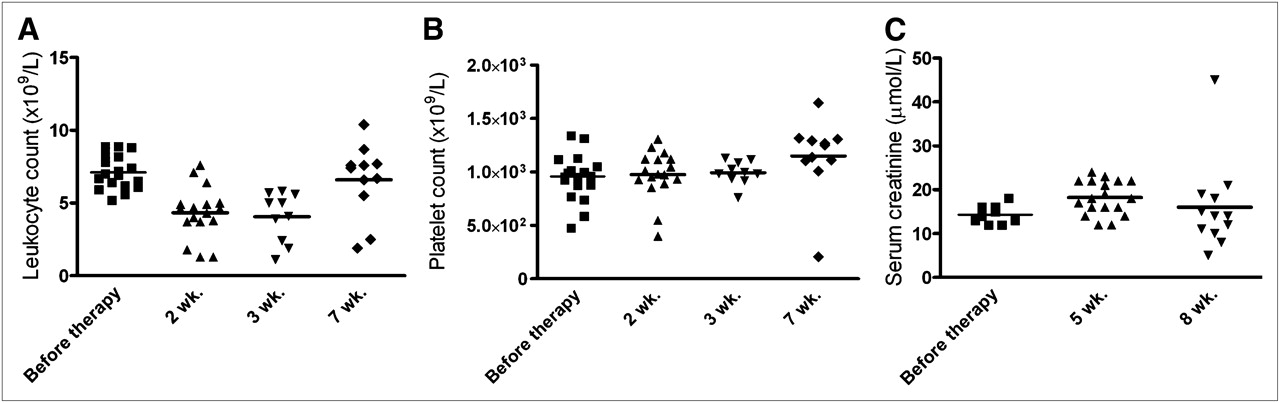
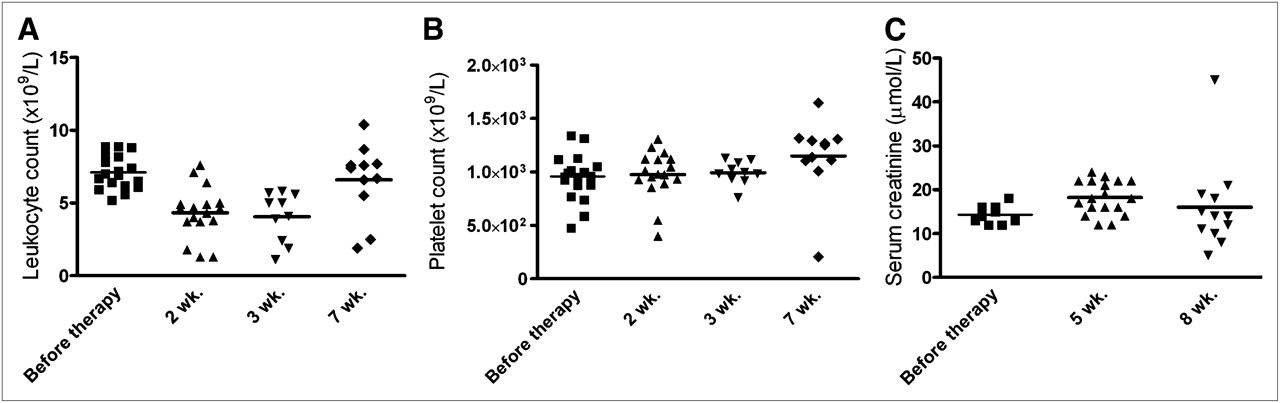
Toxicity due to PRIT was minimal. For all groups at all times, body weight remained greater than 93% of baseline, with no significant changes in mean body weight between the control and treated animals (P = 0.85, Kruskal–Wallis test). Hematologic toxicity in the PRIT groups is illustrated by the leukocyte and platelet counts at 2, 3, and 7 wk after injection (Figs. 6A and 6B). Seven weeks after injection, 5 animals in the group that received 2 cycles had already been euthanized because of excessive tumor growth, whereas all mice that received 3 cycles of treatment were still alive. No differences in leukocyte and platelet counts were observed between the 2- or 3-cycle groups; thus, data of both groups were combined. A minimal but statistically significant decrease in leukocyte counts was measured at 2 and 3 wk after the start of treatment (median baseline counts, 6.9 × 109/L; 2 and 3 wk after injection, 4.5 × 109/L and 4.6 × 109/L, respectively; P < 0.001, Mann–Whitney exact test, 2-tailed). Full recovery was observed at 7 wk after treatment (median counts 7 wk, 7.4 × 109/L, compared with baseline; P = 0.95, Mann–Whitney exact test, 2-tailed). No decrease in platelet counts was observed (median baseline counts, 972 × 109/L; 2 and 3 wk after injection, 964 and 991 × 109/L, respectively, compared with baseline; P = 0.94, Mann–Whitney exact test, 2-tailed). Serum creatinine levels at 5 and 8 wk after therapy were not increased significantly, as shown in Figure 6C (median, 14.0, 18.0, and 14.0 μmol/L at baseline and 5 and 8 wk after therapy, respectively; Kruskal–Wallis, P = 0.94), indicating no evidence of acute nephrotoxicity in the treated mice.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Leukocyte counts (A), platelet counts (B), and serum creatinine levels (C) in mice that were treated with 2 or 3 cycles of TF2 (5.0 nmol) and 177Lu-IMP288 (26 MBq/0.28 nmol per cycle). Blood cell counts were measured before therapy and at 2, 3, and 7 wk after injection. Serum creatinine levels were measured before therapy and at 5 and 8 wk after therapy. Data represent individual determinations.
DISCUSSION
In this study, we investigated the optimization of the pretargeting procedure for radioimmunotherapy using the new humanized recombinant DNL–bsMAb TF2, with binding to CEACAM5 and the hapten HSG, using the 177Lu-labeled di-HSG-DOTA peptide IMP288. We demonstrate that using the optimal dose schedule, this modality can be effective for the treatment of CEA-positive human colonic tumors growing in mice.
All targeted therapies are limited by the amount and accessibility of the antigen. In this model system, in which small tumor xenografts were used, the amount of radioactivity that could be delivered in a single treatment was limited by the amount and specific activity of IMP288 that could be given with antigen-saturating amounts of the bsMAb. We recently reported an evaluation of this same pretargeting system for PET using 68Ga- and 18F-labeled peptides (18) and found, as in that study, that 5 nmol (800 μg) of TF2 saturated the amount of antigen in LS174T tumors. Thus, this amount of bsmAb is the upper limit that should be used in this pretargeting setting (28). Using 5 nmol of TF2, we next determined the optimal IMP288 dose: 0.28 nmol of IMP288 essentially saturated the amount of bsMAb-binding sites. At the maximum specific activity of IMP288 (90 MBq/nmol), no more than 26 MBq (0.700 mCi) of 177Lu could be administered. However, because pretargeting greatly minimizes radiation exposure, this amount was much lower than the MTD. Therefore, other strategies that could increase the total amount of radioactivity administered were evaluated.
Biodistribution studies revealed that the amount of radioactivity delivered to a tumor from a single 177Lu-IMP288 injection could be increased nearly 2-fold by the administration of an additional 2 successive doses of the IMP288 after a single TF2 injection. However, further improvements could be achieved by the administration of successive cycles of the bsmAb, followed by IMP288, and therefore this latter approach was selected. As expected, higher-activity doses, administered in successive cycles, correlated with longer survival. Despite this clear trend, the survival achieved with 3 cycles was not significantly better than with 2 cycles. Furthermore, tumor volume curves in the groups of mice that received 2 or 3 cycles were quite similar. This similarity could be because by the third cycle, tumor uptake had declined (11 %ID/g), compared with uptake at the first 2 administrations (14 %ID/g).
The limited toxicity observed indicates that the MTD had not been reached in these experiments. Three subsequent treatment cycles did not compromise body weight, blood counts, or acute kidney function. The decrease in leukocyte counts (40% of baseline levels) was moderate, with rapid recovery in most animals. The kidneys had the highest 177Lu activity concentration of the normal organs, because the peptide is cleared by urinary elimination. A small fraction of the dose is reabsorbed by the proximal tubular cells and thus retained in the kidney. Serum creatinine levels were not increased after treatment, suggesting the absence of radiation nephritis; however, because radiation effects on the kidneys can take many months to manifest, further studies would be required to assess the risk for chronic renal toxicity, which has been observed in preclinical and clinical testing of peptide radionuclide radioimmunotherapy (29–31).
We initiated studies with 177Lu primarily because the most promising results with radiolabeled antibodies in solid tumors have been observed in the treatment of small-volume disease (≤3 cm) or in an adjuvant setting (32). 177Lu is well suited for the treatment of small-volume disease with minimal radiation, given the medium-energy β-emissions (maximum energy, 0.497 MeV; mean energy, 0.149 MeV; maximum penetration depth, 2.5 mm). With an 11% abundance of 208-keV photons, the biodistribution of the 177Lu-labeled IMP288 peptide by γ-camera imaging will be possible in a clinical setting, providing important insights into the kinetics of tumor binding and retention in patients.
In the current animal study, the washout of 177Lu-IMP288 from the tumor was similar to that of the TF2 bsMAb. In this pretargeting approach, the kinetics of tumor binding might not take full advantage of the 6.7-d physical half-life of 177Lu. In contrast, the half-life of 90Y (64.1 h) is more consistent with the pharmacokinetics of IMP288, but its longer tissue penetration range (maximum, 12.0 mm) would be more appropriate for treating tumors greater than 0.5 cm in diameter (33,34). Because of its high energy (maximum energy, 2.28 MeV; mean energy, 0.935 MeV), a single dose of a 90Y-labeled di-HSG peptide can be given to nude mice at or near MTD level, with significant therapeutic effects (23,31). In contrast, in this model the lower energy of 177Lu is less toxic, which allows for a multiple-dosing strategy, improving therapeutic outcome well below the MTD. On the other hand, because multiple treatment cycles might add to the complexity of PRIT, one should aim at administering the MTD in 1 therapy cycle. The in vivo effects of these β-emitters should be compared in the same animal model, or ultimately in clinical trials, to reveal the optimal combination for therapeutic effects and bone marrow and kidney toxicity.
Attempts to further enhance the activity dose in radioimmunotherapy for solid tumors are restricted by myelotoxicity. Therefore, PRIT was developed as an alternative primarily to circumvent problems associated with the long residence time of the antibody in the blood (11). Several pretargeting approaches have been developed, with preclinical evidence universally indicating the benefits of PRITs over direct targeting in terms of reduced toxicity, with similar or improved efficacy (11,19). The clinical experience with pretargeting has been limited. In patients with metastasized CEA-expressing tumors, Kraeber-Bodéré et al. optimized PRIT with a chemically conjugated bsmAb using a humanized anti-CEACAM5 Fab′ and a murine anti–diethylenetriaminepentaacetic acid Fab′ and a 131I-di-diethylenetriaminepentaacetic acid peptide (1.9–5.5 GBq) (35,36). The tumor responses in the 8 patients with colorectal cancer were modest, much better therapeutic responses were seen in patients with medullary thyroid cancer. However, this last group of patients experienced dose-limiting leuko- or thrombocytopenia, most likely due to diffuse metastatic involvement of the bone marrow, whereas the patients with colorectal cancer had only mild hematologic toxicity (grade II). These results illustrate the relevance of determining the most effective and tolerable treatment strategy and dosing schedule for each different patient population.
CONCLUSION
In this study, we showed that pretargeted radioimmunotherapy with an anti-CEA bsmAb and a 177Lu-labeled peptide could be an effective treatment modality for the treatment of CEA-positive colonic tumors.
Acknowledgments
This study was supported in part by the Dutch Cancer Society (KWF Kankerbestrijding), grant KUN 2008-4038. William J. McBride, David M. Goldenberg, and Edmund A. Rossi are employed by or have financial interest in Immunomedics, Inc., or IBC Pharmaceuticals, Inc.
- © 2010 by Society of Nuclear Medicine
REFERENCES
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.
- 12.
- 13.
- 14.
- 15.
- 16.
- 17.
- 18.
- 19.
- 20.
- 21.
- 22.
- 23.
- 24.
- 25.
- 26.
- 27.
- 28.
- 29.
- 30.
- 31.
- 32.
- 33.
- 34.
- 35.
- 36.
- Received for publication May 21, 2010.
- Accepted for publication July 15, 2010.