

Abstract
The aim of this work was to develop reagents and methods potentially useful in PET, using 68Ga in a 2-step pretargeting protocol. Methods: We prepared bispecific antibodies (bsAbs) for disease-specific targeting of carcinoembryonic antigen-positive cells and recognition of later-administered bivalent hapten-peptide conjugates. The secondary antibody arm (antibody 679) recognizes a histaminyl-succinyl-glycine (HSG) structural subunit. The bsAbs were prepared as Fab′ × Fab′ conjugates using chemical cross-linking methods and as bispecific diabodies using recombinant DNA technologies. A HSG-bivalent hapten conjugate bearing the macrocyclic ring chelating agent 1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid (DOTA) was designed to be readily radiolabeled with 68Ga taken directly from a 68Ge/68Ga generator system. Reagents were tested in vitro and, then, for their targeting properties in a preclinical animal model of human cancer. Results: A chemically cross-linked hMN-14 × 679 F(ab′)2 and a fully humanized bispecific diabody construct (BS1.5H), expressed in Escherichia coli, were prepared for this work. We synthesized the bivalent peptide termed IMP 241 [DOTA-Phe-Lys(HSG)-d-Tyr-Lys(HSG)-NH2] and labeled it with 68Ga and 67Ga at temperatures from 45°C to 100°C, over times of 15 min to 1 h, establishing 15 min at 95°C as a useful condition for 68Ga labeling. When we formulated the IMP 241 bivalent hapten-peptide with ammonium acetate buffer at pH 4–5 and eluted the 68Ga from the generator directly into the peptide solution, we achieved an almost quantitative incorporation of the 68Ga into IMP 241, as analyzed by size-exclusion high-performance liquid chromatography, after mixing the complex with the 679 antibody. For in vivo studies we used 67Ga-IMP 241 as a surrogate for 68Ga-IMP 241, in view of the short, 68-min half-life of the 68Ga nuclide. The 67Ga-IMP 241 was successfully pretargeted to human colon tumor xenografts in athymic mice with both the chemical and the diabody bispecific proteins. High tumor-to-normal tissue ratios for 67Ga uptake were found for all tissues at 1 to 6 h after injection of 67Ga-IMP 241. When using the BS1.5H diabody for pretargeting, tumor-to-blood, tumor-to-liver, and tumor-to-lung ratios of 67Ga-IMP 241 at 1 and 3 h after injection were 41:1 and 137:1, 51:1 and 106:1, and 16:1 and 46:1, respectively. Conclusion: The general approach described, along with the new compositions and the labeling methods we have developed, may eventually allow for use of 68Ga-labeled specific targeting agents in a routine clinical PET application.
- 68Ga
- 67Ga
- PET
- bispecific antibodies
- 2-step pretargeting
Clinical PET application has the potential to dramatically improve disease detection—a potential that, in turn, depends on the successful development of new technologies, reagents, and application methods. Although there are about 20 nuclides of theoretic utility for PET, the 2 most commonly considered nuclides, 18F and 68Ga, retain the best combination of features. 68Ga has 2 major advantages over 18F. First, it can be derived from an in-house generator, making 68Ga supply independent of the need for a nearby cyclotron, and, second, it is a radiometal enabling radiolabeling chemistry via a chelate. The relatively complex and specialized chemistry needed for 18F attachment to disease-specific targeting agents means that processes can only be performed in custom-designed dedicated facilities located near a cyclotron that produces the 18F raw material.
68Ga is available from a long-lived parent nuclide (68Ge; half-life, 271 d) that can be adsorbed to various solid phases, from which the 68Ga can then be selectively eluted. Thus, a 68Ga generator can be made, and several have been described (1–4). The most-developed generator is one based on adsorption of the parent 68Ge to a stannic oxide bed (2), from which the 68Ga is eluted with dilute HCl. Despite the availability of 68Ga generators over many years, it is surprising that no 68Ga-labeled targeting agents have been developed past the point of research article material toward routine clinical use. One object of this work was to overcome radiolabeling issues that have prevented routine clinical preparation of 68Ga-labeled imaging agents. 68Ge/68Ga generators of the stannous oxide type are usually eluted with a 10- to 20-mL portion of ultrapure 1N HCl, providing the 68Ga daughter in highly dilute form and in the presence of a large volume of acid, possibly containing other metal ions and anionic stannates. Once the 68Ga is obtained, there is then the challenge to efficiently bind it to a disease-targeting species, and this has been approached in numerous ways (5–14).
In radiolabeling studies of 67Ga and 68Ga with various chelating agents, several different structural types of chelate have been proposed as stable conjugates for in vivo work (15–18), with the macrocyclic chelator 1,4,7-triazacyclononane-N,N′,N″-triacetic acid (NOTA) derivatives being among the most popular (19). Despite published literature on NOTA-type chelates suggesting that this small macrocycle would be perfect for the relatively small gallium 3+ ion, more recent literature has also described use of the larger 1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid ((DOTA)-type chelates for gallium labeling (13,20). This involved gallium-labeled peptides, such as octreotide, and positive targeting suggested that DOTA-type chelates might prove just as useful for the 68-min half-life 68Ga, as it is for actinium (21), lutetium (22), and yttrium or indium radionuclides (23).
We, together with our collaborators (24–26), and others before us (27–31), have investigated 2-step targeting strategies involving bispecific antibodies (bsAbs) followed by radiolabeled haptens for radioimmunoimaging with various SPECT nuclides, such as 131I, 111In, and 99mTc. Additionally, and specifically for 68Ga imaging using bsAbs, several articles have appeared with secondary arms directed against the gallium complexes of the N,N′-di[(2-hydroxy-5-ethylene-β-carboxy)benzyl]ethylenediamine N,N′-diacetic acid (HBED) class of chelating agents (6,7). Several factors, unfortunately, contributed to produce less than ideal results when these agents were tested recently in a clinical trial in breast cancer patients (14), with 3 false-negative findings reported in patients with a total of 17 known breast lesions. Shedding of the MUC-1 antibody against which the targeting antibody was directed was suggested as part of the problem, although the comparatively hydrophobic nature of the 68Ga-HBED complex and its monovalency toward the bsAb used might be other factors resulting in higher than expected background levels and, consequently, lower than anticipated tumor-to-normal breast tissue ratios.
Raising a specific antibody against a defined Ga-chelate complex results in a system with a very specific binding relationship between the said 68Ga-chelate and the corresponding bsAbs recognizing such Ga-chelate complex. In contrast, a system wherein the moiety recognized by the bsAb is independent of the imaging complex to be targeted is superior in versatility, in that almost any radionuclide, including 68Ga, can be targeted. In addition, rational chemical design can be used to ensure that the radiolabeled chelate has desirable pharmacokinetic and clearance properties. An antibody termed 679 that binds to the addend histaminyl-succinyl-glycyl- (HSG-) has been described previously as useful for in vivo targeting of several radionuclides (32–34). The 679/HSG antibody/antigen recognition system was designed so that the 2 HSG subunits on any bivalent hapten complex would additionally impart a hydrophilic nature to the bivalent hapten, which, in turn, would result in agents that had low levels on nonspecific tissue binding and rapid, and desirably complete, clearance, through the renal system, if the agent did not bind to the bsAb pretargeted at the site of disease.
We have developed the bivalent hapten tetra amino acid peptide IMP 241 [DOTA-Phe-Lys(HSG)-d-Tyr-Lys(HSG)-NH2] for binding yttrium, indium, and iodine radionuclides and as a multinuclide delivery reagent. The 2 HSG subunits recognized by the 679 antibody are combined in this peptide with the DOTA chelate, which was recently reported to be useful for binding 68Ga to peptides (13). As companion bsAbs, we have also developed chemically cross-linked bispecific F(ab′)2 entities and, more recently, recombinant molecular constructs that incorporate at least one 679 anti-HSG binding arm (26). The aim of this work was to extend binding studies of the IMP 241 peptide to 67Ga/68Ga and then determine how well this agent performed in vivo within the context of a bsAb pretargeting protocol in a preclinical xenograft model of human colon cancer.
MATERIALS AND METHODS
67GaCl3 in 0.1N HCl was obtained from Nordion, 111InCl3 was purchased from IsoTex Diagnostics, and sodium (125I) iodide was bought from Perkin-Elmer Life Sciences Inc. An ionic 68Ge/68Ga generator was purchased from DuPont Radiopharmaceuticals and eluted with 0.5N–1.0N solutions of ultrapure HCl prepared using OPTIMA grade concentrated HCl (Fisher Scientific) and Millipore 18-MΩ water. All radiolabelings were performed in 3N HCl acid-washed and Millipore water-rinsed CZ resin vials (West Pharmaceutical Services). Radiolabeled products were analyzed on a size-exclusion high-performance liquid chromatography (SE-HPLC) GF-250 column (Bio-Rad) attached to a Millennium system (Waters Corp.) equipped with a Packard FLO-ONEβ-in-line radiomatic detector. The SE-HPLC columns were equilibrated in 0.2 mol/L sodium phosphate buffer, pH 6.8, and samples were run at 1 mL/min.
The peptide designated IMP 241 [DOTA-Phe-Lys(HSG)-d-Tyr-Lys(HSG)-NH2] (35) was synthesized using an Fmoc-based solid-phase strategy on an Advanced ChemTech automated peptide synthesizer. Briefly, IMP 241 was synthesized on a Sieber amide resin (2.1 g, 0.62 mmol/g) with the following amino acids (6 equivalents per coupling) added in the order shown: Fmoc-Lys(Aloc)-OH, Fmoc-d-Tyr(But)-OH, Fmoc-Lys(Aloc)-OH, Fmoc-Phe-OH, and DOTA-tris(t-butyl) ester (Macrocyclics, Inc.). A single diisopropylcarbodiimide (DIC) coupling overnight was used with the DOTA. Each amino acid was double-coupled for 1 h using DIC as the activating agent in the presence of N-hydroxybenzotriazole (HOBt), followed by a second 1-h coupling using HOBt, O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate and diisopropylethylamine. The Aloc side chains were removed with the Pd catalyst in the usual way (36) and the trityl-HSG-OH (6 equivalents) was double-coupled to the lysine side chains, as described above. The peptide was cleaved from the resin with trifluoroacetic acid, precipitated in ether, and purified by reverse-phase HPLC to obtain the desired peptide (ESMS MH+ 1471).
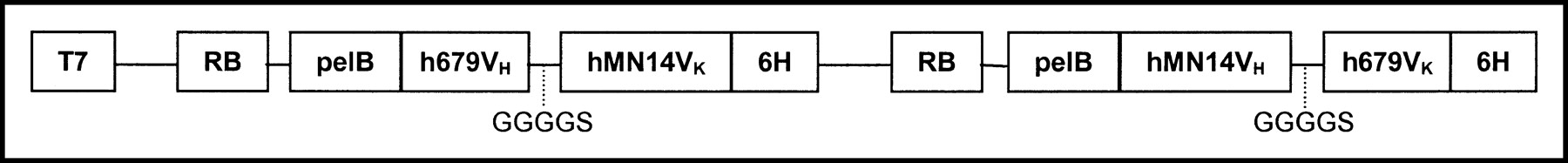
The preparation of a bsAb with specificities against carcinoembryonic antigen (humanized anti-CEA) and histaminyl-succinyl glycine (murine anti-HSG), prepared as Fab′ × Fab′ dimaleimido- chemically cross-linked F(ab′)2 fragment, has been described (37). The bispecific diabody referred to as BS1.5H was constructed from the variable domains of hMN-14 and humanized variable domains of m679 using established methods (38). Briefly, VH and VK sequences of hMN-14 were amplified by polymerase chain reaction (PCR) from a vector that was constructed for expressing hMN-14 Fab′ (39). The VH and VK domains of m679 were amplified by PCR from a plasmid containing the complementary DNA sequences of m679 heavy and light chains (Z. Qu, unpublished data, 2000). Humanized versions of m679VH and m679VK were generated by PCR-based mutagenesis. The strategy used for humanization was to retain all CDR amino acid sequences and any framework residues known to interact with CDR regions. Amino acid residues of mouse framework regions that are not represented in the database of human frameworks were replaced with the most common amino acid found in human sequences at that position. Thirteen amino acid substitutions were made to humanize VH and VK of m679. The dicistronic expression cassettes for BS1.5H are shown in Figure 1. Chemically competent Escherichia coli BL21-pLysS cells (Novagen) were transformed with the expression plasmid for BS1.5H following the manufacturer’s recommendations. Transformants were plated overnight at 37°C on LB agar plates supplemented with kanamycin sulfate (100 μg/mL) and chloramphenicol (34 μg/mL). Transformed colonies were used to inoculate shaker flask cultures of Difco 2× YT broth (Becton Dickinson) supplemented with kanamycin sulfate (100 μg/mL) and chloramphenicol (34 μg/mL). Cultures were shaken at 37°C to an OD600 of 1.6–1.8. An equal volume of room temperature 2× YT media supplemented with antibiotics and 0.8 mol/L sucrose was added to the cultures, which were then transferred to 20°C. After 30 min of shaking at 20°C, expression was induced by the addition of isopropyl thiogalactose to a final concentration of 40 μmol/L and the incubation was continued at 20°C for 15–18 h. Bacteria were pelleted by centrifugation at 10,000g. The cell pellets were frozen and thawed and then resuspended in lysis buffer (2% Triton X-100, 300 mmol/L NaCl, 10 mmol/L imidazole, 5 mmol/L MgSO4, 25 units/mL benzonase, 50 mmol/L NaH2PO4, pH 8.0) using an amount equal to 1% of the culture volume. The suspension was homogenized by sonication and clarified by centrifugation at 40,000g. The soluble extracts were loaded onto Ni-NTA agarose (Qiagen, Inc.) columns using about 1.0 mL Ni-NTA resin per liter of culture. Columns were washed with a buffer containing 20 mmol/L imidazole and eluted with 250 mmol/L imidazole. The Ni-NTA eluate was loaded directly onto a Q-Sepharose (Amersham Pharmacia Biotech) anion-exchange column, and the flow-through fraction containing the products was collected. The yield of BS1.5H was about 0.5 mg/L of culture.
Dicistronic expression cassette for production of BS1.5H in E. coli.
For a typical radiolabeling with 68Ga, IMP 241 (11.4 μL of a 2.2 × 10−3 mol/L stock solution in 0.5 mol/L ammonium acetate, pH 4.0; 2.5 × 10−8 mol) was mixed with 1.0 mL of 2.5 mol/L ammonium acetate, pH 5.5, in an acid-washed 10-mL CZ-Resin vial. The vial was sealed with an acid-washed Flurotec stopper, crimped closed, and swirled to mix. The 68Ge/68Ga generator was then eluted directly into the IMP 241 vial using a nonmetallic catheter (VWR International), and vent needle, with 2 mL 1N HCl, after running the first milliliter of generator eluent to waste. The final pH of the labeling mixture was 4.0. The vial was heated at 95°C for 15 min. After cooling for 5 min, an aliquot was withdrawn and diluted to 1.85 MBq/mL with sterile saline for radioanalysis. Ten to 12 μL of the diluted radiolabeled peptide containing 18–22 kBq 68Ga were mixed with a 20-fold molar excess m679 IgG and applied to the SE-HPLC column. Column recovery of radioactivity was estimated by collecting the entire eluate from the 20-min run and counting 3 × 1-mL aliquots against 3 × 1-mL aliquots of a standard prepared by diluting the same volume of labeled peptide in 20 mL elution buffer. Averages of the triplicate counts were taken and compared with estimate column recovery of applied radioactivity.
For a typical radiolabeling with 67Ga, 200 μL 2N HCl (OPTIMA) were drawn up into the barrel of a 1-mL syringe and, with a vent needle in place, the acid was added to a crimp-cap vial of 104 MBq (2.8 mCi) 67Ga chloride (Nordion) using a nonmetallic catheter. The vial was mixed by inversion and vortex and then spun in a clinical centrifuge at ∼3,000 rpm for ∼2 min, after which it was a left at room temperature for 1 h. In an acid-washed 1.8-mL Eppendorf vial, 500 μL 2.5 mol/L NH4OAc, pH 6.9, was mixed with 11.4 μL of a stock solution of IMP 241 peptide (2.2 × 10−3 mol/L, 2.5 × 10−8mol). This diluted solution of IMP 241 in the acetate buffer was drawn into the barrel of a 1-mL syringe and added to the vial of 67Ga through the nonmetallic catheter. The vial and contents were then heated at 95°C for 15 min to effect radiolabeling. Radioanalyses were performed as described above for the 68Ga analog. 111In-IMP 241 was prepared as described previously (35).
For purification of 67Ga-IMP 241, a Waters Oasis HLB 1-mL extraction cartridge was fitted with a 1–1/2 in. (3.8 cm) 19-gauge needle and placed in a syringe shield fixed to a ring stand. The needle end was inserted through the septum of an empty crimp-cap 2-mL glass collection vial. Also inserted through the septum of the vial was a 1–1/2 in. 18-gauge needle attached to one end of a 24-in. (61.0 cm) double male monitoring line. The other end of the line was attached to a 3-way stopcock fitted to a 5-mL syringe. In this manner, liquid was drawn through the cartridge into the collection vial by use of vacuum. The cartridge was prepared for use by drawing through 500 μL methanol, followed by 500 μL water, and, finally, 500 μL 2.5 mol/L NH4OAc, pH 6.9, buffer. The 67Ga-IMP 241 labeling solution was drawn into a 3-mL syringe and applied to the top of the prepared cartridge. The labeling liquid was drawn onto and through the column and into the collection vial. This first vial contained nonpeptide-bound 67Ga waste. A second empty collection vial was then attached and a wash volume of 1.0 mL 2.5 mol/L NH4OAc, pH 6.9, was drawn through the column. The 67Ga-labeled peptide was eluted into a third empty collection vial with 500 μL methanol and water, 1:1.
In vitro stability on gallium-labeled IMP 241 was assessed by dilution of an original 67Ga-IMP 241 sample 10-fold in human serum and incubation at 37°C. Aliquots were withdrawn at time points from 30 min through 4 h, mixed with a 50-fold molar excess of m679 IgG, and applied to SE-HPLC for analysis. Column recoveries of applied radioactivity were determined.
All animal experiments were conducted in accordance with National Institutes of Health guidelines on the humane use of animals and with prior approval of the institutional animal care and use committee. In vivo targeting experiments were performed with the 67Ga-IMP 241 analog, due to the 68-min half-life of the 68Ga. Athymic nude mice bearing GW-39 human colon tumor xenografts in groups of 4 or 5 per time point were used. For an experiment with the chemically produced construct, animals were given an injection of 15 μg (1.5 × 10−10 mol) hMN-14 × m679 F(ab′)2 bsAb, radioiodinated with 125I (Perkin-Elmer) using the standard IODO-GEN method (Pierce Chemical Co.), 24 h before administration of the 67Ga-IMP 241 peptide. Animals were then given 1.5 × 10−11 mol of peptide, a 10-fold molar deficit of peptide (25,35), equivalent to about 92.5 kBq (∼2.5 μCi) 67Ga per animal. Five animals were killed at each of 1, 2, 3, 4, and 6 h after injection, with tissues (tumor, blood, liver, kidney, spleen, lungs, stomach, small and large intestine, and bone) quantified for both 125I and 67Ga uptake, using dual-energy counting with cross-over correction. A separate series of animals was given the 67Ga-IMP 241 alone, without bsAb pretargeting.
In a second pretargeting experiment in the same xenograft model system, the construct BS1.5H was administered first, followed by IMP 241 labeled with either 67Ga or 111In. We had previously established the preferred targeting and blood clearance time of the 125I-BS1.5H before radiolabeled peptide administration at 15 h (data not shown). The same molar dose of the BS 1.5H (1.5 × 10−10 mol) as the chemical construct was given 15 h before the same 10-fold molar deficit of ∼ 92.5 kBq 67Ga-IMP 241 peptide (1.5 × 10−11 mol). Animals were killed at 1, 3, and 24 h in the 67Ga-IMP 241 groups and at 3 and 24 h in the 111In-IMP 241 groups, with the same range of tissues (except bone) taken and counted in appropriate energy windows for each nuclide.
RESULTS
The synthesis of the IMP 241 peptide on solid phase was routine. Interestingly, the presence of the protected DOTA did not interfere with the aloc group cleavage, which used a palladium catalyst. Also, there was no palladium left in the final purified product, so there was no inhibition of metal-DOTA binding due to nonremoved palladium metal.
The dicistronic expression cassette shown in Figure 1 encodes 2 heterologous polypeptides designed to pair with each other and form 1 functional binding site each for CEA and HSG via domain swapping (39). BS1.5H was obtained from the soluble fraction and purified by immobilized metal affinity chromatography using Ni-NTA followed by Q-Sepharose anion-exchange chromatography. The characterization of BS1.5H included the determination of purity by SE-HPLC and sodium dodecyl sulfate polyacrylamide gel electrophoresis, molecular size by SE-HPLC, binding affinity for CEA by competitive enzyme-linked immunosorbent assay, binding affinity for HSG by BIAcore, and binding valency for CEA by BIAcore.
At receipt, the nominal 740-MBq (20 mCi) 68Ge/68Ga generator contained 1.23 GBq (33.2 mCi) 68Ge and eluted at 70.6% efficiency according to the manufacturer. That should have yielded 868 MBq (23.44 mCi) at first elution, but only 696 MBq (18.82 mCi) were obtained (yield ∼ 57%). With suitable in-growth time allowed, recoveries remained between 55% and 65% during >1 y of constant use and hundreds of elutions. Although 20 mL 1N HCl was recommended, equivalent yields were recovered using 10 mL, and little loss was evident even when using only 5 mL of eluent, since most 68Ga eluted in the 2- to 4-mL volume fraction, as measured from the beginning of the elution time. In addition, the generator could usefully be eluted with 0.5N rather than 1.0N HCl, with only about 10% loss in recovered 68Ga over what one would expect with 1.0N HCl (2).
A SE-HPLC radiomatic analysis of the 68Ga-IMP 241 after mixing with the hMN-14 × 679 F(ab′)2 bsAb is shown in Figure 2. Also shown, for in vitro stability, is a typical radiomatic SE-HPLC analysis of a trace taken after 4 h at 37°C. This trace is unchanged from the original SE-HPLC analysis of the 67Ga-IMP 241-m679 IgG complex or from analyses taken at the earlier times from 30 min through 3 h. Essentially 100% of the detected activity elutes at the retention time of 67Ga-IMP 241-m679 IgG, with column recoveries of applied radioactivity between 74% and 82% at each time interval tested.

{kind=link}
{kind=link}
SE-HPLC analyses of 68Ga-IMP 241 after mixing with 50× molar excess of hMN-14 × 679 bsAb (top), and 67Ga-IMP 241 after a 4-h incubation in human serum and mixing with 50× molar excess of anti-HSG antibody 679 IgG (bottom).
In a targeting experiment with the chemically cross-linked bsAb trace-radiolabeled with 125I, 125I-hMN-14 × 679-F(ab′)2 was given 24 h before 67Ga-IMP 241 to nude mice bearing GW-39 tumors at specified times after injection (5 animals per group). Data for the bispecific protein are given in Table 1, and data for the 67Ga-IMP 241 are given in Table 2. The biodistribution of 67Ga-IMP 241 administered alone, without bsAb pretargeting, is in Table 3, with all data in Tables 1–3 given in terms of percentage injected dose per gram (%ID/g) of tissue (±SD). Biodistribution data for 67Ga-IMP 241 from this experiment are reported in terms of tumor-to nontumor ratio in Table 4. Tumor uptake was between 8.3 and 16.0 %ID/g over the 6 h of the experiment and, at each time point and for every tissue, exceeded uptake in all normal tissues. Blood and kidney activities averaging 8.2 and 6.3 %ID/g at 1 h dropped to 2.1 and 3.1 %ID/g, respectively, at 2 h after injection. Residual activity at the later time points averaged around 3.5 %ID/g in the kidney and about 1.5 %ID/g in the blood out to 6 h after injection. Tumor uptake remained high throughout the duration of the experiment, indicating that targeting was specific and due to the prelocalized bsAb, evidenced by the lack of tumor uptake when injecting the 67Ga-IMP 241 alone (Table 3). Tumor-to-normal tissue ratios are given in Table 4, and only tumor-to-kidney ratios are <5:1 from 2 h after injection onward.
Biodistribution of 125I-hMN-14 × 679-F(ab′)2 bsAb Given 24 Hours Before 67Ga-Labeled Peptide
Biodistribution of 67Ga-IMP 241 Given 24 Hours After 125I-hMN-14 × 679-F(ab′)2 bsAb
Biodistribution of 67Ga-IMP 241 Given Alone, with No bsAb Pretargeting
Biodistribution of 67Ga-IMP 241 Given 24 Hours After 125I-hMN-14 × 679-F(ab′)2 bsAb
In a second targeting experiment, using the construct BS1.5H for pretargeting, we were able to reduce levels of circulating bsAb still further than that achievable with the chemical construct, to 0.36 ± 0.07 %ID/g at 15 h after injection, which was chosen as the optimum time point for injection of the 67Ga-IMP 241. In this experiment, 67Ga-IMP 241 blood retention was 1.6 and 0.1 %ID/g at 1 and 3 h after administration, respectively, of the peptide, and at 16 and 19 h after administration of the BS1.5H. Tumor uptake was 7.9 and 13.2 %ID/g at 1 and 3 h, respectively (Table 5), leading to tumor-to-blood ratios of 41:1 and 137:1, at 1 and 3 h after administration of 67Ga-IMP 241, respectively (Table 6). 67Ga uptake in other normal tissues was close to zero and, in any event, ≤0.5 %ID/g, with the exception of the kidney, which had uptakes of 5.8 and 5.7 %ID/g at 1 and 3 h after injection, respectively.
Biodistribution of 67Ga- and 111In-IMP 241 at Times Indicated After Injection, When Administered 15 Hours After Bispecific Construct BS1.5H
Biodistribution of 67Ga- and 111In-IMP 241 at Times Indicated After Injection, When Administered 15 Hours After Bispecific Construct BS1.5H
DISCUSSION
Targeting with bsAbs followed by radiolabeled bivalent haptens offers a unique opportunity to produce an entirely new generation of radioimaging agents, advantageously coupling disease specificity with exceptional target-to-nontarget ratios of disease-localized radionuclide (26,35). A particular advantage is to be expected in the delivery of PET nuclides using this technology, since the short half-lives of 18F and 68Ga make them incompatible with longer circulating primary targeting species such as intact antibodies. The major disadvantage is the added complexity of the approach, which involves preparation and dosing of 2 separate reagents and consequential timing and ratio issues. These are in addition to other aspects, such as making a judicious choice of reagents to take into development as well as solving more practical radiolabeling matters.
Regarding the bsAb component, the poor yields obtained when preparing Fab′ × Fab′ bsAbs with chemical combination techniques (often in the region of 20%–30%) have been known for >20 y, and a satisfactory process that overcomes this problem has not been developed to date. Also, this approach tends to result in <100% pure, fully structurally defined protein products, which is problematic for clinical development. Therefore, constructs that can be prepared in high yield in bacteria, yeast, or mammalian cells, in a fully defined structural manner at ≥95% purity, are preferred (26).
In an animal pretargeting study performed with the chemically cross-linked bsAb, the agent performed well in targeting the 67Ga-IMP 241 specifically and maintaining it in the xenograft throughout the course of the experiment. The tumor-to-tissue ratios in Table 4 suggest that imaging with this agent from 2 h through 6 h after injection would be feasible. However, this F(ab′)2 behaves in circulation and clearance as one would expect, and the residual bsAb remaining in blood (Table 1) serves to capture a relatively large amount (1.3–2.1 %ID/g between 2 and 6 h after injection; Table 2) of later injected 67Ga-IMP 241, which itself shows no blood retention when given alone (Table 3). In turn, this translates into useful, but less than optimal, tumor-to-blood ratios of 6:1 to 12:1 between 2 and 6 h after injection of the 67Ga-IMP 241.
We have the view that a bsAb used for 2-step imaging needs to use a species with an almost complete serum clearance in a reasonable time period after administration. We chose 24 and 15 h as time intervals (before injection of 68Ga) for the bispecific chemical conjugate and the BS1.5H, respectively, based on previously determined optimum clearance times for each species. Rapid and complete serum clearance obviates the need for bsAb clearing agents or blocking agents, which would engender considerably more complexity regarding doses and timings (35). Without a clearing agent, and with less than optimal bsAb blood clearance, one would have a radiolabeled peptide displaying lower than ideal target-to-tissue ratios due to radiolabel capture by circulating bsAb. In this work, the BS1.5H construct has been shown to be more effective than the corresponding Fab′ × Fab′ chemically manufactured bsAb in targeting 67Ga, when tested in the same xenograft model, primarily due to its more complete clearance, with <1 %ID/g in circulation at the time of 67Ga-IMP 241 injection. Full details of BS1.5H properties will be published elsewhere. Despite the near-complete blood clearance, the BS1.5H retained strong tumor uptake, leading to outstanding tumor-to-blood ratios of 41:1 and 137:1 at 1 and 3 h after injection, respectively. As shown in Table 6, all tumor-to-normal tissue ratios were >40:1 by 3 h after injection, with the exception of the kidney, which contained 5.7–5.8 %ID/g at 1 and 3 h, respectively. Due to the much lower 67Ga-IMP 241 uptake when given alone, this suggests that some renal capture by residual bsAb may be taking place, although some loss of 67Ga from the complex may also be involved, since 111In-IMP 241 renal uptake is lower in the same targeting system. In any event, these relatively low levels may not present much of a problem for imaging purposes.
As a primary requirement, the radiolabeled entity that is administered after the bsAb should be designed from the outset to have desirable biophysical properties. Ideally, it should not accumulate in any normal tissues and should be rapidly flushed via the urine, if not captured by pretargeted bsAb. 67Ga-IMP 241, administered alone, deposited between 1.7 and 2.6 %ID/g in the kidney over the 6-h course of the experiment, with all other tissues having very low uptakes, which suggests at least adequate stability (Table 3).
For this bsAb targeting system, the most valid comparison for the data we obtained with that previously reported probably involves the work of Schuhmacher et al. (40). These investigators injected xenografted mice with an anti-MUC-1 × anti-HBED-CC-gallium F(ab′)2 × Fab′ bsAb, followed by a dose of a Ga-HBED-CC-apotransferrin blocker, and then followed by the 67Ga-HBED-CC complex in a carefully determined set of timings and dosages. They obtained tumor-to-blood, tumor-to-kidney, tumor-to-lung, and tumor-to-liver ratios of 67Ga of around 2.5:1, 2.5:1, 3:1, and 6:1 at 3 h after injection, which they state are comparable with work by others for early time points after injection. Although these are good results, they do not approach the ratios reported herein, with tumor-to-blood, tumor-to-kidney, tumor-to-lung, and tumor-to-liver ratios of 67Ga of around 137:1, 2.3:1, 41:1, and 106:1 at the same 3 h after injection time point when using BS1.5H pretargeting followed by 67Ga-IMP 241. Reasons for the lower ratios observed previously in this general pretargeting area are multifactorial, with choice of antibody, antibody affinities, antigen-shedding tendencies, inadequate blood clearances, the added complexity inherent in 3-step systems, less than ideal radiolabels, use of radiolabeled agents that are too hydrophobic for excellent tissue (particularly renal) clearance, and use of monovalent rather than bivalent radiolabeled haptens as some of the factors involved.
Further improvement in the system as described here may be sought by making a bsAb that retains 2 binding sites for the target cell antigen and 1 for the radiolabeled moiety (40), but which retains the complete serum clearance properties, at some defined interval between 15 and 48 h after injection. This might be expected to increase the absolute amount of bsAb targeted and retained at the tumor. In reference to this point, it must be borne in mind that an ideal agent would also be capable of delivering a therapeutic radionuclide. In this respect, use of a bivalent CEA-binding bsAb may be preferred, and use of a minimally residualizing radiolabeled agent in the kidney may be mandatory. With regard to the chelating moiety itself, though a purist might claim that NOTA would be a better choice than DOTA as the chelate to ensure maximal 68Ga stability, the stability of the 67Ga-IMP 241 complex appears adequate for early time-point imaging, while use of the DOTA chelate enables an easier development path for IMP 241 analogs of indium, yttrium, lutetium, bismuth, and actinium and other similar metals of interest.
Successfully making a clinically useful set of reagents for radioimaging generally requires that radiolabeling processes remain relatively simple. This has not been true in the past for procedures involving 68Ga, with generator-eluent drying, concentration, or ethereal extraction of highly dilute 68Ga being developed to overcome radiolabeling problems. However, given the short-lived, high-energy emissions, these procedures are not practical for widespread adoption in a routine clinical setting. The 68Ge/68Ga generator used in this work was of the type first described by Loc’h et al. in 1980 (2), and our review of the original article suggested that handling and elution of the generator could be changed to obtain the 68Ga in a smaller volume of less concentrated HCl. In turn, this might enable a facile labeling to be developed, with the only drawback being a small, and acceptable, reduction in elutable 68Ga. It proved possible to obtain the 68Ga in 2- to 4-mL volumes, allowing use of a kit containing the peptide and sufficient buffer to neutralize the 0.5N–1.0N HCl and thereby allow 68Ga labeling of IMP 241 by direct elution of the generator.
Radiolabeling of the IMP 241 peptide with 67Ga or 68Ga could be accomplished in about 15 min at an elevated temperature with apparent yields of at least 80%, as analyzed by SE-HPLC of bsAb-bound radiogallium. Given the inconsistencies in measuring column recoveries in this complex system, the real incorporation may be >80%. For the animal studies, we purified 67Ga-IMP 241 from possible contamination by other 67Ga species, using an extraction cartridge process that took 5–10 min and gave the desired agent in an aqueous alcohol solution. Using ethanol in lieu of methanol, this certainly would not be incompatible with the 68-min half-life of 68Ga in a clinic or radiopharmacy, although further process work designed to guarantee >95% incorporation and dispensing with the purification would be desirable. As with the SE-HPLC, recoveries from the cartridge as the desired radiolabeled peptide were again generally in the 80% range, with losses spread throughout the system (i.e., in tubing, other fractions, syringes, and so forth), most probably due to handling losses.
Handling and use of the 68Ge/68Ga generator was simple, with its reliability established by hundreds of elutions over the course of this work. The finding that the generator could be usefully eluted in much smaller volumes of HCl enabled us to devise reagents that might be adapted to a kit formulation for ready and easy 68Ga labeling in the future. Three or 4 elutions of the generator are readily made in any 6- to 8-h period, and the generator has ingrowth of 68Ga to 50% of its maximum after just only over 1 h since the previous elution. The major cold metal contamination detected by atomic absorption was tin (data not shown), probably in the stannate form, as described earlier (12), but which did not significantly affect 68Ga-labeling procedures. This stannic oxide generator has been available for 23 y, although a new type using 68Ge adsorbed onto a titanium dioxide support has recently become commercially available and may replace the Loc’h generator in time. However, the newer generator’s performance has not been adequately described in the literature as yet.
CONCLUSION
These studies demonstrate that the interweaved issues involved in bringing a bsAb targeting system for PET to clinical fruition can be addressed successfully. The outstanding target-to-tissue ratios obtained in these experiments encourage us to continue to refine and develop our agents still further, in the hope that they may soon realize their promise clinically, not only for improved cancer imaging but also for other diseases that have appropriate molecular targets for bsAbs.
Footnotes
Received Apr. 24, 2003; revision accepted Sep. 25, 2003.
For correspondence or reprints contact: Gary L. Griffiths, Immunomedics, Inc., 300 American Rd., Morris Plains, NJ 07950.
E-mail: ggriffiths{at}immunomedics.com