


Abstract
Random bacteriophage (phage) display peptide libraries have traditionally been used for the selection of clones that bind specific tissues, tumors, and antigens. However, once the targeting peptide is synthetically produced, it often displays a lower affinity than the original phage because of a lack of avidity effects and removal from the virion surface. We hypothesized that multivalent bifunctional phage displaying peptides that target novel molecular biomarkers would facilitate the in vivo imaging of cancer. This study provides proof of principle for the use of phage displaying multiple melanocortin-1 receptor–homing peptides for the pretargeting and subsequent imaging of murine melanomas in vivo. Methods: A 2-step melanoma pretargeting–imaging system was developed by first generating and biotinylating phage that displayed up to 5 copies of α-melanocyte–stimulating hormone (α-MSH) peptide analogs. Second, streptavidin was conjugated to diethylenetriaminepentaacetic acid for the purpose of radiolabeling with 111In. Results: The specificity of the MSH2.0 phage for the B16-F1 melanoma was demonstrated both in vitro and in vivo. In vitro micropanning assays with phage at inputs of 107 and 106 transducing units per milliliter resulted in ∼200- and ∼1,000-fold-greater recovery of the MSH2.0 phage over the background, respectively. In vivo distribution studies indicated that melanoma uptake values were 2.6 ± 1.1, 0.6 ± 0.2, and 1.0 ± 0.1 (mean ± SD) percentage injected dose per gram at 0.5, 6, and 24 h after the injection of 111In-radiolabeled streptavidin (111In-SA). The accumulation of radioactivity within the tumor was 1.8 times greater for the biotinylated MSH2.0 phage than for the biotinylated wild-type phage. These data, combined with reduction by 2.4-fold through competition with a nonradiolabeled α-MSH peptide analog, indicated the specific targeting of melanoma tumors in vivo. SPECT/CT image analysis of B16-F1 melanoma–bearing mice showed that intravenously injected biotinylated α-MSH phage were retained within melanoma tumors at 4 h after injection of 111In-SA. Conclusion: This study demonstrated the use of multivalent bifunctional phage in a 2-step pretargeting–imaging system.
Bacteriophage (phage) display is a well-established combinatorial chemistry technique that uses a population of fd filamentous phage genetically modified to display a library of random peptides on their surfaces (1). One such library that is commonly used is the fd-tet fUSE5 library. This library contains phage with modified coat protein III displaying up to 5 copies of a fused random peptide on the tip of the phage particle. Affinity selection of phage constructs is performed through multiple rounds of stringent washing and amplification (1). Traditionally, the selected targeting peptides are synthesized, and their in vitro and in vivo binding characteristics are then determined (1–3). Phage display has been proven useful in the identification of novel binding ligands for proteins, carbohydrates, and nucleic acids (4–8).
Recently, phage display was used for the discovery of new and unique cancer-imaging peptides (9–11). Of these tumor-homing peptides, only a handful have been developed into noninvasive cancer-imaging agents (12,13), perhaps because of differences between peptides fused to phage coat proteins and chemically synthesized peptides (10,14). The conformational structures of the selected fused peptides are affected by the microenvironment created by the surface of the phage particle. Thus, synthetic peptides derived from sequences selected on phage can have binding characteristics different from those of peptides displayed on the surface of the phage particle. Another disadvantage of the use of selected synthetic peptides for the imaging of cancer is that the covalent addition of radiochelators or fluorescent tags to a small peptide can often have deleterious effects on the affinity of the peptide for binding to its target (15). Conversely, the covalent addition of imaging tags to a phage particle has minimal effects on the ability of the displayed peptide to bind to its target (16). It would therefore be advantageous to use a labeled phage for validation of the tumor-targeting propensity of a selected peptide.
Direct imaging with very high-molecular-weight phage particles presents its own set of challenges. Previous studies of the distribution and clearance profiles of fd filamentous phage in mice (17,18) demonstrated clearance of phage through the reticuloendothelial system; therefore, the accumulation of large amounts of phage in the liver, spleen, and lungs was observed. Phage intravenously injected into a mouse are completely cleared from the tissues in ∼48 h, although they have an average blood half-life of ∼15 min (17). Implementation of a 2-step pretargeting system could allow for the clearance of the majority of the phage before injection of the imaging label. We propose that the use of biotinylated phage displaying tumor-homing peptides in combination with radiolabeled streptavidin may offer a novel approach to cancer imaging in animals.
Here we used a well-described, high-affinity peptide–receptor system, namely, the melanocortin-1 receptor (MC1 receptor) and its ligand, the peptide α-melanocyte–stimulating hormone (α-MSH) (19,20), for the development of a phage-based pretargeting strategy for melanoma imaging. The MC1 receptor possesses a nanomolar affinity for its ligand and has been found to be overexpressed on up to 80% of human melanomas from patients with metastatic lesions (21). α-MSH peptide analogs were engineered and fused with phage coat protein III by use of the fUSE5 vector for use as melanoma-imaging agents. A biotinylated fUSE5 phage displaying an α-MSH peptide analog (bio-MSH2.0 phage) was able to target the cell surface receptor, the MC1 receptor (20), on B16-F1 mouse melanoma cells both in vitro and in vivo. Analysis of in vivo distribution data revealed selective tumor uptake and retention within syngeneic grafted B16-F1 melanoma tumors in C57BL/6 mice over a 24-h time period (after injection of 111In-radiolabeled streptavidin [111In-SA]). Imaging studies revealed that intravenously injected bio-MSH2.0 phage resulted in tumor targeting at 4 h after injection of 111In-SA. These studies provided proof of principle that phage display technology coupled with a pretargeting approach could offer a powerful and efficient means for enhancing the discovery of novel imaging peptides and assessing their tumor-targeting propensities in vivo.
MATERIALS AND METHODS
Materials
Cell culture reagents were purchased from Invitrogen. All other chemicals were purchased from Sigma Chemical Co., unless otherwise stated.
Cell Line
B16-F1 mouse melanoma cells (American Type Culture Collection) were grown in complete medium (RPMI 1640 medium, 10% fetal bovine serum, 2 mM l-glutamine, and gentamicin at 48 μg/mL) at 37°C in 5% CO2. The cell line was tested for pathogens before injection into mice.
Generation of Clonal Phage Populations
Clonal phage populations of 2 α-MSH peptide analogs, MSH1.0 (ASYSMEHFRWGRPVAG) and MSH2.0 (AMEHFRWGRPVGSGSGSGSVWYAG), were generated by use of the fUSE5 vector (generous gift from George Smith, University of Missouri, Columbia, MO) as previously described (22). Briefly, the fUSE5 vector was digested with the SfiI restriction endonuclease. Equal molar amounts of sense and antisense phosphorylated DNA oligonucleotides were hybridized in a buffer containing 50 mM NaCl, 10 mM Tris(hydroxymethyl)aminomethane (Tris) (pH 8.0), and 1 mM ethylenediaminetetraacetic acid by heating to 94°C and slowly cooling to room temperature. A room-temperature DNA ligation reaction was performed for insertion of the hybridized DNA into the fUSE5 vector. Cells of Escherichia coli K91 Blue Kan (a host strain for filamentous phage) were electroporated in the presence of the ligated vector and amplified in 2 mL of NZY broth (NZ amine with yeast extract) with tetracycline at 20 μg/mL. The proper DNA sequence was verified by DNA sequencing at the University of Missouri DNA Core Facility. Infected cultures were expanded to 1 L, and the amplified phage were precipitated with polyethylene glycol. The polyethylene glycol–precipitated phage were further purified by cesium chloride ultracentrifugation. After dialysis in Ca2+- and Mg2+-free phosphate-buffered saline (PBS), the phage particle concentrations in virions were determined spectrophotometrically. E. coli K91 Blue Kan was used in a titration assay to quantitate the concentrations of infectious units (transducing units [TU]) in the purified samples. The resulting phage titers were expressed in terms of TU per milliliter.
Phage Dot Blots
Two sequential 1-μL dots of phage solution (MSH1.0, MSH2.0, or wild type [WT]) containing 1011 virions per milliliter (23) were allowed to air dry on a 0.2-μm nitrocellulose membrane. This step was followed by blocking overnight at 4°C with 6% bovine serum albumin in Tris-buffered saline (TBS; pH 7.0). The presence of phage particles was detected by the addition of a rabbit polyclonal antiphage antibody (courtesy of George Smith) at a dilution of 1:1,000. The primary antibody was incubated with the membrane at room temperature for 1 h, this step was followed by extensive washing with TBS plus 1% polysorbate 20. Next, an antirabbit antibody conjugated to horseradish peroxidase (Santa Cruz Biotechnology) was added at a 1:1,500 dilution and incubated with the membrane at room temperature for 1 h. The membrane was washed with TBS plus 1% polysorbate 20, rinsed with distilled H2O, and immersed in 4CN peroxidase substrate solution (KPL). This same procedure was followed for the detection of the C terminus of α-MSH with a sheep antibody to the α-MSH C terminus (Chemicon) and an antisheep antibody conjugated to horseradish peroxidase (Santa Cruz Biotechnology).
Micropanning of Biotinylated and Nonbiotinylated Phage
B16-F1 melanoma cells were grown to 80% confluence in 35-mm tissue culture plates (Fisher Scientific) and removed with PBS plus 2 mM ethylenediaminetetraacetic acid. The suspension of cells was incubated in complete medium with biotinylated or nonbiotinylated MSH2.0 phage or WT phage at inputs of 108, 107, and 106 TU/mL for 1.5 h at 37°C. Next, the cells were washed 5 times with PBS. The phage titers in the final cell suspension, containing 2.5% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), were determined with E. coli K91 Blue Kan (recovered phage).
Biotinylation of Phage
NHS-PEO4-biotin (Pierce Biotechnology; 588.67 Da) was dissolved in dimethyl sulfoxide and added to a suspension of phage in 100 mM NaH2PO4 (pH 7.4 achieved with NaOH) at a 1,000-fold molar excess relative to the phage particles, and the mixture was rotated at room temperature for 2 h. The reaction was stopped with a final concentration of 400 mM ethanolamine (pH 9.0), and the mixture was rotated at room temperature for 1 h. Next, the phage preparation was extensively dialyzed against TBS to remove excess free biotin.
Covalent Attachment of DTPA to Streptavidin
2-(4-Isothiocyanatobenzyl)-diethylenetriaminepentaacetic acid (Macrocyclics) was dissolved in carbonate buffer (0.5 M sodium carbonate; pH 9.5) and added to streptavidin suspended in PBS, yielding a mixture containing 50-fold more diethylenetriaminepentaacetic acid (DTPA) than streptavidin. The mixture was rotated overnight at 4°C. Excess DTPA was removed with Zeba Desalt Spin Columns (Pierce Biotechnology).
Stability of DTPA-Streptavidin
DTPA-streptavidin was radiolabeled with 111In by the addition of 0.37 MBq to 0.1 μg of DTPA-streptavidin in 10 mM N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (HEPES; pH 7.0); this step was followed by incubation at 37°C for 1.5 h. Excess 111In was removed with Zeba Desalt Spin Columns. The resulting purified samples were divided into aliquots, which were placed in 1 of 3 different buffers: HEPES (10 mM; pH 7.0), PBS, and normal mouse serum. The samples were incubated either at 25°C or at 37°C for 1, 4, 8, or 24 h. Zeba Desalt Spin Columns were used to remove free 111In, and the resulting amounts of radioactivity in the final samples and the previously purified sample were compared.
Biodistribution and SPECT/CT of Biotinylated Phage and DTPA-Streptavidin
All animal studies were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (24) and the policy and procedures for animal research at the Harry S. Truman Veterans Memorial Hospital. C57BL/6 mice were obtained from Harlan. The animals were provided with water ad libitum and fed biotin-free rodent chow for 5 d before injection. C57BL/6 mice receiving 106 B16-F1 mouse melanoma cells were subcutaneously inoculated in the right flank or intrascapular region. At 10 d after inoculation, tumors weighed approximately 0.05–0.1 g. Groups of 3 mice received tail vein injections of 5 × 1012 virions of biotinylated phage. Phage were allowed to circulate for 4 h (23), and then a tail vein injection of 1.85 MBq of 111In-SA was given. Mice were sacrificed by cervical dislocation at 0.5, 2, 4, 6, or 24 h after injection for the purpose of harvesting the organs of interest. Each organ was weighed, and counts were determined with a Wallac 1480 automated γ-counter (Perkin-Elmer). Five mice were used for competition studies. These mice received tail vein injections of 5 × 1012 virions of the bio-MSH2.0 phage mixed with 100 μg of [Nle4,d-Phe7]α-MSH (NDP); 4 h later, an injection of 1.85 MBq of 111In-SA was given.
Mice used for the purpose of imaging received tail vein injections of 5 × 1012 virions of biotinylated phage, and then an injection of 7.40 MBq of 111In-SA was given. The mice were euthanized with carbon dioxide at 4 h after injection of the radiolabel. Imaging was performed in the USVA Biomolecular Imaging Center at the Harry S. Truman Veterans Memorial Hospital by use of a microSPECT/CT system (Siemens Preclinical Solutions) equipped with a high-resolution 2-mm pinhole collimator. At the time of the SPECT scan, there was approximately 2.81 MBq of 111In-SA remaining in the mouse. A total of 1,349,826 counts were acquired by SPECT imaging. The 78 × 78 × 102 volumetric isotropic voxel image matrix data were reconstructed by use of a 3-dimensional ordered-subset expectation maximization algorithm. This instrument was also equipped with an 80-kVp x-ray source and software for 360° CT data acquisition. A fanbeam (Feldkamp) filtered backprojection small-animal CT image reconstruction algorithm was used for the production of a 512 × 512 × 960 isotropic voxel image matrix. Coregistered SPECT and CT data were visualized by use of Amira 3.1 software (TGS).
RESULTS
The MC1 receptor–α-MSH receptor ligand system was used to develop and characterize a new phage-based pretargeting strategy for melanoma imaging with biotinylated phage and 111In-SA. Phage displaying α-MSH peptide analogs were generated by use of the fUSE5 phage display vector (Table 1). These phage were then sequenced to confirm that the coding inserts were within the proper reading frame. The MSH1.0 phage clone was designed to display the native α-MSH peptide sequence displayed at the N terminus of phage coat protein III, whereas the MSH2.0 phage clone contained a modified α-MSH sequence. MSH2.0 lacked 3 nonessential amino acids at the amino terminus of the α-MSH sequence and contained a linker peptide between it and phage coat protein III. The phage clones were probed with an anti–α-MSH antibody to determine whether the peptide sequences were accessible and reactive with an antibody that recognizes the core receptor–binding residues for the MC1 receptor. A phage particle dot blot assay was performed with the anti–α-MSH antibody (Fig. 1A). The results of the phage immunoassay revealed that the native α-MSH sequence displayed on the MSH1.0 phage clone was not immunoreactive, whereas MSH2.0 was recognized by the antibody. These results demonstrated that the α-MSH sequence on the MSH2.0 phage clone had greater reactivity and predicted more biologic activity than the peptide on the MSH1.0 phage clone.

(A) Presence of displayed α-MSH peptide analogs was probed by ability of antibody to C terminus of α-MSH peptide to recognize and bind to phage-displayed peptide. Two 1-μL dots of phage solution containing 1011 virions per milliliter were immobilized on 0.2-μm nitrocellulose, and presence of phage particles was demonstrated with antiphage antibody (1:1,000 dilution). Presence of recognizable α-MSH peptide was determined with antibody to C terminus of α-MSH peptide (1:1,000). Complexes were detected with secondary antibody conjugated to horseradish peroxidase (1:1,500). (B) Ability of displayed peptide to be recognized and bound by MC1 receptor on cultured B16-F1 melanoma cells was determined by micropanning. Suspension of B16-F1 melanoma cells was incubated with bio-MSH2.0 phage or nonbiotinylated MSH2.0 phage at inputs of 108, 107, and 106 TU/mL for 1.5 h on rotator at 37°C. Cells were then extensively washed with PBS, and final wash containing 2.5% CHAPS was used for determination of phage titers with E. coli. Bio = biotinylated.
Amino Acid Sequences of Native α-MSH Peptide and α-MSH Peptide Analogs Displayed on Phage Particles
A micropanning assay was used to test the specific binding of the MSH2.0 phage (Fig. 1B). After incubation of various amounts of the phage with B16-F1 cells, the cells were washed and lysed, and the phage titers were determined with E. coli K91 Blue Kan cells to quantify the amounts of phage still bound to or internalized by the cells. Micropanning of the MSH2.0 phage at an input of 108 TU/mL against B16-F1 mouse melanoma cells and then extensive washing resulted in an 18-fold-higher recovery of the MSH2.0 phage than of the WT phage. The WT phage nonspecific background decreased to 200-fold below that of the MSH2.0 phage at an input of 107 TU/mL. When a phage input of 106 TU/mL was incubated with the B16-F1 cells, no WT phage was recoverable. These results indicated the selective binding of the phage displaying the MSH2.0 peptide to B16-F1 mouse melanoma cells.
The MSH2.0 and WT phage were biotinylated with NHS-PEO4-biotin as part of the biotin-streptavidin pretargeting strategy. The biotin linker used contained an extended hydrophilic polyethylene oxide (PEO4) spacer arm that may minimize possible steric hindrances involved with streptavidin binding to biotin molecules on the surface of a large phage particle. Biotin on the surface of the virion was verified by inhibition of phage infection of E. coli in the presence of streptavidin at 0.05 μg/mL (data not shown).
To determine whether biotinylation of the MSH2.0 phage affected MC1 receptor binding, we repeated the assay with biotinylated phage constructs (Fig. 1B). Figure 1B shows that micropanning of the bio-MSH2.0 phage and the biotinylated WT phage (bio-WT phage) against cultured B16-F1 mouse melanoma cells mirrored the findings obtained with the nonbiotinylated phage. These results demonstrated that the addition of PEO4-biotin to the surface of the phage particle resulted in no significant alteration of phage binding to cultured B16-F1 mouse melanoma cells in vitro.
Streptavidin, the second component of the 2-step targeting system, was conjugated to the metal chelator DTPA through an isothiocyanate linkage. The DTPA-streptavidin complex exhibited greater than 90% labeling efficiency. The stability of the 111In-SA compound was investigated to ensure the feasibility of extended in vivo use. 111In-SA was incubated in 3 different solutions, PBS, 10 mM HEPES, and mouse serum, and at 2 different temperatures, 25°C and 37°C (data not shown). After 24 h, samples incubated in mouse serum maintained ∼80% of their original activity, whereas all other samples exhibited no statistically significant change in their retained activity. Thus, the 111In-SA complex was sufficiently stable for in vivo applications.
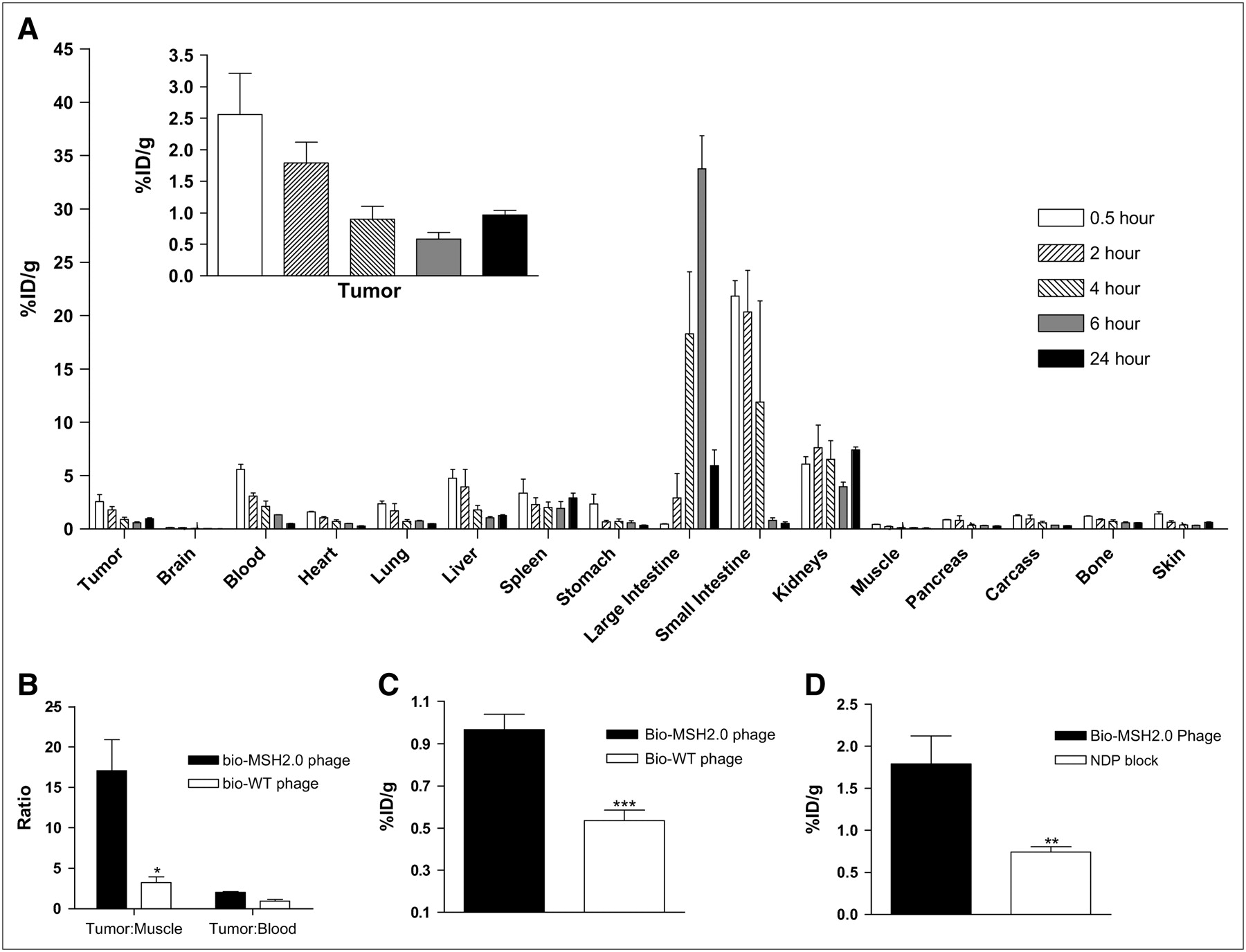
The distribution and tumor-targeting properties of the pretargeted bio-MSH2.0 phage followed by 111In-SA were investigated in vivo with the B16-F1 mouse melanoma model. The bio-MSH2.0 phage was injected into C57BL/6 mice bearing syngeneic grafted B16-F1 melanoma tumors and allowed to circulate for 4 h. At predetermined times after the injection of 111In-SA, mice were sacrificed, and counts in their organs and tissues were determined to examine the distribution of the radioactivity. The clearance of 111In radioactivity was found to be primarily through the urinary and hepatobiliary systems (Fig. 2A). The tumor uptake of 111In-SA appeared initially to be attributable to the blood volume within the tumor. However, at 24 h after injection, the retention of 111In-SA within the tumor was clearly observed, with a percentage injected dose per gram (%ID/g) of 1.0 ± 0.1 (mean ± SD) (Fig. 2A) and a ratio of activity in the tumor to activity in the blood of 2.0 ± 0.2 (Fig. 2B).

(A) Biodistribution was determined through the use of C57BL/6 mice with syngeneic grafted B16-F1 melanoma tumors. These mice received tail vein injections of 5 × 1012 virions of bio-MSH2.0 phage. Four hours later, mice received tail vein injections of 1.85 MBq of 111In-SA. Mice were sacrificed at 0.5, 2, 4, 6, and 24 h after injection, and their organs were harvested for counting. (B) Tumor-to-muscle ratios and tumor-to-blood ratios for both bio-MSH2.0 phage and bio-WT phage were compared. C57BL/6 mice bearing B16-F1 tumors received tail injections of 5 × 1012 virions of bio-WT phage and, 4 h later, 1.85 MBq of 111In-SA. (C) At 24 h after injection of 111In-SA, levels of accumulation of radioactivity within tumor were compared for bio-MSH2.0 phage and bio-WT phage. (D) C57BL/6 mice with syngeneic grafted B16-F1 melanoma tumors received tail vein injections of either 5 × 1012 virions of bio-MSH2.0 phage or 5 × 1012 virions of bio-MSH2.0 phage mixed with 100 μg of NDP. Four hours later, mice received tail vein injections of 1.85 MBq of 111In-SA. Mice were sacrificed at 2 h after injection, and levels of accumulation of radiolabel within tumor were compared for both phage. *P = 0.001; **P = 0.002; ***P = 0.003.
To validate that the radioactivity found within the tumor at 24 h after injection was attributable to binding and retention of the bio-MSH2.0 phage, we examined the distribution of the bio-WT phage followed by 111In-SA. The in vivo distribution of 111In-SA alone was also examined. Tumor uptake at 24 h after injection was found to be 0.4 ± 0.1 %ID/g, and whole-body clearance was found to be similar to previously reported results (25). Examination of the ratio of activity in the tumor to activity in the muscle (tumor-to-muscle ratio) for the various phage constructs revealed a preferential localization of 111In-SA within the B16-F1 tumor because of the presence of the bio-MSH2.0 phage. The tumor-to-muscle ratio for the bio-MSH2.0 phage was 17.5 ± 3.7 at 24 h after injection (Fig. 2B). In comparison, the tumor-to-muscle ratio for the bio-WT phage was 3.0 ± 0.6 at 24 h after injection (P = 0.001). The accumulation of radioactivity within the tumor at 24 h after injection was 1.8 ± 0.2 times greater in mice injected with the bio-MSH2.0 phage than in mice injected with the bio-WT phage (P = 0.003) (Fig. 2C). These data suggested that the targeting of the bio-MSH2.0 phage to the B16-F1 tumor in vivo was α-MSH peptide mediated.
To validate that the radioactivity found within the tumor was attributable to specific binding of the bio-MSH2.0 phage, nonradioactive NDP peptide was coinjected with the bio-MSH2.0 phage (Fig. 2D). NDP is a potent, protease-resistant peptide analog of α-MSH that is commonly used in competition assays (26). The presence of the NDP peptide reduced the retention of 111In-SA and the bio-MSH2.0 phage by 2.4 ± 0.4-fold (P = 0.002), demonstrating the specificity of binding for the bio-MSH2.0 phage in vivo.
The 2-step pretargeting approach was used for the in vivo imaging of solid mouse melanoma tumors. C57BL/6 mice bearing B16-F1 melanomas were imaged at 4 h after the injection of 111In-SA by small-animal SPECT/CT. SPECT of the mice clearly demonstrated the accumulation of 111In-SA and thus of the bio-MSH2.0 phage within the tumor. SPECT/CT images representative of melanoma-bearing mice injected with the bio-MSH2.0 phage are shown in Figure 3. These results strongly suggested that the use of biotinylated phage displaying tumor-homing peptides combined with radiolabeled streptavidin in a 2-step imaging process is a valuable approach for the validation of the ability of selected peptides to target and image tumors.

C57BL/6 mice with syngeneic grafted B16-F1 melanoma tumors received tail vein injections of 5 × 1012 virions of bio-MSH2.0 phage and, 4 h later, 7.40 MBq of 111In-SA. Mice were sacrificed at 4 h after injection, and image data were acquired with small-animal SPECT/CT system.
DISCUSSION
Two multivalent bifunctional phage constructs, MSH1.0 and MSH2.0, were developed for the in vivo imaging of solid B16-F1 mouse melanoma tumors. MSH1.0, with the native α-MSH peptide sequence (Table 1), was not immunoreactive, possibly because of steric hindrance or conformational changes induced by the direct fusion with coat protein III (Fig. 1A). In MSH2.0, the first 3 amino acids of the native α-MSH sequence were removed in an effort to place the conserved receptor-binding sequences closer to the amino terminus, where they may be more readily accessible. A spacer sequence of 4 Gly-Ser amino acid repeats and a Val-Trp-Tyr tripeptide were then added to the carboxy terminus of the MSH2.0 sequence to protect the configuration of the peptide from any influence of secondary structures from coat protein III. MSH2.0 was found to be immunoreactive, and both biotinylated and nonbiotinylated MSH2.0 phage constructs were recognized and bound by the appropriate receptor, the MC1 receptor, in vitro. Surprisingly, the ability to locate a functioning α-MSH targeting sequence on the phage particle was not as straightforward as expected. Directly fusing the targeting sequence to coat protein III appeared to alter the peptide conformation and limit ligand recognition. A spacer sequence had to be added between the α-MSH peptide and the coat protein to allow the targeting sequence to adopt a bioactive conformation. Had the α-MSH peptide sequence undergone phage display–based affinity maturation, the optimal active conformation of the peptide on the phage coat protein would have been accounted for in the affinity selection process.
α-MSH peptide sequences bind to their cognate MC1 receptors with high affinity and specificity. Numerous α-MSH peptide analogs have been chemically synthesized, radiolabeled, and examined for melanoma imaging and therapy. In comparison with pretargeted phage, 99mTc-radiolabeled linear α-MSH peptide analogs displayed higher tumor uptake values at early time points but similar tumor uptake values at 4 h after injection in B16-F1 melanoma–bearing mice (27). Tumor uptake values for 99mTc-radiolabeled CGC-NDP and MAG2-NDP (Cys-Gly-Cys = CGC; mercaptoacetylglycylglycyl-α-aminobutyrate = MAG2) at 4 h were 0.56 ± 0.14 and 0.74 ± 0.15 %ID/g; the corresponding value for the pretargeted phage was 0.9 ± 0.4 %ID/g. Conjugates of linear α-MSH peptide analogs and 1,4,7,10-tetraazacyclododedane-N,N′,N″,N‴-tetraacetic acid (DOTA) displayed tumor uptake values ranging from 0.65 ± 0.05 to 7.77 ± 0.35 %ID/g at 4 h after injection and from 0.23 ± 0.01 to 2.32 ± 0.15 %ID/g at 24 h (28); the pretargeted phage exhibited tumor uptake values of 0.9 ± 0.4 and 1.0 ± 0.1 %ID/g at 4 and 24 h after injection, respectively. Pretargeted phage exhibited tumor-targeting properties similar to those of 99mTc-labeled linear α-MSH peptide analogs but less notable than those of the optimal radiolabeled DOTA-conjugated peptides. Additional peptide structural engineering is likely to be required to achieve tumor uptake values of 10–20 %ID/g, which have been reported for metal-cyclized α-MSH analogs (29,30).
Upon analysis of the in vivo distribution of 111In-SA, it was evident that the clearance of the activity through the urinary tract led to kidney uptake and retention. The kidney retention of 111In-SA even at 24 h after injection was expected (Fig. 2A). It is known that streptavidin is a proteolytically stable protein and that the retention of streptavidin in the kidneys is largely attributable to its resistance to degradation (31). However, the observed kidney uptake value, about 5 %ID/g, was lower than many of the values reported in the literature (25,31–33). This finding is likely a result of the lysine residues within streptavidin being altered after covalent attachment of the chelator DTPA. Previous work showed that arginine and lysine residues within the streptavidin protein contribute to the accumulation of streptavidin in the kidneys (25). Modification of the lysine residues shifts the biodistribution of streptavidin so that the accumulation of streptavidin is also seen in the liver and spleen (32).
Comparison of the in vivo distributions of 111In-SA in mice injected with either the bio-MSH2.0 phage or the bio-WT phage revealed no change in the clearance of the radiolabel. However, tumor accumulation of the radiolabel was 1.8-fold higher with the bio-MSH2.0 phage than with the bio-WT phage (Fig. 2C) and 2.7-fold higher than with 111In-SA alone. Furthermore, there was a noteworthy difference in the tumor-to-muscle ratios of 17.5 and 3.0 for the bio-MSH2.0 phage and the bio-WT phage at 24 h after injection, respectively (Fig. 2B). The in vivo targeting specificity of the bio-MSH2.0 phage for the MC1 receptor was examined in an in vivo competitive binding study with the high-affinity α-MSH peptide analog NDP (Fig. 2D). The competitive binding study was performed at 2 h after injection because of the pharmacokinetics of the NDP blocking peptide (29). Finally, SPECT/CT of B16-F1 tumor–bearing mice confirmed the accumulation of the radiolabel within the tumor. These mice also had activity in the kidneys and intestines, a finding that is reflective of the distribution data, which indicated the clearance of activity through the urinary and hepatobiliary systems. Further analysis of the mice used for imaging revealed an uptake value (5.2 ± 0.8 %ID/g) for 111In-SA and the bio-MSH2.0 phage within the tumor that was higher than the uptake value (0.9 ± 0.3 %ID/g) obtained in the biodistribution studies. The increased tumor accumulation was probably attributable to the higher dose of 111In-SA administered. These data provided the proof necessary to demonstrate that the combination of phage display technology and the pretargeting strategy allows for assessment of the in vivo tumor-targeting ability of a peptide. This newly described technique will prove useful for the in vivo screening of newly discovered phage displaying selected tumor-targeting peptides.
It has been envisioned that a pretargeting strategy can be used to rapidly assess the tissue-targeting properties of novel peptide sequences discovered through phage display selection in vivo. The phage described in this report were developed as a proof-of-principle model for pretargeted-phage imaging with a well-characterized biologic system. The strength of the pretargeting approach is that it supports the efficient discovery of novel peptide imaging agents. There is a need to expand the repertoire of peptides that bind to novel cellular targets. At present, a few peptide-based imaging agents are used in clinics (34), and numerous radiolabeled peptides are undergoing preclinical research, but they bind to only a few unique cellular targets (15). Phage display technology is a powerful tool for the discovery of peptide ligands for novel molecular targets, and pretargeted-phage imaging will have a positive impact on its efficiency and success.
CONCLUSION
This study demonstrated the ability of a pretargeted bio-MSH2.0 phage to target and image melanomas in vivo. This approach provides a simple, cost-effective, and flexible imaging strategy that is useful for various applications, including the in vivo characterization of peptides and their tumor-targeting propensities.
Acknowledgments
This work was supported in part by a Merit Review Award from the Veterans Administration, by grant NIH P50 CA103130-01, and by the University of Missouri Radiosciences Institute and the Department of Energy (grant ER 61661). The authors would like to acknowledge the contributions of Tiffani Shelton, Lisa Watkinson, Terry Carmack, Marie T. Dickerson, Said Daibes Figueroa, and George P. Smith.
References
- Received for publication May 26, 2006.
- Accepted for publication November 21, 2006.





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.