


Abstract
Prostate-specific membrane antigen (PSMA)–targeted radiotherapy of prostate cancer (PCa) has emerged recently as a promising approach to the treatment of disseminated disease. A small number of ligands have been evaluated in patients, and although early tumor response is encouraging, relapse rate is high and these compounds localize to the parotid, salivary, and lacrimal glands as well as to the kidney, leading to dose-limiting toxicities and adverse events affecting quality of life. We envision that dual-target binding ligands displaying high affinity for PSMA and appropriate affinity for human serum albumin (HSA) may demonstrate a higher therapeutic index and be suitable for treatment of PCa by targeted α-therapy. Methods: Six novel urea-based ligands with varying affinities for PSMA and HSA were synthesized, labeled with 131I, and evaluated by in vitro binding and uptake assays in LNCaP cells. Four compounds were advanced for further evaluation in a preclinical model of PCa. The compounds were compared with MIP-1095, a PSMA ligand currently in clinical evaluation. Results: The compounds demonstrated affinity for PSMA on the order of 4–40 nM and affinity for HSA in the range of 1–53 μM. Compounds with relatively high affinity for HSA (≤2 μM) showed high and sustained blood-pool activity and reduced uptake in the kidneys. 131I-RPS-027, with a 50% inhibitory concentration (PSMA) of 15 nM and a dissociation constant (HSA) of 11.2 μM, cleared from the blood over the course of 48 h and showed good tumor uptake (10 percentage injected dose per gram) and retention and a greater than 5-fold decrease in kidney uptake relative to MIP-1095. The tumor-to-kidney ratio of 131I-RPS-027 was greater than 3:1 at 24 h after injection, increasing to 7:1 by 72 h. Conclusion: RPS-027 shows dual targeting to PSMA and albumin, resulting in a high tumor uptake, highly favorable tissue distribution, and promising therapeutic profile in a preclinical model of prostate cancer. In comparison to existing ligands proposed for targeted therapy of prostate cancer, RPS-027 has tumor-to-tissue ratios that predict a significant reduction in side effects during therapy. Using iodine/radioiodine as a surrogate for the radiohalogen 211At, we therefore propose dual-target binding ligands such as RPS-027 as next-generation radiopharmaceuticals for targeted α-therapy using 211At.
Targeted radiotherapy (TRT), the systemic application of radiolabeled drugs or biologicals to treat disseminated cancers, has reached a tipping point with the pending approval of Lutathera (177Lu-DOTATATE; Advanced Accelerator Applications Inc.) for the treatment of neuroendocrine cancer (1) and with the publication of promising clinical results for the treatment of metastatic castrate-resistant prostate cancer with radiolabeled small-molecule inhibitors of prostate-specific membrane antigen (PSMA) (2–8). These low-molecular-weight peptidomimetic structures rapidly distribute throughout the body and clear rapidly from the blood (9). In contrast to larger targeting constructs such as immunoglobulins, the low molecular weight facilitates capillary transport and penetration into solid tumors (10,11). Such rapid and systemic tissue access is desirable for targeting of tumors and enabling access to widely disseminated tumor lesions (12). However, rapid tissue access and distribution may also enable off-target interactions in normal tissues that express physiologic levels of the target or may lead to concentration of the therapeutic radiopharmaceutical in excretory organs such as the kidney. These interactions may lead to normal tissues receiving radiation doses that cause potentially fatal irreversible tissue damage (13–15).
To date, TRT of prostate cancer with low-molecular-weight PSMA inhibitors has not required nephroprotection schemes despite demonstrable renal accumulation of the radiotherapeutics, though renal radiation dose must be considered and tracked because modest response rates and high rates of relapse mean that patients receive several cycles of β-emitting compounds over their course of treatment (2,3). PSMA therapy is associated, however, with concentration of therapeutic radiopharmaceuticals in the lacrimal, salivary, and parotid glands of patients; irradiation of these tissues can cause glandular dysfunction leading to xerostomia or dry eye (2,3). Off-target effects may become severe when patients receive multiple cycles of β-emitter or in the case when the therapeutic radionuclide chosen is an α-emitter (16).
Preliminary experiments in preclinical models have suggested that uptake in these structures can be reduced by treatment with 2-(phosphonomethyl)-pentandioic acid, a high-affinity PSMA inhibitor (17,18). An alternative to such a pharmacologic displacement strategy is to modulate normal tissue access via compartmentalization of the radiopharmaceutical in safe-haven tissues. The blood pool becomes an attractive space to consider compartmentalizing α-emitters because it would allow access to the tumor vascular bed but should hinder rapid diffusion into normal tissues. At any one time, about 60%–70% of the entire blood volume in a human is contained in the veins (19). Hence, an α-emitter compartmentalized to the blood pool will reduce off-target effects because most veins have diameters considerably greater (200–5,000 μm) than the α-particle length of radionuclides such as 211At (25–100 μm) (20). Albumin targeting is ideal for such an application because the protein is abundant in blood serum, has a long physiologic half-life, and is known to reversibly bind small negatively charged or hydrophobic molecules (21–23). Reversible binding to albumin has been shown to extend blood clearance and change tissue distribution of biologics (24), peptides (25), drugs (26), contrast agents (27), and radiopharmaceuticals (23,28).
We report herein the results of our efforts to design a new class of dual-target binding small molecules that display high affinity (low nanomolar) chemical targeting to prostate cancer cells through interaction with PSMA, and display a range of moderate (micromolar) affinities for albumin. We show that modulation of albumin affinity among potent PSMA ligands has a demonstrable effect on tumor targeting and normal organ compartmentalization and residence time. To study this phenomenon, we use iodine/radioiodine as a surrogate for the α-emitting radiohalogen 211At (29–31), and our results support investigation of such dual-target binding ligands as next-generation radiopharmaceuticals for α-targeted therapy of prostate cancer using 211At.
MATERIALS AND METHODS
Chemistry
A full description of the synthesis of compounds RPS-001, RPS-005, RPS-020, RPS-022, RPS-023, RPS-025, RPS-026, and RPS-027 (Fig. 1) and the corresponding organostannane precursors 11–17 (the organostannanyl analog of RPS-026 was not prepared) can be found in the supplemental materials (available at http://jnm.snmjournals.org).

Structures of RPS series compounds and their affinities for PSMA and HSA.
Radiosynthesis
Radiolabeling was performed as previously described with modification (2). One hundred microliters of a 250 μg/mL solution of organnostannane precursor 11–17 in EtOH was added to a vial containing 74–740 MBq (2–20 mCi) of Na124I or Na131I in 30–60 μL of aqueous NaOH. A 15% v/v H2O2/AcOH solution was prepared, and 50 μL were transferred immediately to the reaction vial. The reaction was mixed for 20 s and let stand for 5 min at room temperature. It was then diluted with 3 mL of H2O and passed through a preactivated SOLA cartridge (Thermo Scientific). The cartridge was washed with H2O (3 mL) and dried with air. The radiolabeled intermediate was eluted into a second vial with 1 mL of a 4 M HCl/dioxane solution. The reaction was mixed for 20 s and let stand for 40 min. It was then diluted with H2O (9 mL) and passed through a preactivated Bond Elut Plexa cartridge (Agilent Technologies, Inc.). The cartridge was washed with 5 mL of a 20% v/v EtOH/H2O solution and dried with air. The radiolabeled product was eluted with dimethyl sulfoxide (100–300 μL).
Human Serum Albumin (HSA) Affinity Determination
HSA was immobilized to high-performance liquid chromatography–grade silica by the Schiff base method as described previously and was packed into 10 × 2.1 mm inner diameter microcolumns (32,33). The protein content of these columns was approximately 60 mg of HSA per gram of silica (34). Control microcolumns were prepared in the same manner but with no HSA being added during the immobilization step. The retention factor for each compound was measured on both an HSA microcolumn and a control column by injecting 5-μL samples that contained approximately 50 μM of the compound in 0.067 M potassium phosphate buffer (pH 7.4). All samples were injected in triplicate at room temperature and at 1.0 mL/min, with the phosphate buffer used as the mobile phase. Similar injections were made with samples containing sodium nitrate, which was used as a void volume marker. Elution of the injected compounds was monitored by absorbance detection. The dissociation constant (Kd) for each compound with HSA was determined using the measured retention factors, after correction for any observed retention on the control column, along with the estimated content of active HSA in the column, as based on injections made with warfarin and l-tryptophan (i.e., probes for Sudlow sites I and II of HSA) (34,35). The estimated precision of the Kd values was ±2%–14%.
Cell Culture
The PSMA-expressing human prostate cancer cell line LNCaP was obtained from the American Type Culture Collection. Cell culture supplies were from Invitrogen unless otherwise noted. LNCaP cells were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (Hyclone), 4 mM l-glutamine, 1 mM sodium pyruvate, 10 mM N-2-hydroxyethylpiperazine-N-2-ethanesulfonic acid, 2.5 mg/mL d-glucose, and 50 μg/mL gentamicin in a humidified incubator at 37°C/5% CO2. Cells were removed from flasks for passage or for transfer to 12-well assay plates by incubating them with 0.25% trypsin/ethylenediaminetetraacetic acid.
In Vitro Determination of 50% Inhibitory Concentration (IC50)
IC50 values of the nonradioactive iodine–containing ligands were determined by screening in a multiconcentration competitive binding assay against 99mTc-((7S,12S,16S)-1-(1-(carboxymethyl)-1H-imidazol-2-yl)-2-((1-(carboxymethyl)-1H-imidazol-2-yl)methyl)-9,14-dioxo-2,8,13,15-tetraazaoctadecane-7,12,16,18-tetracarboxylic acid technetium tricarbonyl complex (99mTc-MIP-1427) for binding to PSMA on LNCaP cells, according to methods previously described (36). Briefly, LNCaP cells were plated 48 h before the experiment to achieve a density of approximately 5 × 105 cells/well (in triplicate) in RPMI-1640 medium supplemented with 0.25% bovine serum albumin. The cells were incubated for 1 h with 1 nM 99mTc-MIP-1427 in serum-free RPMI-1640 medium in the presence of 0.1–10,000 nM test compounds. Radioactive incubation medium was then removed by pipette, and the cells were washed twice using 1 mL of ice-cold N-2-hydroxyethylpiperazine-N-2-ethanesulfonic acid buffer. Cells were harvested from the plates and transferred to tubes for radioactive counting using a Packard Cobra II γ-Counter. IC50 values were determined by nonlinear regression using GraphPad Prism software (GraphPad Software).
Inoculation of Mice with Xenografts
All animal studies were approved by the Institutional Animal Care and Use Committee of Weill Cornell Medicine and were undertaken in accordance with the guidelines set forth by the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals. Animals were housed under standard conditions in approved facilities with 12-h light–dark cycles. Food and water were provided ad libitum throughout the course of the studies. Male inbred athymic nu/nu mice were purchased from The Jackson Laboratory. For inoculation in mice, LNCaP cells were suspended at 4 × 107 cells/mL in a 1:1 mixture of phosphate-buffered saline:Matrigel (BD Biosciences). Each mouse was injected in the left flank with 0.25 mL of the cell suspension. The mice were imaged when the tumors reached approximately 200–400 mm3, whereas biodistributions were conducted when tumors were in the range 100–400 mm3.
Tissue Distribution Studies
A quantitative analysis of the tissue distribution of 131I-labeled compounds was performed in separate groups of male NCr-nu/nu mice bearing LNCaP cell xenografts administered via the tail vein as a bolus injection (∼370 kBq [10 μCi]/mouse) in a volume of 0.05–0.1 mL of saline solution containing 2.5% v/v dimethyl sulfoxide. The animals (n = 3–5/time point) were euthanized by asphyxiation with CO2 at the indicated time points after injection. Tissues, including blood, heart, lungs, liver, spleen, pancreas, kidneys, stomach, large and small intestines (with contents), skeletal muscle, bone, and tumor, were dissected, excised, weighed wet (Sartorius analytic balance), and counted in a Wizard automated γ-counter (Perkin Elmer). A 1 percentage injected dose per gram (%ID/g) standard was counted with the tissue samples. Tissue time–radioactivity levels expressed as %ID/g were determined. Blood pharmacokinetics were modeled as a dual-compartment system using biexponential least-squares regression fit to the data. The regression method was implemented in MATLAB R2015b (The MathWorks). An independent-samples t test was used to compare different time points and between different compounds, and in all analyses the statistical significance (α-level) was set at a P value of less than 0.05.
Imaging Studies
LNCaP xenograft tumor–bearing mice (2 per compound) were injected intravenously via the tail vein as a bolus injection of 7.03–7.77 MBq (190–210 μCi) of 124I-RPS-027. The specific activity of 124I-RPS-027 was in the range of 3–10 GBq/μmol. The mice were imaged by small-animal PET/CT (Inveon; Siemens Medical Solutions, Inc.) at 1, 3, 6, 24, and 48 h after injection. Total acquisition time was 30 min, and a CT scan was obtained either immediately before or immediately after the acquisition for both anatomic coregistration and attenuation correction. The data were reconstructed using the commercial Inveon software supplied by the vendor. Image-derived tumor uptake was estimated by drawing a region of interest.
RESULTS
Chemistry and Radiochemistry
Reference compounds and their trimethylstannyl precursors were synthesized as described in Supplemental Figure 1. Synthesis yields ranged from 4% to 20% in 5–8 steps from L-Glu(OtBu)-OtBu•HCl. Trimethylstannylation was accomplished in 20%–82% yield from the protected 4-iodophenyl precursors.
Radiolabeling was accomplished by an iododestannylation reaction, followed by deprotection of the labeled intermediate in acidic conditions (Fig. 2). Total reaction time was 60 min, radiochemical yield ranged from 43% to 72%, and radiochemical purity was greater than 90% for all compounds tested. Specific activity varied from 2 to 10 GBq/μmol according to the starting 124I or 131I activity. The deprotection step proved to be time sensitive: for reaction times below 40 min, incomplete deprotection was observed, whereas reaction times greater than 45 min led to the formation of an unidentified impurity that could not be removed during purification by solid-phase extraction. The characteristics of the radiosyntheses are described in Table 1. No significant difference in labeling yield was observed when 124I was used in place of 131I.

Radiosynthesis of dual-target binding ligands via iododestannylation reactions. 131I-RPS-001 and 131I-RPS-025 were synthesized by same procedure from their corresponding organostannane precursors.
Characteristics of Radiosynthesis of Dual-Target Binding Ligands with 131I
In Vitro Studies
The range of affinities for PSMA was determined to be 4–40 nM, with most of the compounds clustered between 10 and 15 nM (Fig. 1). In the same assay RPS-001, also known as MIP-1095, was found to have an IC50 of 0.3 nM. Compounds bearing the p-(iodophenyl)butyric acid moiety were found to have a high affinity (1–2 μM) for HSA (Fig. 1). RPS-025 could not be eluted from the column, which prevented the calculation of a precise Kd. The compound RPS-027 had a modest affinity of 11 μM, whereas RPS-001, RPS-022, and RPS-026 were in the range 19–26 μM. RPS-023 (Kd = 53.2 μM) was determined to have a weak affinity.
Biodistribution and Small-Animal PET/CT Imaging
The biodistribution studies of the 6 ligands demonstrated that albumin binding affinity contributed markedly to the different pharmacokinetics observed in mice. RPS-001 (Kd = 20 μM for HSA) demonstrated relatively rapid clearance from the blood (Fig. 3A). The measured changes in blood activity became statistically significant by 72 h. Initial kidney uptake was high (>100 %ID/g), and from 24 to 96 h after injection 131I-RPS-001 activity in the kidney cleared from 65.24 ± 22.61 %ID/g at 24 h to 2.12 ± 2.47 %ID/g at 96 h. The measured changes in kidney activity became statistically significant by 48 h. Tumor uptake was high, with gradual tumor washout observed over several days (19.81 ± 6.16 %ID/g at 24 h vs. 10.21 ± 4.30 %ID/g at 96 h) (Fig. 3A), such that the tumor-to-kidney ratio increased with time. Low, PSMA-mediated uptake was observed at 24 h after injection in the large intestine (2.35 ± 1.26 %ID/g) and the spleen (2.41 ± 1.40 %ID/g), and accumulated activity was negligible in all tissues except for the tumor and kidney by 48 h after injection. The tissue uptake at earlier time points was extrapolated using previously reported data (37).

Biodistribution of dual-target binding ligands in nude mice bearing PSMA+ LNCaP human tumor xenografts. Error bars represent SD of measured values. (A) 131I-RPS-001 at 24, 48, 72, and 96 h after injection. (B) 131I-RPS-005 at 24, 48, 72, and 96 h after injection. (C) 131I-RPS-020 at 1, 6, 12, 24, and 48 h after injection. (D) 131I-RPS-022 at 1, 6, 12, 24, and 48 h after injection. Int = intestine.
Accumulation of 131I-RPS-005 (Kd = 0.89 μM for HSA) in the blood was exceptionally high, and clearance was slow and statistically insignificant over the 96-h observation window (Fig. 3B). At 24 h after injection, blood activity was 21.35 ± 3.99 %ID/g, which decreased to 15.57 ± 3.98 %ID/g by 96 h. This slow blood clearance is likely to be responsible for the non–PSMA-mediated uptake observed in tissues such as the heart (4.40 ± 0.74 %ID/g at 24 h), lungs (8.87 ± 0.74 %ID/g at 24 h), and liver (3.41 ± 0.56 %ID/g at 24 h). Kidney uptake (39.09 ± 2.96 %ID/g) was lower at 24 h after injection than observed for 131I-RPS-001, but clearance was considerably slower. In combination with the comparatively low tumor uptake of approximately 10% (9.37 ± 1.56 %ID/g at 24 h; 10.82 ± 2.64 %ID/g at 96 h), these pharmacokinetics result in poor tumor-to-background ratios at all time points.
Because the tissue uptake of 131I-RPS-005 appeared to peak within the first 24 h, 131I-RPS-020 (Kd = 2.1 μM) was investigated at early time points as well. Prolonged blood retention was also observed, with an initial accumulation of 16.20 ± 4.68 %ID/g at 1 h after injection only decreasing to 12.71 ± 1.55 %ID/g at 48 h (Fig. 3C), which was not statistically significant. This was associated with high off-target uptake, most notably in the lungs (6.12 ± 0.95 %ID/g at 1 h after injection) and kidneys (16.21 ± 0.97 %ID/g at 1 h). Activity continued to accumulate in the kidneys, peaking at 22.26 ± 2.48 %ID/g at 24 h after injection. Tumor uptake (4.81 ± 1.27 %ID/g) was lower than observed for RPS-005, as a comparison of PSMA affinities might predict, but with no statistically significant change over the course of 48 h.
In contrast, 131I-RPS-022 (Kd = 22.9 μM) showed a rapid and significant drop in blood activity over time, with negligible activity detected as early as 12 h after injection (Fig. 3D). Considerable uptake was observed in the liver (12.46 ± 1.63 %ID/g) and small intestine (13.09 ± 2.98 %ID/g) at 1 h after injection, though clearance from each organ was rapid. Kidney uptake, which reached (52.18 ± 5.35 %ID/g) at 1 h after injection, demonstrated a significant clearance of activity, reaching 2.27 ± 1.41 %ID/g by 24 h, leading to favorable tumor-to-kidney and tumor-to-background ratios at later time points. However, tumor uptake peaked at 7.20 ± 0.10 %ID/g at 1 h after injection and decreased significantly to 3.35 ± 1.70 %ID/g by 6 h.
131I-RPS-027 showed a promising biodistribution profile over the time period studied (Fig. 4). Activity in the blood at 1 h after injection was 3.91 ± 0.48 %ID/g, which decreased to 0.58 ± 0.17 %ID/g by 24 h. Statistically significant decreases in blood activity were observed over the entire observation period. Initial uptake was also observed in the liver (6.79 ± 0.70 %ID/g; 1 h), small intestine (8.01 ± 0.78 %ID/g; 1 h), large intestine (8.56 ± 1.67 %ID/g; 3 h), spleen (4.13 ± 1.37 %ID/g; 1 h), and heart (1.30 ± 0.04 %ID/g; 1 h). Clearance from these tissues decreased largely in proportion to blood clearance, suggesting that the normal organ activity is related to blood-pool activity rather than tissue uptake. Minimal activity was detected in the tissue by 12 h after injection with the exception of the kidneys (15.12 ± 2.82 %ID/g) and the tumor (9.73 ± 1.01 %ID/g). Although maximum tumor uptake (12.41 ± 0.84 %ID/g; 3 h) was lower than for 131I-RPS-001, tumor uptake remained as high as 8.13 ± 2.03 %ID/g at 24 h and showed no significant change over 24 h period but dropped significantly to 3.05 ± 1.30 %ID/g at 72 h, leading to excellent tumor-to-background and tumor-to-kidney (>2) ratios as early as 18 h after injection. Although tumor uptake of 131I-RPS-027 was roughly 50% of that for 131I-RPS-001 at all time points studied, the kidney concentration of 131I-RPS-027 was 5-fold less than that for 131I-RPS-001.
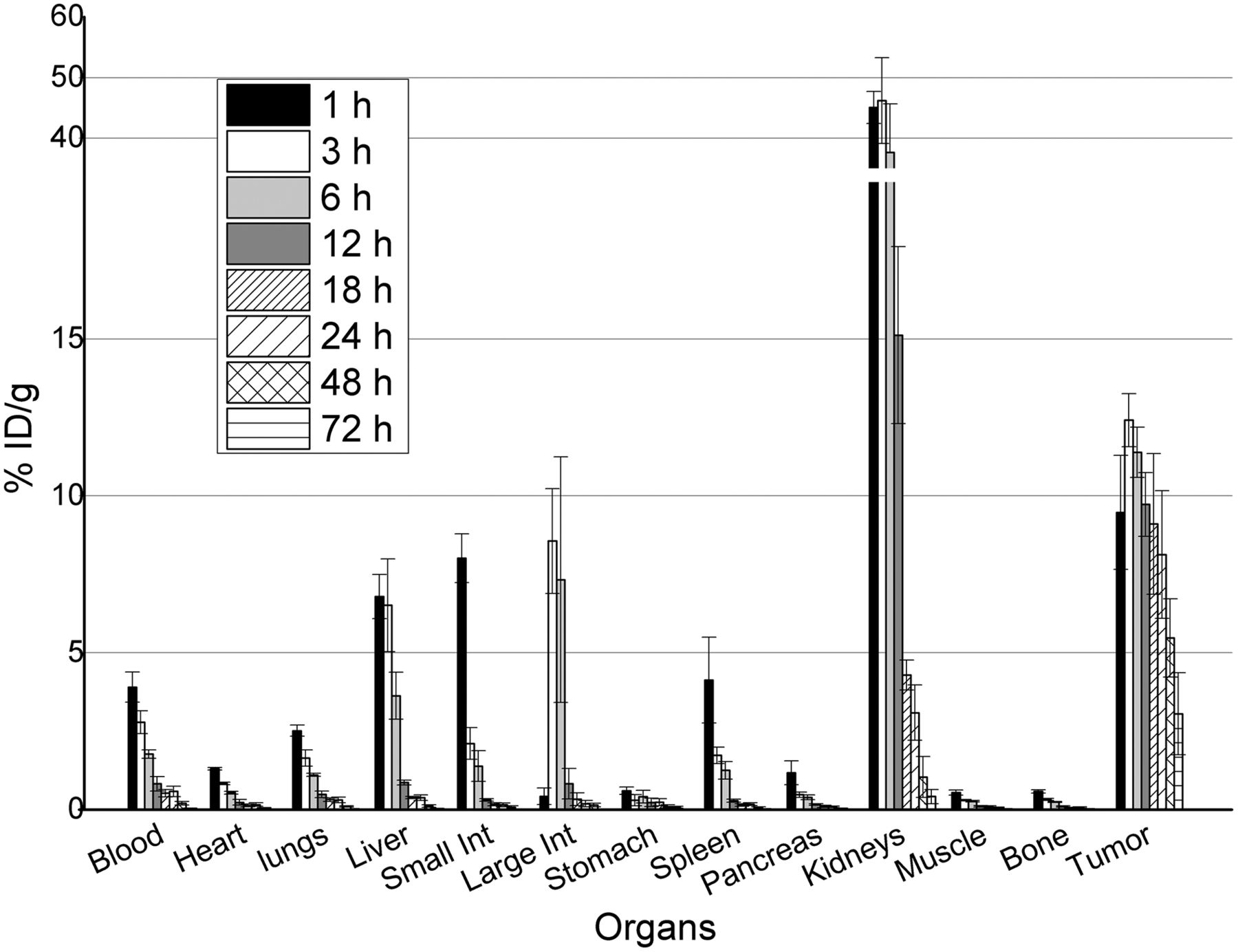
Biodistribution of 131I-RPS-027 in nude mice bearing PSMA+ LNCaP human tumor xenografts. Error bars represent SD of measured values. Int = intestine.
Desirable pharmacokinetics continued to be observed at longer time points for 131I-RPS-027 (Fig. 4). Activity in the blood remained detectable up to 48 h after injection (0.20 ± 0.06 %ID/g), whereas tumor–to–normal-tissue ratios continued to increase because of rapid clearance from the kidneys (1.04 ± 0.65 %ID/g at 48 h). These in vivo findings were visually recapitulated by small-animal PET/CT imaging of LNCaP xenograft mice using 124I-RPS-027. Initial uptake in the tumor, kidneys, and hepatobiliary system is evident at 1 h (Fig. 5), with clearance from nontarget tissue resulting in highly specific tumor targeting at 24 and 48 h after injection.

Small-animal PET/CT imaging of LNCaP xenograft mice by small-animal PET/CT using 124I-RPS-027 (7.4 MBq [200 μCi]).
Time–activity curves were plotted to facilitate a greater understanding of the pharmacokinetic profile of the dual-target binding ligands. Uptake in the tumor, blood, and kidneys is plotted in Figure 6. The highest tumor uptake is observed for 131I-RPS-001, and the kinetics of tumor washout are similar for the 3 compounds with lower affinity for albumin (Fig. 6A). The level of activity in the tumor remains most steady in the 2 ligands with highest albumin binding (Fig. 6A). Striking differences are apparent in the rate of blood clearance among the various compounds studied, with 131I-RPS-005 and 131I-RPS-020, both having high affinity for albumin, showing minimal blood clearance over 48 h and beyond (Fig. 6B). The clearance kinetics of 131I-RPS-001, 131I-RPS-022, and 131I-RPS-027 reflect their relative affinities for albumin, with 131I-RPS-022 clearing most quickly and 131I-RPS-027 clearing most gradually. Blood curves showed a rapid initial distribution phase followed by a slower elimination phase. In this model, the half-life for the distribution phase was 2.17, 2.1, and 3.15 h for 131I-RPS-001, 131I-RPS-022, and 131I-RPS-027, respectively, whereas the corresponding half-life for the elimination phase was 20.38, 17.3, and 21.66 h. In comparison, the half-lives for distribution and elimination phases for 131I-RPS-020 were 3.85 and 3,300 h, respectively.

Time–activity curves for blood (A), tumor (B), and kidneys (C) derived from biodistribution data. Error bars represent SD of measured values.
Absolute kidney uptake of RPS-027 and RPS-022 was dramatically reduced compared with RPS-001 at all time points studied. Kidney clearance is approximately described by an exponential decay function for 131I-RPS-001, 131I-RPS-022, and 131I-RPS-027 (Fig. 6C). In contrast, 131I-RPS-005 and 131I-020 show prolonged retention and much flatter clearance curves. Tumor-to-kidney and tumor-to-blood ratios were calculated as a function of time. The tumor-to-kidney ratio of 131I-RPS-027 reached approximately 3 by 24 h and continued to increase significantly with time (Fig. 7A). In comparison, the tumor-to-kidney ratio of 131I-RPS-001 did not reach 3 until nearly 96 h after injection. The tumor-to-kidney ratio of 131I-RPS-022 also increased rapidly, particularly at earlier time points and significantly by 12 h after injection (>1), but this was driven more by rapid kidney clearance than by tumor uptake. The tumor-to-blood ratio of 131I-RPS-027 was lower than 131I-RPS-001 (Fig. 7B), predominantly reflecting enhanced albumin binding.
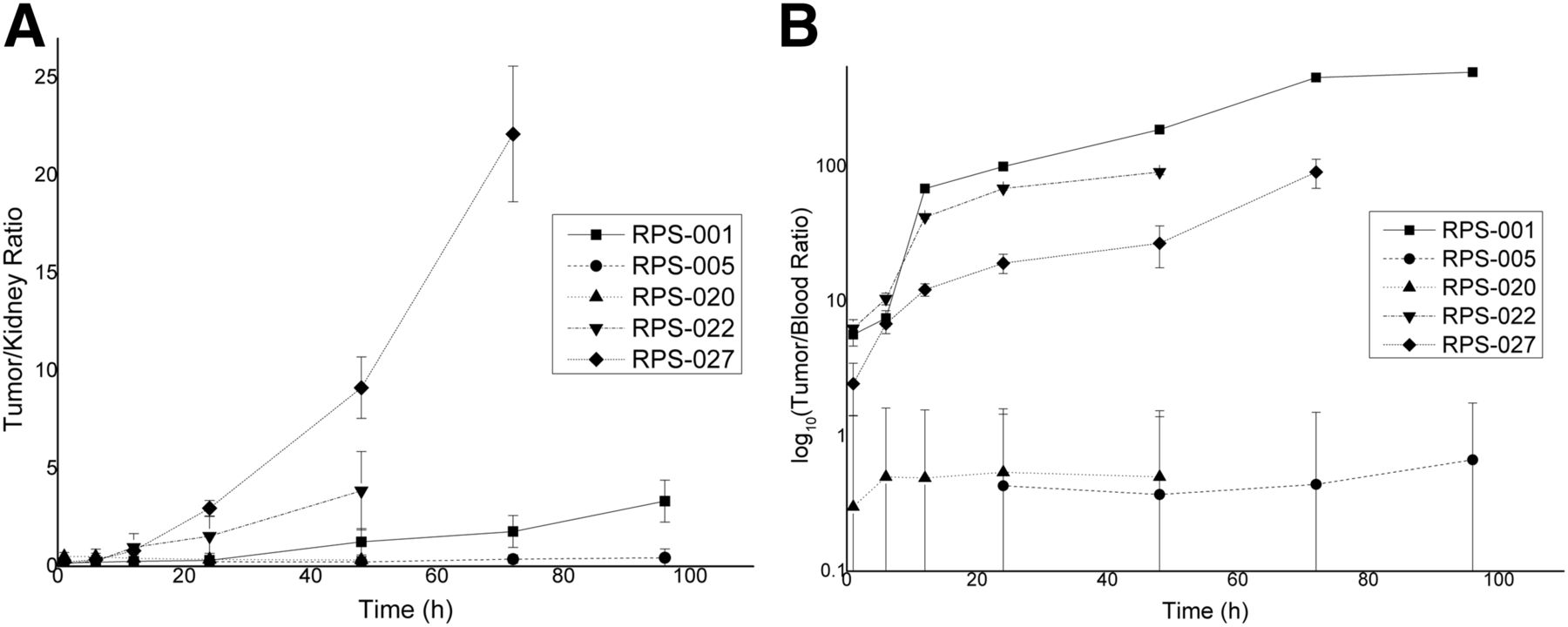
Tumor-to-background ratios show effect of enhanced albumin binding. Error bars were computed from SD of measured values. (A) Tumor-to-kidney ratio. (B) Tumor-to-blood ratio.
DISCUSSION
In the absence of stable isotopes of astatine, iodine has been proposed as a useful surrogate for drug development and for predicting radiation dosimetry. Recent work has confirmed that the pharmacokinetics of a small-molecule PSMA inhibitor, 131I-DCIBzL, and its astatinated analog (2S)-2-(3-(1-carboxy-5-(4-211At-astatobenzamido)pentyl)ureido)-pentanedioic acid (211At-6) (38) were similar in a preclinical prostate cancer model, although small but statistically significant differences were observed. These differences included an apparent decrease in PSMA affinity for the astatinated analog, as evidenced by a 10%–30% reduction of uptake in PSMA-expressing tissues, and an apparent increase in blood binding (38).
Modulation of blood binding has been demonstrated preclinically to increase tumor uptake and decrease kidney uptake (28,39) without causing a corresponding increase in hematologic toxicity. In a recent study of DOTA-conjugated albumin labeled with the α-emitter 213Bi (half-life = 45 min), doses up to 3.7 MBq could be administered to mice safely, with no plasma enzyme modifications or histologic abnormalities detected over 1 y of observation (40). This is in sharp contrast to recently reported findings of a small-molecule PSMA inhibitor labeled with 211At (half-life = 7.2 h) in which the maximum tolerated single dose in immunocompetent mice was 2 orders of magnitude lower, at 0.037 MBq, due to severe, irreversible renal toxicity (38). High kidney uptake was also observed for 131I-MIP-1095 in mice, although this did not prove to be dose-limiting during initial clinical evaluation in humans of a single therapy cycle (2). In this context, the increased blood-pool half-life of 131I-RPS-027 and significantly lower renal uptake and retention, relative to 131I-MIP-1095, facilitated by enhanced binding to albumin, represent an important component to an increased radiotherapeutic index.
It is likely that the reduction in kidney uptake on albumin binding is due to a decrease in the rate of filtration and reabsorption in proximal renal tubules (41,42). Because PSMA is also expressed in proximal renal tubules (43,44), it is possible that decreases in kidney uptake might be a function of reduced affinity for PSMA. However, the observation that RPS-027 (IC50 = 15 nM) shows higher initial uptake than the strongly albumin-associating RPS-020 (IC50 = 8 nM) and lower initial uptake than the weakly associating RPS-022 (IC50 = 10 nM) suggests that differences in kidney uptake are indeed mediated by albumin binding.
In comparison to 131I-MIP-1095 (45), 131I-DCIBzL (46), and 211At-6 (38), 131I-RPS-027 shows considerably lower kidney uptake at all time points from 1 to 72 h after injection. The difference in uptake between 131I-MIP-1095 (131I-RPS-001) and 131I-RPS-027 at 24 h is 20-fold. In the context of the dosimetry reported for MIP-1095 in patients, the lower kidney uptake of 131I-RPS-027 projects to a significantly lower absorbed dose to the kidneys, and a reduced risk of relevant nephrotoxicity at therapeutic doses or under a multiple treatment cycle regime. Moreover, the tumor-to-kidney ratio for 131I-RPS-027 is greater than 2 as soon as 18 h after injection, rises to 3 by 24 h, and exceeds 7 by 72 h. This compares favorably with 211At-6, and a projection of the comparison between 131I-DCIBzL and 211At-6 to RPS-027 predicts that astatination will further increase the ratio (38). As such, the dose-limiting, irreversible nephrotoxicity of 211At-6 might be resolved by 211At-RPS-027. 131I-RPS-022 also shows higher tumor-to-background ratios than 131I-RPS-001 and 211At-6 at early time points, but this is driven by rapid excretion rather than by tumor uptake.
Dose-limiting toxicity to the salivary glands was observed for 131I-MIP-1095 (2). Moderate xerostomia has been reported for 177Lu-PSMA-617 (3,6), but translation of this ligand to targeted α-particle therapy as 225Ac-PSMA-617 led to severe and sustained xerostomia (18). It has been reported that PSMA is expressed at low levels in the parotid and lacrimal glands (47), and recent experiments have shown that 2-(phosphonomethyl)-pentandioic acid can be used to displace 68Ga-PSMA-HBED-CC from rat salivary glands (48). These findings suggest that uptake of radiopharmaceuticals in these structures is PSMA-mediated. Uniquely to the class of glutamate-urea heterodimers found in most clinically investigated radiolabeled PSMA inhibitors, the fluorinated small-molecule PSMA inhibitor 18F-DCFBC showed tumor localization with moderately persistent blood-pool activity and a corresponding decrease in signal intensity in the salivary glands (49), suggesting that albumin-binding is a potential strategy for reducing salivary gland uptake that, a priori, does not rule out adequate tumor uptake in humans. Plasma binding has been demonstrated to reduce salivary gland uptake of cyclosporine (50) and warfarin (51) in rats. Because the small size of murine salivary glands precludes detection of uptake by imaging or biodistribution studies, work is ongoing to confirm the effect of modulated albumin binding on salivary gland uptake in larger animals.
CONCLUSION
A low-molecular-weight PSMA inhibitor (IC50 = 15 nM), RPS-027, was developed as part of a structure–activity relationship study on dual-target binding ligands with unequal affinity for both PSMA and serum albumin. 131I-RPS-027 was prepared in 49.5% radiochemical yield from its organostannane precursor and showed good tumor uptake and retention in a LNCaP xenograft model up to 72 h after injection. Rapid washout was observed in other tissues, including the kidneys. These pharmacokinetics appear to be a consequence of an appropriate level of binding to albumin (Kd = 11 μM) that extends blood clearance, increases tumor uptake by increasing the number of passes through the tumor bed, and reduces kidney uptake by decreasing renal filtration and reuptake. With a highly favorable tumor-to-kidney ratio (>3 by 24 h after injection), the projected dosimetry of RPS-027 compares favorably with MIP-1095. Using iodine/radioiodine as a surrogate for the radiohalogen 211At, we propose dual-target binding ligands such as RPS-027 as a next-generation radiopharmaceutical for targeted α-therapy of prostate cancer using 211At.
DISCLOSURE
No potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Dr. Yeona Kang for her assistance in determining blood clearance kinetics and statistical analysis.
Footnotes
Published online Apr. 27, 2017.
- © 2017 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication December 16, 2016.
- Accepted for publication April 11, 2017.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Albumin Binder-Conjugated Fibroblast Activation Protein Inhibitor Radiopharmaceuticals for Cancer Therapy
- The History of Prostate-Specific Membrane Antigen as a Theranostic Target in Prostate Cancer: The Cornerstone Role of the Prostate Cancer Foundation
- Improving Theranostic Gallium-68/Lutetium-177-Labeled PSMA Inhibitors with an Albumin Binder for Prostate Cancer
- Albumin-Binding PSMA Ligands: Implications for Expanding the Therapeutic Window
- A Single Dose of 225Ac-RPS-074 Induces a Complete Tumor Response in an LNCaP Xenograft Model
- Dual-Targeted Molecular Imaging of Cancer