


Abstract
Vascular endothelial growth factor-A (VEGF-A) acts via 2 vascular endothelial growth factor receptors, VEGFR-1 and VEGFR-2, that play important and distinct roles in tumor biology. We reasoned that selective imaging of these receptors could provide unique information for diagnostics and for monitoring and optimizing responses to anticancer therapy, including antiangiogenic therapy. Herein, we report the development of 2 first-in-class 89Zr-labeled PET tracers that enable the selective imaging of VEGFR-1 and VEGFR-2. Methods: Functionally active mutants of scVEGF (an engineered single-chain version of pan-receptor VEGF-A with an N-terminal cysteine-containing tag for site-specific conjugation), named scVR1 and scVR2 with enhanced affinity to, respectively, VEGFR-1 and VEGFR-2, were constructed. Parental scVEGF and its receptor-specific mutants were site-specifically derivatized with the 89Zr chelator desferroxamine B via a 3.4-kDa PEG linker. 89Zr labeling of the desferroxamine B conjugates furnished scV/Zr, scVR1/Zr, and scVR2/Zr tracers with high radiochemical yield (>87%), high specific activity (≥9.8 MBq/nmol), and purity (>99%). Tracers were tested in an orthotopic breast cancer model using 4T1luc-bearing syngeneic BALB/c mice. For testing tracer specificity, tracers were coinjected with an excess of cold proteins of the same or opposite receptor specificity or pan-receptor scVEGF. PET imaging, biodistribution, and dosimetry studies in mice, as well as immunohistochemical analysis of harvested tumors, were performed. Results: All tracers rapidly accumulated in orthotopic 4T1luc tumors, allowing for the successful PET imaging of the tumors as early as 2 h after injection. Blocking experiments with an excess of pan-receptor or receptor-specific cold proteins indicated that more than 80% of tracer tumor uptake is VEGFR-mediated, whereas uptake in all major organs is not affected by blocking within the margin of error. Critically, blocking experiments indicated that VEGFR-mediated tumor uptake of scVR1/Zr and scVR2/Zr was mediated exclusively by the corresponding receptor, VEGFR-1 or VEGFR-2, respectively. In contrast, uptake of pan-receptor scV/Zr was mediated by both VEGFR-1 and VEGFR-2 at an approximately 2:1 ratio. Conclusion: First-in-class selective PET tracers for imaging VEGFR-1 and VEGFR-2 were constructed and successfully validated in an orthotopic murine tumor model.
Vascular endothelial growth factor-A (VEGF-A) is the main regulator of angiogenesis in cancer and other pathologies (1). For this reason, vascular endothelial growth factor (VEGF) and its 2 main receptors, VEGFR-1 and VEGFR-2, are the principal targets of current antiangiogenic therapy (2). Unfortunately, only a small fraction of cancer patients benefits from such a therapy, whereas serious side effects are possible (2). Therefore, it is critically important to identify and validate reliable biomarkers for optimization of antiangiogenic therapy for individual patients, and VEGF receptors (VEGFRs) could play this role. As a step in this direction, we have recently demonstrated that in mouse cancer models imaging of VEGFRs allows for real-time noninvasive monitoring of the progress and failure of antiangiogenic therapy (3,4). In these studies, we used a 99mTc SPECT tracer based on an engineered single-chain version of human VEGF-A, which binds to both VEGFR-1 and VEGFR-2 of human or mouse origin (3–5). Although the pan-receptor tracer did not discriminate between VEGFR-1 and VEGFR-2 responses, our findings suggested that VEGFRs could be valuable imaging biomarkers in the course of antiangiogenic therapy.
We reasoned that independent imaging of VEGFR-1 and VEGFR-2 may provide more detailed information on responses to antiangiogenic therapy. According to current understanding, VEGFR-1 and VEGFR-2 play different and, possibly, even opposite roles in endothelial cell biology and angiogenesis (1,6,7). VEGF induces robust tyrosine autophosphorylation in the cytoplasmic domain of VEGFR-2, followed by a cascade of the downstream signaling that supports growth and survival of endothelial cells (1,6,7). In contrast, having much higher affinity binding to VEGFR-1, VEGF induces only weak tyrosine autophosphorylation in this receptor and, presumably, less downstream signaling via canonical phosphotyrosine-mediated pathways, leading to suggestions that VEGFR-1 might serve as a negative regulator of angiogenesis via sequestering VEGF (6,7). This function was further supported by the discovery of an alternatively spliced circulating form of VEGFR-1 (sVEGFR-1) capable of sequestering VEGF (1,6,7).
In addition to endothelial cells, VEGFR-1 and VEGFR-2 are also expressed on a variety of other cells where they are playing not yet fully understood roles. For example, VEGFR-1 could be found on tumor cells of different tissue origin, where it promotes tumor growth rather than stimulates tumor angiogenesis (8). In animal tumor models, both VEGFR-1 and -2 are implicated in the development of established micrometastases (9); however, it is VEGFR-1 that is critically involved in the preparation of metastatic niche (10). In view of such differential involvement of VEGFR-1 and VEGFR-2 in the onset and progression of cancer, it would be logical to expect that each receptor might be an important biomarker for the selection of an appropriate cancer treatment or for monitoring the response to therapy in individual patients.
Here, we report the development of first-in-class 89Zr-labeled PET tracers for selective imaging of either VEGFR-1 or VEGFR-2. To enable such selectivity, we describe here the engineering of 2 novel mutant versions of single-chain VEGF (scVEGF) with enhanced affinity to either VEGFR-1 or VEGFR-2 and a site-specific derivatization of these proteins with PEGylated 89Zr chelator, desferroxamine B (DFO). We reasoned that using 89Zr tracers would allow for much longer observation time than using more short-lived isotopes and could provide logistical convenience of the centralized tracer production for clinical use. Both tracers rapidly accumulated in 4T1luc orthotopic mouse breast tumors in immunocompetent BALB/c mice, allowing for the successful PET imaging of tumors. Blocking experiments with cold proteins of the same or opposite receptor specificity provided in vivo confirmation of either VEGFR-1 or VEGFR-2 selectivity of the novel tracers.
MATERIALS AND METHODS
General Procedures
All experiments using laboratory animals were performed in accordance with the Memorial Sloan Kettering Institutional Animal Care and Use Committee. Detailed information regarding all reagents and general experimental procedures, including the used animal model, production of receptor-specific VEGF mutants, and in vivo and ex vivo evaluation of novel tracers is provided in the supplemental materials (available at http://jnm.snmjournals.org).
Statistical Analysis
Quantitative data are expressed as mean ± SD. Means were compared using the Student t test, with P values of less than 0.05 considered statistically significant.
RESULTS
Enhanced VEGFR-1 and VEGFR-2 Specificity of scVEGF Mutants and DFO Conjugates
Previous studies indicated that certain point mutations in VEGF confer significant receptor specificity (11,12). On the basis of these reports, we constructed mutants of pan-receptor scVEGF, scVR1 and scVR2, with anticipated selective binding to VEGFR-1 and VEGFR-2 (Fig. 1A), respectively, and evaluated them in vitro. First, we assessed the specificity of the mutants to VEGFR-2 (Figs. 1B and 1C). Once engaged with a cognate ligand, VEGFR-2, unlike VEGFR-1, displays robust tyrosine autophosphorylation followed by internalization (1), which can be reliably detected in 2 previously reported tissue culture assays with 293/KDR cells overexpressing VEGFR-2 (3–5). As expected, scVEGF and scVR2 induced VEGFR-2 tyrosine autophosphorylation at the same concentration range, whereas the activity of scVR1 was barely detected (Fig. 1B). Next, we tested the mutants for their ability to protect 293/KDR cells from VEGFR-2–mediated cytotoxicity of shiga-like toxin (SLT)–VEGF fusion toxin. We found that both scVEGF and scVR2 successfully competed out SLT-VEGF, protecting cells in a similar dose-dependent manner with the same inhibitory concentration of 50% of approximately 2 nM, whereas scVR1 was not active in this assay (Fig. 1C). To test the affinity of the mutants to VEGFR-1 and to VEGFR-2, we used a pull-down assay (affinity capture) with soluble human VEGFR1-Fc and VEGFR2-Fc chimeras. We found that scVR1 and scVR2 were preferentially captured by the corresponding soluble receptor Fc chimeras, whereas pan-receptor scVEGF, as expected, was captured by both (Fig. 1D).
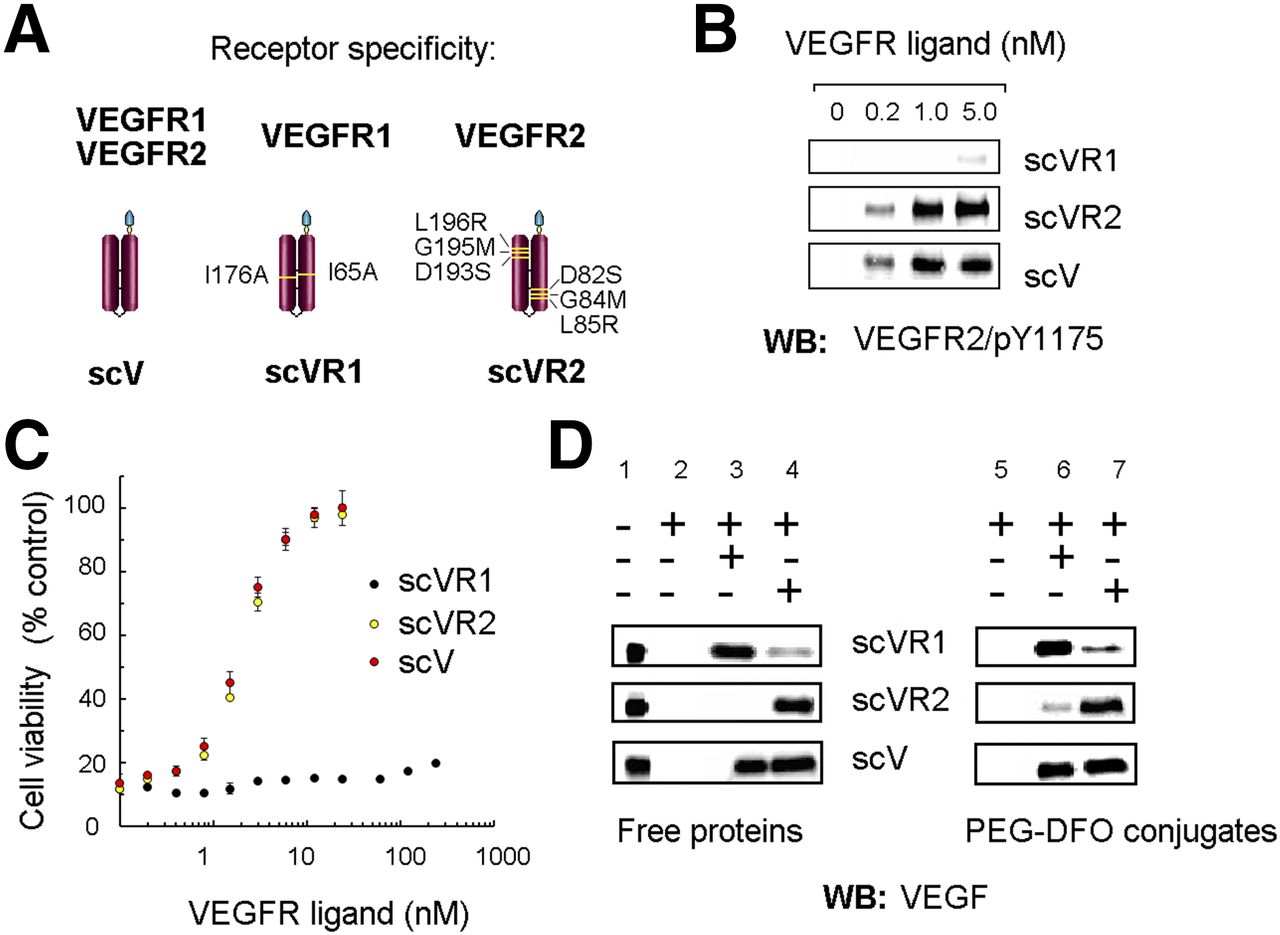
Receptor specificity of VEGFR ligands. (A) Schematic representation of VEGFR ligands. (B) Induction of VEGFR-2 tyrosine autophosphorylation in 293/KDR cells. (C) Competition of VEGFR ligands with SLT-VEGF toxin for binding to VEGFR-2 and protection of 293/KDR cells from SLT-VEGF–induced toxicity. (D) Protein G agarose, with or without VEGFR-Fc chimeras (7.5 pmoles, each), was incubated in presence of free VEGFR ligands (3 pmoles, each, lanes 1–4) and their DFO conjugates (3 pmoles, each, lanes 5–7), then washed expensively with radioimmunoprecipitation assay buffer, separated by 15% gels, and analyzed by WB.
Because both mutants displayed the expected receptor preference in vitro, we proceeded to making PEGylated DFO conjugates. The synthesis of DFO conjugates targeted to VEGFRs followed a 2-step procedure (Supplemental Fig. 1), previously developed for preparation of other scVEGF-based conjugates (5,13). Although we have reported earlier that site-specific modification via Cys-tag did not affect the receptor-binding ability of scVEGF-based conjugates (3–5), it was important to confirm this for the mutant-based conjugates. Soluble receptor pull-down assay confirmed that site-specific conjugation of PEGylated DFO did not affect the receptor specificity of the corresponding conjugates (Fig. 1D).
Altogether, the results of these 3 assays indicated that there is a preferential binding of scVR1 and scVR2 to cognate versus noncognate receptors, and site-specific modification with PEGylated DFO does not affect their ability to preferentially bind to cognate receptors. However, given the limitations of in vitro assays, the most reliable assessment of VEGFR specificity could be obtained only in vivo.
Blood Clearance, Biodistribution, and Dosimetry
Considering that in mouse tumor models more than 90% of scVEGF-based PET and SPECT tracers are cleared from the blood pool within 1–2 h after injection (3–5), we selected the time points 2, 6, 12, 24, and 48 h after injection for ex vivo biodistribution and PET imaging studies in 4T1luc orthotopic tumor–bearing BALB/c mice (n = 4/time point). Experiments were initiated when 4T1luc tumors were readily detectable visually (4–5 mm), as well as by bioluminescent imaging on intraperitoneal injection of luciferine (Supplemental Fig. 3). In these experiments, animals (n = 4/time point) were injected with indicated tracer. For blocking studies, animals were injected with a tracer mixed with a 35-fold excess of indicated cold protein.
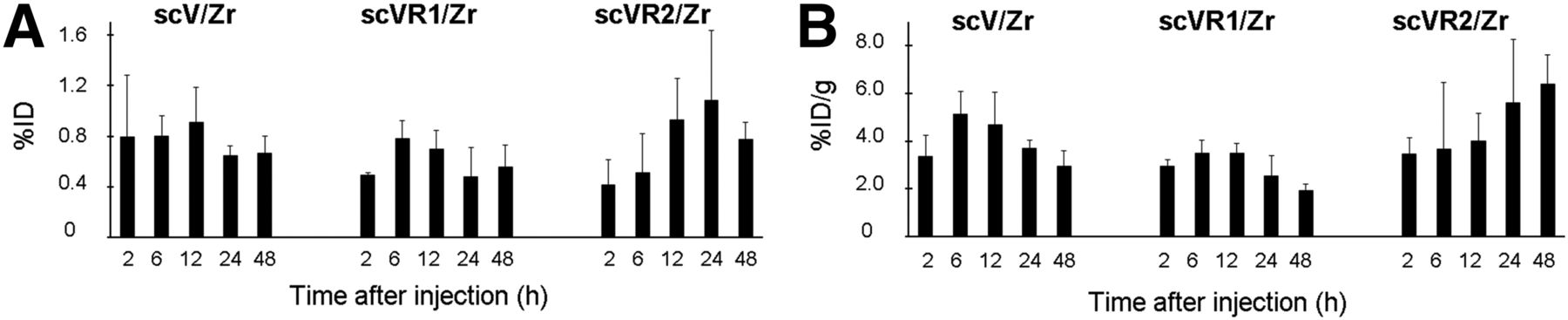
As expected, blood clearance for scV/Zr, scVR1/Zr, and scVR2/Zr was rapid, with less than 8 percentage injected dose per gram (%ID/g) remaining in the blood at 2 h after injection and further declining to more than 0.8 %ID/g at 48 h after injection. Biodistribution data at various time points, as well as biodistribution data for blocking experiment at 24 h after injection are shown in Figure 2 for scVR1/Zr and Supplemental Figures 4A and 4B for scVR2/Zr and scV/Zr, respectively. Although maximum uptake was achieved at 2–6 h after injection for all tracers and organs, there were variations in the kinetics of tracer uptake and clearance between different organs and between different tracers for the same organ. Notably, the variations within each uptake/clearance kinetic were small and only a few reached statistical significance. We found that coinjecting scV/Zr, scVR1/Zr, or scVR2/Zr with a 35-fold excess of corresponding cold protein did not significantly affect tracer uptake in organs at 24 h after injection, indicating that tracer accumulation in healthy tissue is mostly driven by non–VEGFR-mediated processes (Fig. 2; Supplemental Figs. 4A and 4B). This lack of statistically significant effects of blocking was not due to any experimental failures, because, as will be discussed further, in the same animals blocking strongly inhibited tumor tracer uptake. This also explains why an increase in blood activity concentration was not observed: because only tumor uptake is receptor-mediated, the additional fraction of activity that remained in the blood due to tumor blocking was low compared with all other organs combined and hence did not lead to a significant increase in blood activity concentration.

Biodistribution data of scVR1/Zr. 4T1luc tumor–bearing mice were injected with scVR1/Zr tracer (1–1.5 MBq/mouse [27–40 μCi]), alone or in mixture with 35-fold excess of cold scVR1 protein via tail vein for blocking.
As expected for relatively small approximately 32-kDa protein conjugates, the major tracer uptake was found in the kidneys (>50 %ID/g for all tracers), whereas other organs displayed relatively low uptake. However, some retention of activity, observed in all organs, could be explained by residualization of free 89Zr after catabolism of the conjugate (14).
Ex vivo biodistribution data were subsequently used to calculate the expected human dosimetry for all 3 tracers using the organ and total-body masses of the 70-kg standard man anatomic model (15) and the OLINDA software (16) to obtain mean organ absorbed doses and effective dose in rad/MBq and rem/MBq, respectively. These calculations indicated that absorbed doses for kidney would be between 0.27 and 0.3 rad/MBq, whereas all other organ absorbed doses were calculated to be less than 0.05 rad/MBq. The total effective doses for scV/Zr, scVR1/Zr, and scVR2/Zr were 0.035, 0.035, and 0.038 rem/MBq, respectively (Table 1).
Tumor Tracer Uptake
Tumor uptake for the pan-receptor scV/Zr as well as the receptor-specific scR1/Zr and scVR2/Zr was determined at various time points after injection, and the results were calculated as %ID and %ID/g (Figs. 3A and 3B, respectively). The uptake was in the range of 2–8 %ID/g, and there were small but statistically significant increases in scV/Zr uptake between 2 and 6 h after injection and decline in tumor uptake for scV/Zr and scVR1/Zr by 48 h (Figs. 3A and 3B, respectively), with increasing tumor-to-blood ratios over the course of the experiment (Supplemental Fig. 5).

Tumor uptake for all tracers as %ID (A) and %ID/g (B). Tumor-bearing mice received scV/Zr, scVR1/Zr, or scVR2/Zr tracer (1–1.5 MBq/mouse) via tail vein. Columns and error bars represent mean and SD values for indicated tissues.
Notably, there was a statistically significant increase in scVR2/Zr tracer uptake between 2 and 48 h after injection, suggesting either a gradually increased tumor uptake of the residual circulating tracer or tumor infiltration with cells that bound scVR2/Zr prior to tumor recruitment, such as endothelial precursor cells or monocytes/macrophages. As in other organs, activity in tumor tissue at later time points was most likely due to 89Zr that was residualized after internalization and catabolism of the conjugate (14).
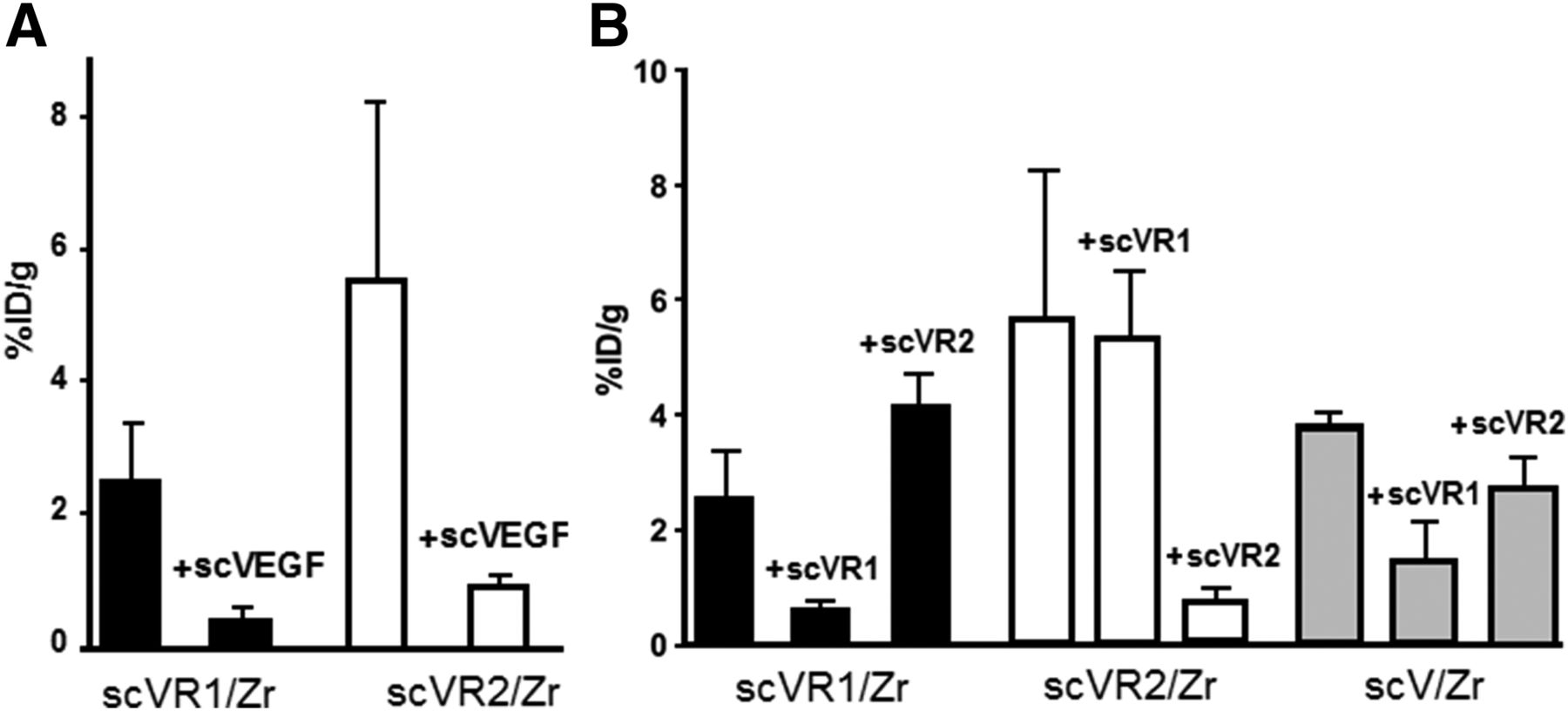
Tumor uptake for scVR1/Zr and scVR2/Zr at 24 h after injection was decreased approximately 6-fold, when tracers were coinjected with a 35-fold excess of cold pan-receptor scVEGF, indicating that in tumor, unlike in the major organs, VEGFR-mediated mechanisms were responsible for more than 80% of uptake (Fig. 4A). To assess the contribution of receptor-specific uptake for receptor-specific tracers, blocking experiments were performed with cold scVR1 and scVR2 proteins. As shown in Figure 4B, uptake of each tracer at 24 h after injection was decreased approximately 6-fold by the cold protein of the same specificity, whereas no blocking was observed with cold protein of the opposite specificity. Interestingly, each cold mutant, albeit to a different extent, partially decreased tumor uptake of pan-receptor scV/Zr tracer, suggesting that its tumor accumulation is mediated by both VEGFR-1 and VEGFR-2 at a ratio of approximately 2:1 (Fig. 4B).

Blocking experiments reveal receptor specificity of scVR1/Zr and scVR2/Zr tracers. Tumor-bearing mice (n = 4) were injected with indicated tracer (1–1.5 MBq/mouse) alone or in mixture with 35-fold excess of cold protein, either pan-receptor scVEGF (A) or receptor-specific scVR1 and scVR2 (B).
PET Imaging
PET imaging experiments of 4T1luc tumor–bearing mice using scV/Zr, scVR1/Zr, and scVR2/Zr tracer were performed at 2, 6, 12, 24, and 48 h after injection. Tumors were readily detectable at all time points with all 3 tracers (Fig. 5A for scVR2/Zr; Supplemental Figs. 5B and 5C for scV/Zr and scVR1/Zr, respectively).
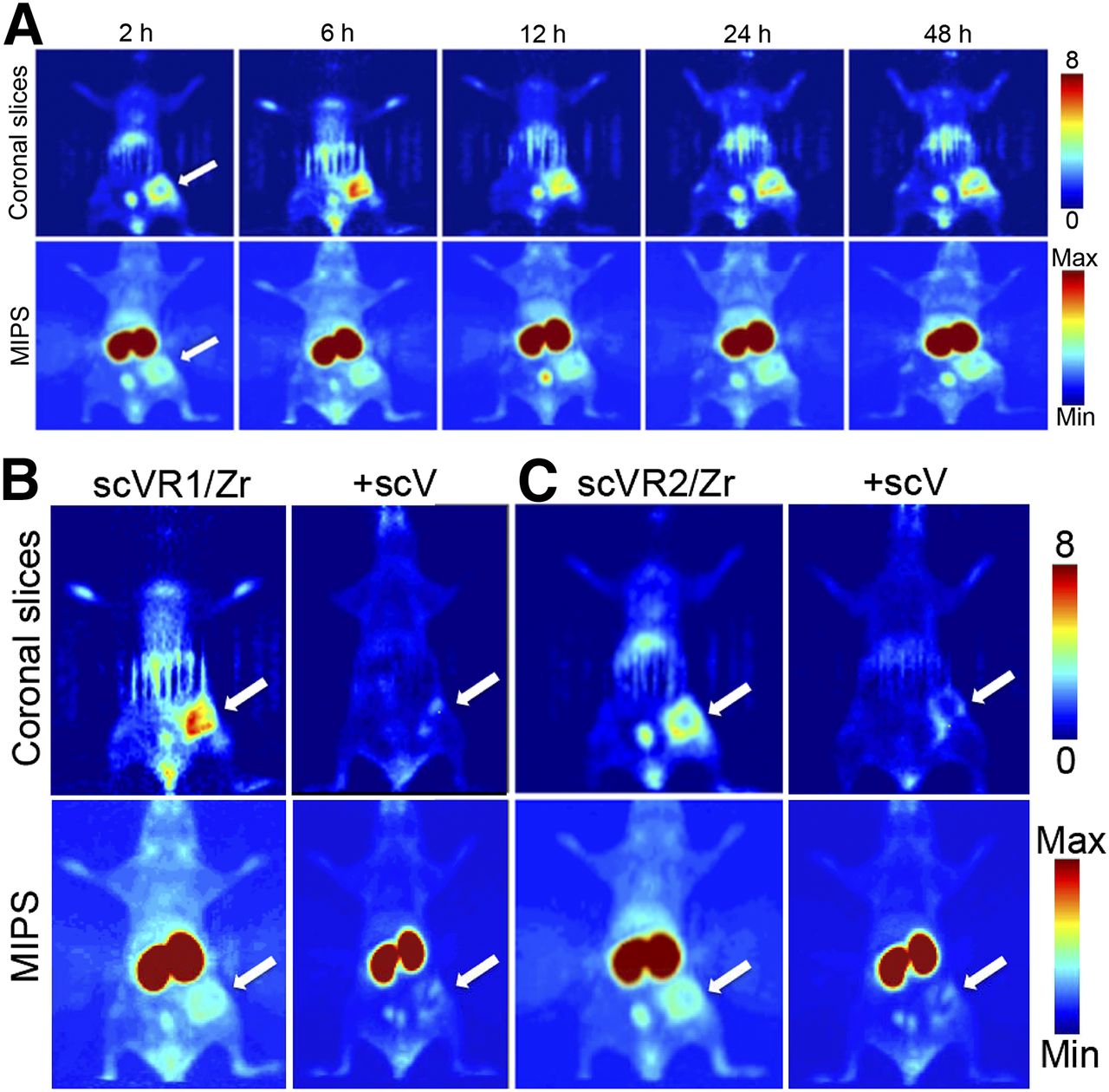
PET imaging with VEGFR-specific targeted tracers. Tumor-bearing mice were injected with indicated tracer (10.4–12.9 MBq/mouse) alone or in a mixture with 35-fold excess of cold scVEGF. (A) PET images acquired for same mice injected with scVR1/Zr at indicated time points. PET images for mice injected with scVR2/Zr and scV/Zr are shown in Supplemental Figures 6A and 6B, respectively. (B and C) PET images of mice at 24 h after injection of indicated tracer alone or in mixture with 35-fold excess of cold scVEGF. Max = maximum; min = minimum; MIPS = maximum-intensity projections.
In agreement with the previous reports on imaging 4T1luc tumors using scVEGF-based PET and SPECT tracers (3–5), 89Zr tracer uptake in tumor area was visibly heterogeneous, with the higher uptake at the tumor periphery (Fig. 5A for scVR1/Zr; Supplemental Figs. 6A and 6B for scVR2/Zr and scV/Zr, respectively). This heterogeneity could not be attributed to the development of central necrotic areas, because tumors were small (4–5 mm) and had not developed necrosis, as judged by histochemical hematoxylin and eosin staining (Supplemental Fig. 7).
Imaging experiments also supported receptor-mediated and receptor-specific mechanism of uptake for scVR1/Zr and scVR2/Zr, of scVR1/Zr, because coinjection of these tracers with a 35-fold excess of cold scVR1 or scVR2 significantly decreased the corresponding tracer uptake (Fig. 5B). Interestingly, quantitative analysis of longitudinal imaging in individual mice supported the notion that for scVR2/Zr tracer, radioactivity might continue to accumulate in tumor on a 48-h time scale (Supplemental Fig. 5C) through yet to be established mechanisms.
Immunohistochemical Analysis of VEGFR1 and VEGFR-2 in 4T1luc Tumors
Most VEGFR-1 was found on cells that do not express a pan-endothelial marker CD31, although a significant subset of VEGFR-1–positive cells was localized close to CD31-positive endothelium (Fig. 6A). VEGFR-2 immunostaining was colocalized with that for CD31 (Fig. 6B), indicating that most VEGFR-2, but only a small subset of VEGFR-1 that colocalizes with VEGFR-2 (Supplemental Fig. 8), are expressed on tumor endothelial cells. Further studies would be necessary to establish which tumor and stromal cells express VEGFR-1 in 4T1luc tumors.
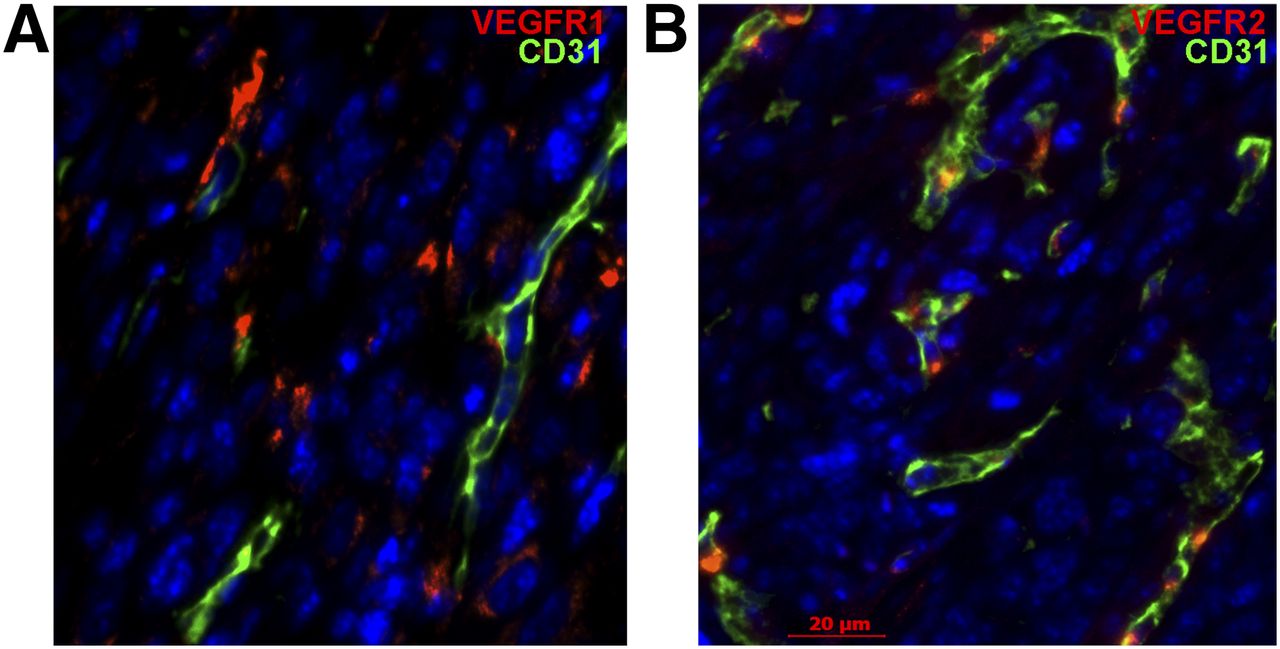
VEGFR-1 and VEGFR-2 are expressed by different cells in 4T1luc tumors. Immunofluorescent staining for VEGFRs (red) and pan-endothelial marker CD31 (green). 4′,6-diamidino-2-phenylindole (blue), staining of nuclei. Scale bar, 20 μm. (A) VEGFR-1 is expressed mostly on CD31-negative cells. (B) VEGFR-2 is predominantly expressed on CD31-positive cells.
Interestingly, in many fields the prevalence of VEGFR-1 in 4T1luc was visibly higher than that of VEGFR-2 (Supplemental Fig. 8, compare left and middle). Although it seemingly contradicts to a rather similar tumor uptake of scVR1/Zr and scVR2/Zr (Fig. 3), direct comparison between immunohistochemistry and imaging data could be misleading, because immunohistochemistry reflects prevalence of all receptors, whereas imaging reflects prevalence of only accessible and functionally active receptors. Indeed, double immunostaining for VEGFR-1 and VEGFR-2 revealed that VEGFR-2 always colocalizes with a subset of VEGFR-1–positive cells (Supplemental Fig. 8, right).
DISCUSSION
We report here the engineering of 2 novel human VEGF-A–based proteins, scVR1 and scVR2, with selective affinity to either VEGFR-1 or VEGFR-2. This selectivity was not affected by site-specific conjugation of PEGylated DFO, allowing for development of 89Zr PET tracers. 89Zr radiolabeling of conjugates yielded 2 novel tracers, scVR1/Zr and scVR2/Zr, in excellent radiochemical yield, high purity, and high specific activities. To validate the receptor selectivity of these tracers in vivo, we envisaged that proof-of-concept experiments performed in the easily accessible, orthotopic tumor model 4T1 using immunocompetent mice would be a reasonable starting point.
Previous findings (3–5,17–19) demonstrated the potential of the in vivo VEGFR or VEGF imaging. Results included heterogeneous distribution of tracer within the tumor paired with different responses of these tumor regions to antiangiogenic therapy (3–5,17–19), even though the tracer was not VEGFR-specific. The independent roles of VEGFRs in normal and pathologic angiogenesis, vasculogenesis, and tumor biology have been recently discovered (1,6,7). In this regard, the ability of selective whole-body imaging of VEGFRs could provide valuable mechanistic insights as well as vital real-time information on response to antiangiogenic therapy, especially when it comes to time-sensitive treatments, such as cancer therapy. We herein report 2 new first-in-class 89Zr PET tracers for independent imaging of VEGFR-1 and VEGFR-2. The receptor-specific tracers scVR1/Zr (Fig. 5A) and scVR2/Zr (Supplemental Fig. 6A) readily accumulated in tumor with preferential localization at the periphery of the tumor in a pattern similar to that of pan-receptor tracer scV/Zr (Supplemental Fig. 6B) and other scVEGF-based tracers (3–5).
Blocking experiments with an excess of cold scVEGF indicate that at least 80% of scVR1/Zr or scVR2/Zr uptake is VEGFR-mediated (Figs. 4A, 5B, and 5C). Critically, blocking experiments with an excess of cold scVR1 or scVR2 established that each mutant could inhibit only uptake of tracer of the same, but not the opposite, receptor specificity (Fig. 4B). In a good agreement with the data on the selective receptor binding in functional assays in tissue culture and in vitro (Fig. 1), these results indicate a high level of in vivo receptor selectivity for both scVR1/Zr and scVR2/Zr and suggest that these tracers can be used for independent assessment of VEGFR-1 and VEGFR-2 dynamics in various pathologies and treatment regimens.
Somewhat surprisingly, we found that contrary to tumor uptake, coinjection of tracer with a large excess of cold protein does not statistically significantly affect biodistribution in the major organs (Fig. 2; Supplemental Figs. 4A and 4B). Considering rather large variations in biodistribution studies (average STD, ∼40%), we can conclude only that unlike in tumors, most of the tracer uptake in major organs is not VEGFR-mediated.
We found that kidney uptake in tumor-bearing BALB/c mice for scVEGF-based 89Zr tracers was somewhat higher than that for scVEGF-PEG-DOTA radiolabeled with 99mTc, 64Cu, or 68Ga (5,18). Although our animal data suggest a tolerable overall human dosimetry, further studies are required to ascertain safety of receptor-specific 89Zr tracers. Further studies are also required on the mechanisms of tracer uptake, such as receptor-mediated internalization versus binding at the surface of the cells and the nature of cells that are involved in uptake of scVR1-based tracer at various tumor compartments. We expect that such studies will provide a firm mechanistic foundation for imaging VEGFR-1 and VEGFR-2 in antiangiogenic therapeutic regimens.
One mechanistic insight that we gained using scVR1 and scVR2 receptor–specific tracers deals with a better understanding that VEGFRs are responsible for uptake of pan-receptor scVEGF-based tracer in a 4T1luc orthotopic model (5). Judging by blocking studies with receptor-specific cold proteins, we demonstrated that uptake of pan-receptor tracer is mediated by both VEGFR-1 and VEGFR-2, however, with a greater involvement of VEGFR-1, with an approximate ratio of 2:1 (Fig. 4B).
The significance and potential applications of receptor-selective tracers for independent imaging of VEGFR-1 and VEGFR-2 are based on the critical, yet different roles that VEGFR-1 and VEGFR-2–positive cells are playing at distinct stages of cancer progression (20). Recent studies indicate that VEGFR-1–positive hematopoietic stem and progenitor cells (HSPCs) are involved in organizing and maintaining protumorigenic immunosuppressive microenvironment in visceral organs, paving the way for metastatic dissemination of tumor cells and growth of metastatic lesions (1,6–10,21–23). Importantly, limited studies indicate that the prevalence of mobilized HSPCs in blood correlates with severity of metastatic disease in human patients (23). The effects of VEGFR-1–positive HSPCs are blocked by anti–VEGFR-1 antibody (21,23). In view of these data, we can expect that VEGFR-1–selective tracer would be most useful in detecting metastatic lesions and guiding anti–VEGR-1 therapy (24).
VEGFR-2, in turn, is expressed mostly on tumor endothelial cells and found at particularly high levels on endothelial cells at the invasive tips of the growing capillaries (25). VEGF/VEGFR-2 signaling is a major regulator of tumor angiogenesis (1,6,7,25) and is currently targeted by several antiangiogenic drugs. Unfortunately, these drugs are only marginally effective, as was underscored by several failures in recent phase III breast cancer clinical trials (26). Current explanation for this inefficiency is that different VEGF/VEGFR-2 inhibitors induce only a transient regression of tumor vasculature followed by vascular rebound that rekindles aggressive tumor growth (27). We have previously reported the feasibility of monitoring the progress of antiangiogenic therapy in mouse tumor models with pan-receptor scVEGF-based SPECT tracer, when scVEGF/99mTc tumor uptake declined during the initial response to therapy and then rebounded as therapy became ineffective (3,4). In these experiments, decline and rebound in pan-receptor tracer uptake followed similar changes. These findings suggest that imaging VEGFR-2 with VEGFR-2–selective tracer that is not wasted on VEGFR-1 could be particularly useful in assessing the success and failure of antiangiogenic regimens in individual cancer patients in real time. Considering how unpredictable and inefficient the current antiangiogenic therapy is (2), we expect that availability of a well-defined molecular VEGFR-2 imaging biomarker for image-guided therapy would meet a real medical need.
CONCLUSION
We report here first-in-class 89Zr PET tracers scVR1/Zr and scVR2/Zr for selective imaging of, correspondingly, VEGFR-1 and VEGFR-2. The specificity of tracers was confirmed by blocking studies with cold proteins of similar and opposite receptor specificity. We expect that new tracers will allow investigation of distinct roles of VEGFR-1 and VEGFR-2 in tumor biology and assessment of the use of these receptors as imaging biomarkers for diagnostic and image-guided anticancer therapy.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. The authors gratefully acknowledge the MSKCC Small Animal Imaging Core Facility as well as the Radiochemistry & Molecular Imaging Probe Core, which are supported in part by NIH grant P30 CA08748. This work was also supported in part by NIH SBIR contract HHSN26801400041C to Joseph M. Backer. We also gratefully acknowledge support from Mr. William H. and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research and The Center for Experimental Therapeutics of Memorial Sloan Kettering Cancer Center. Joseph M. Backer and Marina V. Backer are employees at SibTech Inc. and Joseph M. Backer has an equity interest. No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Jul. 7, 2016.
- © 2016 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication January 28, 2016.
- Accepted for publication May 16, 2016.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}