


Abstract
11C-LY2795050 is a novel κ-selective antagonist PET tracer. The in vitro binding affinities (Ki) of LY2795050 at the κ-opioid (KOR) and μ-opioid (MOR) receptors are 0.72 and 25.8 nM, respectively. Thus, the in vitro KOR/MOR binding selectivity is about 36:1. Our goal in this study was to determine the in vivo selectivity of this new KOR antagonist tracer in the monkey. Methods: To estimate the ED50 value (dose of a compound [or drug] that gives 50% occupancy of the target receptor) of LY2795050 at the MOR and KOR sites, 2 series of blocking experiments were performed in 3 rhesus monkeys using 11C-LY2795050 and 11C-carfentanil with coinjections of various doses of unlabeled LY2795050. Kinetic modeling was applied to calculate regional binding potential (BPND), and 1- and 2-site binding curves were fitted to these data to measure 11C-LY2795050 binding selectivity. Results: The LY2795050 ED50 at MOR was 119 μg/kg based on a 1-site model for 11C-carfentanil. The 1-site binding model was also deemed sufficient to describe the specific binding of 11C-LY2795050 at KOR. The ED50 at KOR estimated from the 1-site model was 15.6 μg/kg. Thus, the ED50 ratio for MOR:KOR was 7.6. Conclusion: The in vivo selectivity of 11C-LY2795050 for KOR over MOR is 7.6. 11C-LY2795050 has 4.7-fold-lower selectivity at KOR over MOR in vivo as compared with in vitro. Nevertheless, on the basis of our finding in vivo, 88% of the PET-observed specific binding of 11C-LY2795050 under baseline conditions will be due to binding of the tracer at the KOR site in a region with similar prevalence of KOR and MOR. 11C-LY2795050 is sufficiently selective for KOR over MOR in vivo to be considered an appropriate probe for studying the KOR with PET.
The κ-opioid receptor (KOR) is one of three major subtypes of the opioid receptor. The endogenous ligand for KOR is dynorphin, which is related to dysphoria. The administration of dynorphin results in conditioned place aversion in animals, and dynorphin activation of KOR results in decreased dopamine release in the brain reward circuitry (1,2). The involvement of the KOR in addiction is thought to be through its ability to modulate dopamine function. Activation of the KOR by dynorphin or administration of KOR agonists inhibits psychostimulant-induced dopamine release. Attenuation of dopamine release has been shown to inhibit both the psychomotor effects and the reinforcing behaviors of psychostimulants. Repeated administration of drugs of abuse leads to the dysregulation of the dynorphin–KOR modulatory system. In addition, KORs are believed to play a role in depression, anxiety, and Alzheimer disease (3).
The potential role of KORs in mediating alcohol drinking behavior has been the focus of several studies in animals and humans. Mice that lack KORs drink more alcohol and have greater release of dopamine in response to alcohol (4). Rodents with an increased propensity to consume alcohol have lower dynorphin levels in several brain regions that are involved in the control of alcohol drinking (5–7). In humans, the presence of the OPRK1 allele (which decreases expression of KOR) is associated with increased risk of alcoholism (8).
Two prior PET studies have examined the μ-opioid receptor (MOR) and the δ-opioid receptor (DOR) and their possible roles in the treatment of alcoholism. The tracer for MOR was 11C-carfentanil and for DOR was 11C-methyl-naltrindole. Weerts et al. (9,10) imaged recently abstinent alcoholics with each tracer before and after administration of the nonspecific opioid antagonist naltrexone. To date, there have been no imaging studies of KOR because a highly selective KOR tracer has not been heretofore available.
Recently, 2 new radioligands were developed for imaging KORs. Ravert et al. (11) introduced 11C-GR103545, a KOR agonist tracer, and Talbot et al. (12) evaluated its kinetics in baboons. Poisnel et al. (13) reported the KOR antagonist tracer 11C-MeJDTic and characterized it ex vivo in mice. Both tracers showed activity uptake patterns consistent with the KOR distribution determined from in vitro autoradiographic studies of displaceable specific binding (SB). 11C-GR103545 has been advanced to applications in humans at our laboratory (14). 11C-MeJDTic appears to be a highly selective antagonist (i.e., selectivity for KOR over MOR = Sκ:μ = Ki(μ)/Ki(κ) = 700; Sκ:δ = Ki(δ)/Ki(κ) > 10,000, where S is selectivity and Ki is binding affinity) (15). However, it is not clear how this tracer will behave in vivo, because its imaging evaluation has not been reported. As an agonist, 11C-GR103545 binds to receptors in the high-affinity state. Therefore, it could be used to assess the potential changes in receptor state in diseases and might also be more sensitive to competition from endogenous opiates. However, a potent KOR agonist tracer will induce pharmacologic effects at relatively low mass doses. Hence, its injected mass will need to be strictly controlled.
Our group recently reported a new KOR antagonist radioligand 11C-LY2795050 (16). Radioligand competition assays using cloned human KOR, MOR, and DOR indicated 11C-LY2795050 displayed in vitro Ki values of 0.72, 25.8, and 153 nM, respectively, for KOR, MOR, and DOR. These in vitro affinities suggest a great selectivity of KOR over DOR (200:1) and moderate selectivity of KOR over MOR (35:1). Therefore, investigation of selectivity for KOR over MOR in vivo is important for proper use of the new selective KOR tracer.
A few studies have compared the in vitro and in vivo selectivity of PET tracers, and there are documented discrepancies between the in vitro and in vivo affinity and selectivity profiles (17,18). In this paper, we report the in vivo selectivity of 11C-LY2795050 derived from PET imaging experiments.
Two series of experiments in rhesus monkeys were performed. First, we measured the ED50 value (dose of a compound [or drug] that gives 50% occupancy of the target receptor) of LY2795050 at MOR using an occupancy model and 11C-carfentanil, a selective MOR tracer (19). Second, we measured the ED50 of LY2795050 at KOR using 1- and 2-site binding models and 11C-LY2795050 as the radiotracer.
MATERIALS AND METHODS
General Experimental Design
PET imaging experiments in rhesus monkeys (Macaca mulatta) were conducted according to a protocol approved by the Yale University Institutional Animal Care and Use Committee. Monkeys were initially immobilized with ketamine (10 mg/kg intramuscularly) and subsequently anesthetized with isoflurane (1%–2% in O2). Vital signs (pulse rate, blood pressure, respirations, electrocardiogram) were monitored continuously.
Two series of occupancy experiments were performed in 3 rhesus monkeys (mean age ± SD, 5.0 ± 2.0 y old; mean weight ± SD, 8.8 ± 3.8 kg) with the radiotracers 11C-carfentanil and 11C-LY2795050. On each scan day, a baseline scan was obtained, followed by a blocking study performed with varying doses of unlabeled LY2795050 (Table 1). Typically, unlabeled LY2795050 was coadministered with the tracer.
Study Design
Radiochemistry
11C-LY2795050 was synthesized as previously described (16). Both the chemical and the radiochemical purity of the 11C-LY2795050 final product were greater than 95%. Specific activity (mean ± SD) at the end of synthesis was 24.6 ± 14.3 MBq/nmol (n = 14). 11C-carfentanil was synthesized with an AutoLoop (Bioscan) in a procedure similar to that used for the synthesis of 11C-P943 (20). In brief, a solution of 0.5–1.0 mg of desmethyl carfentanil precursor in 80–90 μL of anhydrous dimethyl sulfoxide and 2.5 μL of 1N CsOH in MeOH was loaded onto a stainless steel loop and reacted with 11C-MeOTf for 5 min at room temperature. The radiolabeled product was purified with semipreparative high-performance liquid chromatography (column: Prodigy ODS [Phenomenex], 10 μm, 250 × 10 mm; mobile phase: 35% acetonitrile and 65% of 0.1 M ammonium formate with 0.5% acetic acid, pH 4.2 [v/v]; flow rate: 5 mL/min). Fraction containing the product was collected, diluted with water (50 mL), and passed through a Waters C18 SepPak cartridge. The SepPak cartridge was rinsed with water (10 mL) and eluted with ethanol (1 mL) to recover the product. The ethanol solution was diluted with sterile saline (9 mL) and passed through a sterile 0.22-μm membrane filter for terminal sterilization to afford a sterile product solution ready for injection. Radiochemical purity of the product was greater than 95% at the end of synthesis. Specific activity was 160.5 ± 128 MBq/nmol at the end of synthesis (n = 6).
Scans
PET imaging was performed with a Focus 220 scanner (Siemens/CTI), which has a resolution of 1.4 mm at the center of the field of view. After a 9-min transmission scan, acquisition of emission data started when the bolus administration of tracer started. All 11C-LY2795050 and 11C-carfentanil scans were obtained for 120 min. All images were reconstructed with attenuation and scatter correction using a filtered backprojection algorithm and a Shepp–Logan axial filter with a cutoff frequency of 0.15 cycles per pixel. Injected doses of 11C-LY2795050 and 11C-carfentanil were 12.4 ± 8.4 and 4.6 ± 3.0 MBq/kg, respectively, and injected mass doses were 0.34 ± 0.23 and 0.03 ± 0.01 μg/kg, respectively. The mass limits (occupancy < 5%) for each tracer were based on the affinities of the tracers for their respective targets.
MR images were acquired for each rhesus monkey on a 3.0-T Trio scanner (Siemens). T1-weighted images were acquired in the coronal plane with a spin-echo sequence (echo time, 3.34 ms; repetition time, 2,530 ms; flip angle, 7°; section thickness, 0.50 mm; field of view, 140 mm; image matrix; 256 × 256 × 176 pixels; and voxel size, 0.547 × 0.547 × 0.500 mm).
Each monkey’s early summed PET images (typically 0–10 min) were coregistered to the monkey’s MR images via a 6-parameter rigid registration with the multitransform method (21). An affine linear-plus-nonlinear registration was performed (Bioimage Suite 2.5; Yale University [www.bioimagesuite.org]) for each MR image to a high-resolution rhesus monkey template on which regions of interest were defined. The following 15 regions of interest were drawn for both tracers: amygdala, brain stem, caudate, cingulate cortex, frontal cortex, globus pallidus, hippocampus, insula, nucleus accumbens, occipital cortex, pons, putamen, substantia nigra, temporal cortex, and thalamus.
Arterial Input Function of 11C-LY2795050
Arterial samples were obtained for 11C-LY2795050 because there were no previous data regarding a valid reference region for this tracer. Twenty-one blood samples (0.5 mL) were drawn at 0.75, 1.5, 2.25, 3, 4, 5, 6, 8, 9, 12, 15, 20, 25, 30, 40, 50, 60, 75, 90, 105, and 120 min after the injection of 11C-LY2795050. Samples drawn at 4, 12, 30, 60 (3.5 mL), and 90 min (1–5 mL) were also analyzed for radioactive metabolites according to published methods (16). Arterial samples were not taken for 11C-carfentanil because there is an accepted reference region (22).
Estimate of ED50 for LY2795050 at MOR
The simplified reference tissue model (SRTM) (23) was used to estimate binding potential (BPNDMOR) of 11C-carfentanil at MOR using the occipital cortex as the reference region (22). A modified Lassen plot, using BPND values instead of regional total distribution volume (VT) with no intercept, was applied to calculate percentage occupancy comparing each blocking scan to its matching baseline scan (24). The effective dose of LY2795050 to achieve 50% occupancy at MOR (ED50MOR) was estimated using nonlinear fitting of a sigmoidal dose-occupancy curve [y = 100x/(ED50MOR + x)] to the 11C-carfentanil data (GraphPad Prism; GraphPad Software Inc.), where y is percentage occupancy and x is the dose of LY2795050 (μg/kg).
11C-LY2795050 BPND
The regional VT of 11C-LY2795050 was calculated using the multilinear analysis (MA1) (25) method (t*, 40 min) with a metabolite-corrected plasma time–activity curve. VT values in the cerebellum at baseline and during multiple blocking scans were compared to evaluate and validate the use of the cerebellum as a reference region. On the basis of the comparison of VT values, the SRTM was then used to estimate regional binding potential (BPND) of 11C-LY2795050 (with the cerebellum as the reference region).
Binding Models to Estimate ED50 of LY2795050 at KOR
2-Site Model
According to a 2-site model, the observed binding potential (BPNDmeasured) of 11C-LY2795050 in the presence of unlabeled LY2795050 can be modeled as the sum of SB at KOR (BPNDKOR) and SB at MOR (BPNDMOR) (Eq. 1). In essence, the observed BPND is a weighted sum of 2 receptor availabilities at the 2 binding sites that can be expressed in terms of occupancies, Eq. 1where BPNDmeasured is the measured binding potential in each region r = {1, …,8} and Occi the occupancy of binding site i by LY2795050, assuming that occupancy is uniform across brain regions.
Eq. 1where BPNDmeasured is the measured binding potential in each region r = {1, …,8} and Occi the occupancy of binding site i by LY2795050, assuming that occupancy is uniform across brain regions.
Each occupancy value can be expressed in terms of dose and the ED50 at the respective binding sites, Eq. 2
Eq. 2
Equation 2 can be applied to multiple regions, simultaneously. It contains 2 unknown BP terms ( ,
,  ) for each region, r, and 2 unknown terms,
) for each region, r, and 2 unknown terms,  ,
,  , that are assumed to be shared across all regions.
, that are assumed to be shared across all regions.
1-Site Model
As a simpler model, the 1-site model attributes all blockade to drug action at KOR (Eq. 3). Eq. 3
Eq. 3 Eq. 4
Eq. 4
Model Assumption
The maximum occupancy by LY2795050 was constrained to 100% at both the KOR and the MOR sites in Equations 2 and 4, meaning that maximum Occi is 1 in Equations 1 and 3. The parameter ED50KOR was shared across regions and monkeys.
In the 2-site model, the ED50MOR was fixed to the estimated value from the fit of a 1-site model to the 11C-carfentanil data. Thus, a fit of Equation 2 to all 8 regions simultaneously required us to estimate (number of regions × 2) + 1 parameters from 8 BP versus dose curves. In the 1-site model, (number of regions + 1) parameters were estimated.
The binding models were fitted to data from moderate- and high-binding regions (caudate, cingulate cortex, frontal cortex, globus pallidus, insula, nucleus accumbens, putamen, and substantia nigra) from 3 monkeys, simultaneously. Goodness of fit to the 1- versus 2-site model was determined by an F test (significance level, 0.05; degrees of freedom, 8,86). To assess the sensitivity of our estimates of ED50KOR to the choice of ED50MOR, we examined the percentage change in estimated ED50KOR because the fixed value of ED50MOR was varied by ±10% in the 2-site model.
We are assuming that the measured occupancy is entirely due to cold compound and not an increase in endogenous dynorphin because the compound LY2795050 is an antagonist, not an agonist.
RESULTS
Derivation of ED50 Value for LY2795050 at MOR
Occupancies at MOR by multiple doses of LY2795050 are shown in Figure 1. Occupancies were obtained from modified Lassen plots using 12 brain regions (brain stem, caudate, cerebellum, cingulate cortex, frontal cortex, globus pallidus, hippocampus, insula, pons, putamen, temporal cortex, and thalamus), excluding the nucleus accumbens because—although a high-binding region—it lies distant from the line of regression (Supplemental Fig. 1; supplemental materials are available at http://jnm.snmjournals.org). ED50MOR was estimated as 119 ± 21 μg/kg. All tissue time–activity curves were fitted well using SRTM under both the baseline and the blocking conditions.

Occupancy data for LY2975050 at MOR were fitted to occupancy (Occ = 100 × dose/(ED50MOR + dose)).
Assessment of Reference Region for 11C-LY2795050 and Derivation of Regional BPND
We used MA1 for estimating VT because the estimates showed high correlation with VT estimates using the 2-tissue-compartment model (slope = 0.94; r2 = 0.95). Some example fits to the time–activity data using MA1 and correlation plot between the 2-tissue-compartment model VT and MA1 VT are provided in Figure 2.

(A) Time–activity curves of 11C-LY2795050 in monkey. Lines represent fits by MA1 model (t* = 40 min). (B) Correlation between VT values estimated by 2-tissue-compartment model and MA1 (t* = 40 min), y = 0.94x+0.12, r2 = 0.95.
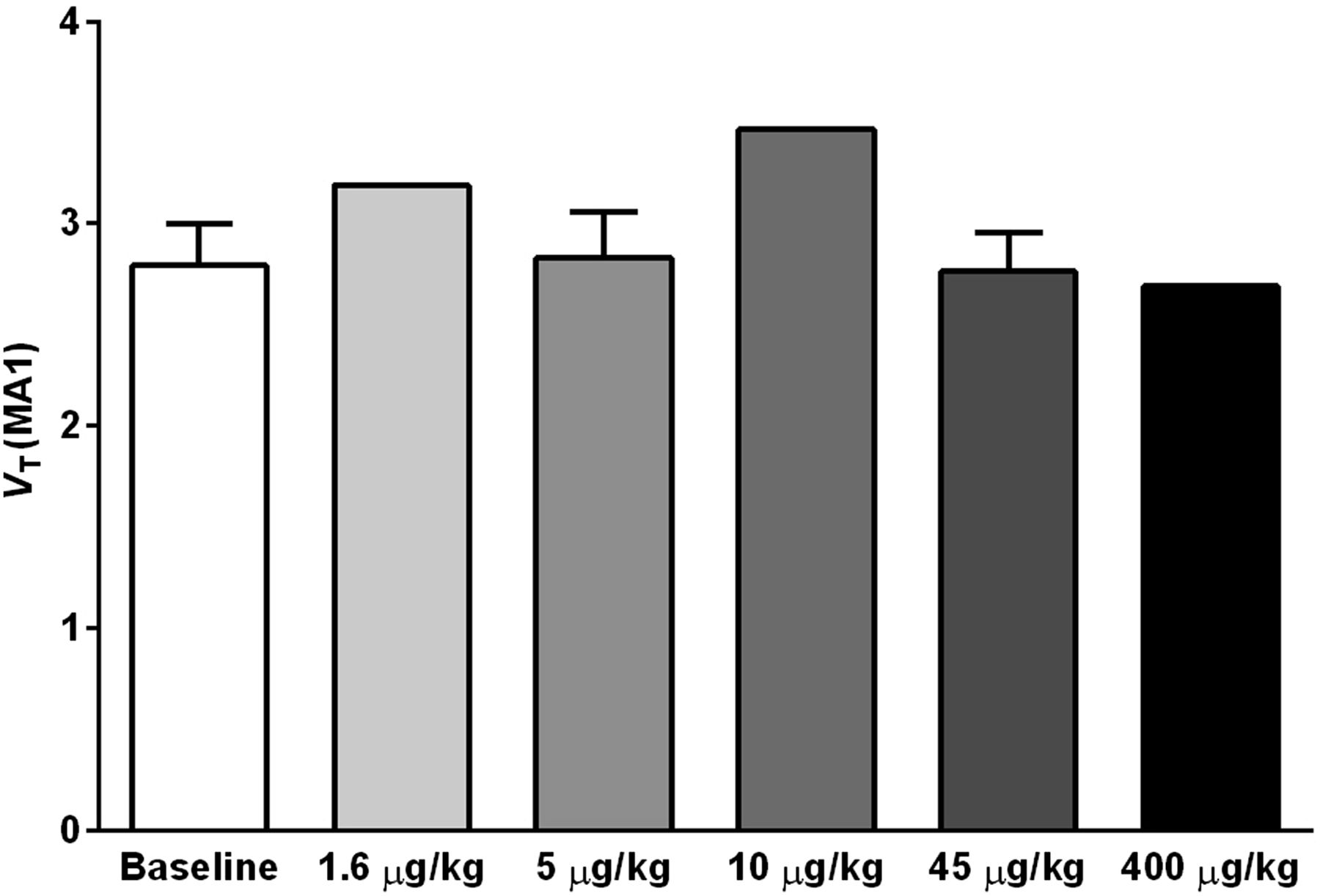
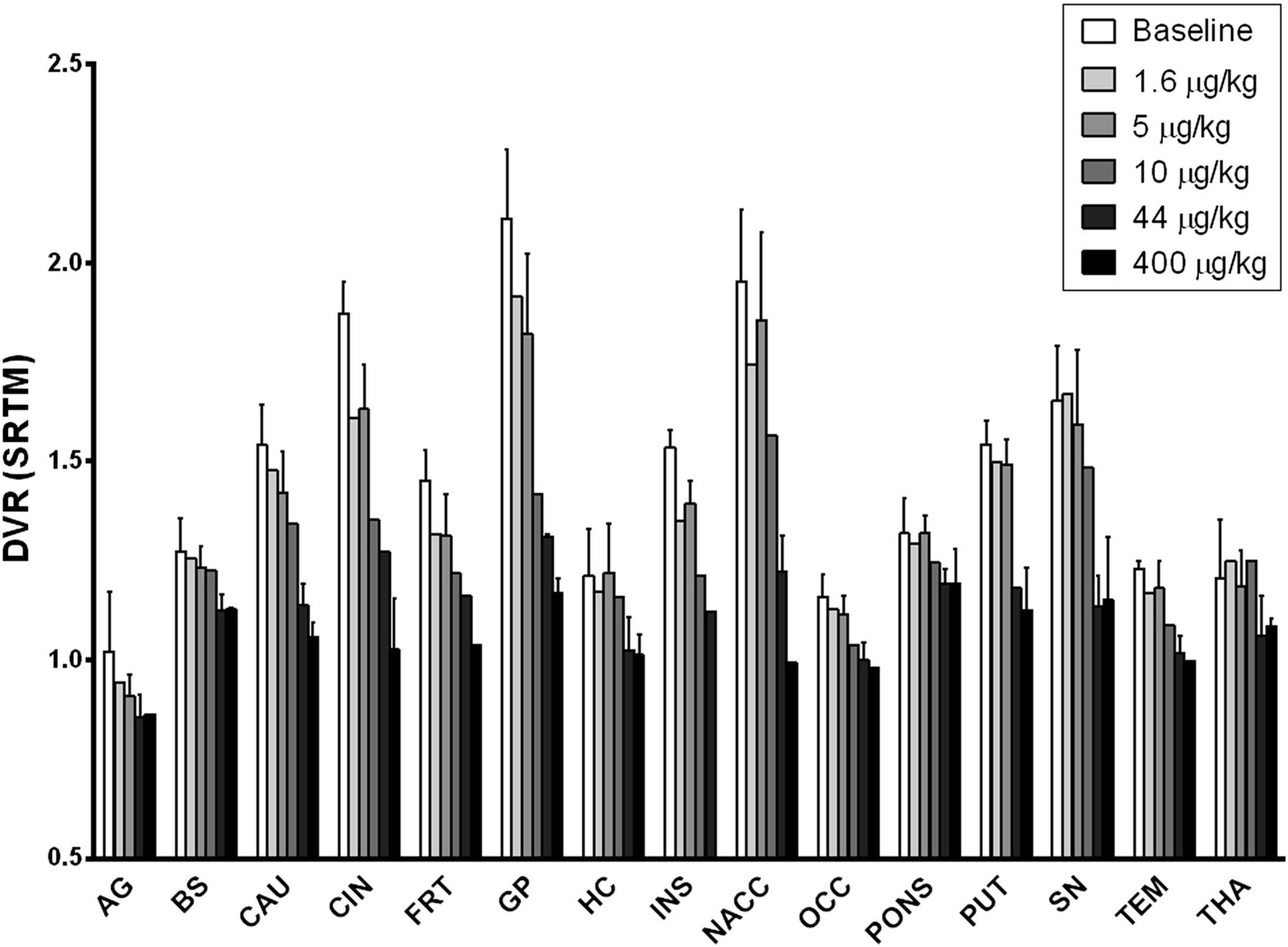
Shown in Figure 3 are VT values of 11C-LY2795050 in the cerebellum at baseline and from multiple blocking scans calculated using MA1. There was no difference between values at baseline and blocking scans with varying doses of LY2795050. Thus, the cerebellum was used as a reference region in calculating BPND. To present positive values, the distribution volume ratio (DVR = BPND+1) for each region is shown in Figure 4 for multiple LY2795050 doses. In the baseline condition, regional binding was highest (DVR > 1.5) in the globus pallidus, cingulate cortex, nucleus accumbens, substantia nigra, caudate, putamen, and insula. This finding is consistent with the reported distribution of KOR in primates (26). Binding in all regions decreased with increasing doses of LY2795050 (Figs. 4 and 5). Unreliable estimates of BPND (percentage SE > 40%) were excluded in binding site analysis in Figures 4 and 6.
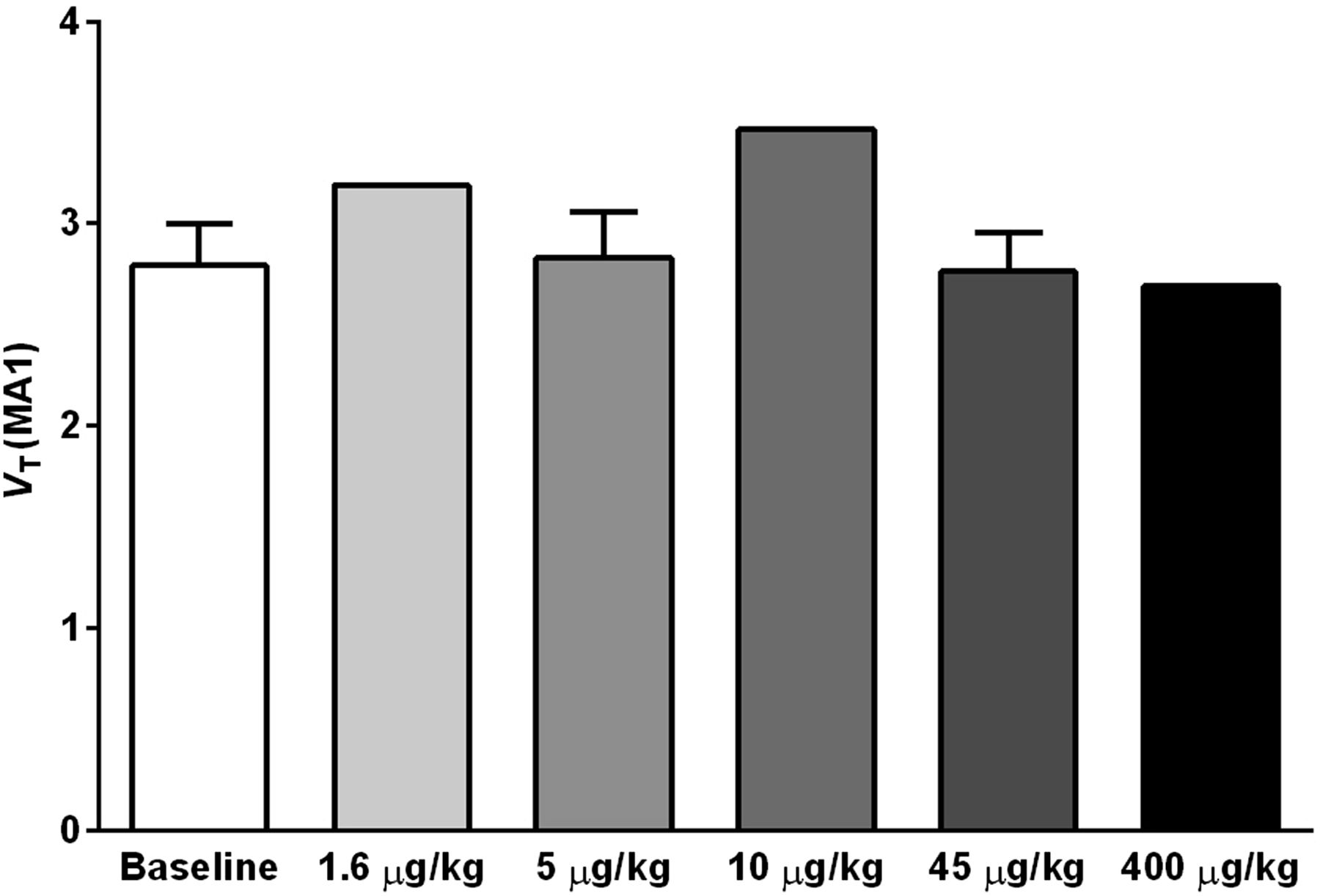
Cerebellum VT of 11C-LY2975050 estimated from baseline and blocking scans with 5 different doses of LY2975050 using MA1 model and metabolite-corrected arterial plasma curve as input function. No difference in VT across these doses indicates that cerebellum is valid reference region for 11C-LY2975050 in rhesus monkeys.

Six different doses of LY2975050 cause dose-dependent decrease in DVR (BPND + 1) in all regions of brain examined. AG = amygdale; BS = brain stem; CAU = caudate; CIN = cingulate cortex; FRT = frontal cortex; GP = globus pallidus; HC = hippocampus; INS = insula; NACC = nucleus accumbens; OCC = occipital cortex; PUT = putamen; SN = substantia nigra; TEM = temporal cortex; THA = thalamus.

Average binding potential (BPND) images: baseline scan (n = 5) (A), blocking scan with 5 μg/kg of LY2795050 (n = 3) (B), blocking scan with 400 μg/kg of LY2795050 (n = 2) (C), and matching MR images (D).
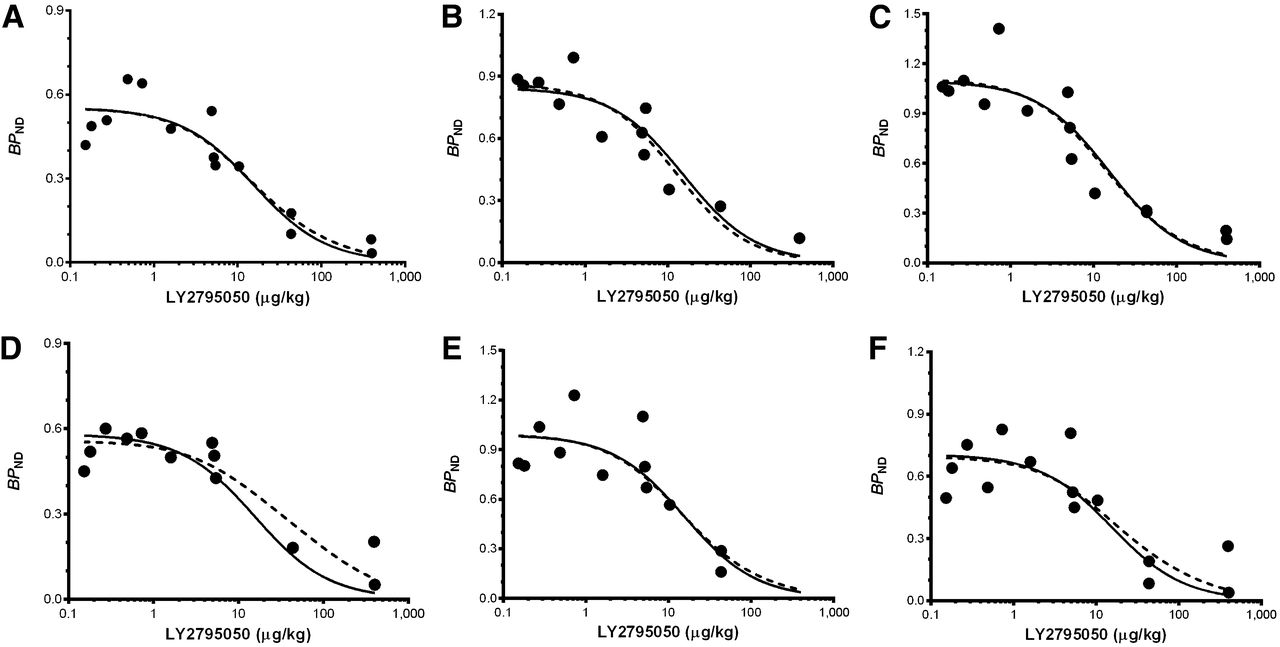
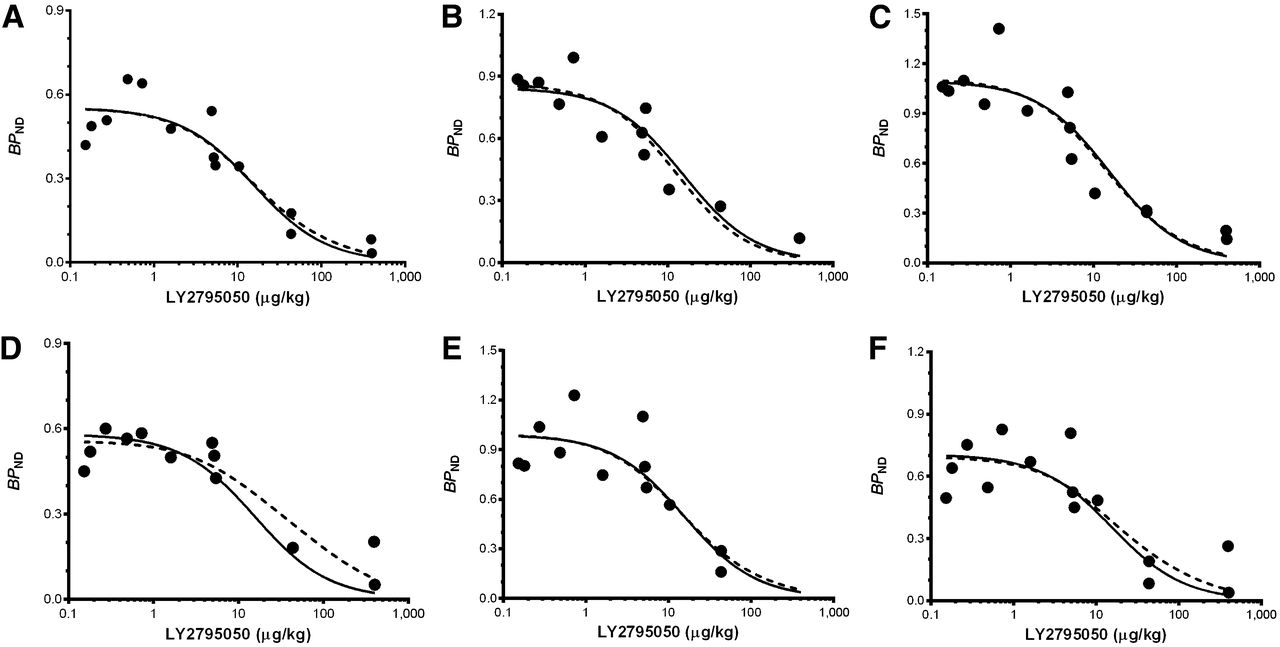
Fits of 1- (solid line) and 2-site (dashed line) binding models. Model fits to 8 brain regions simultaneously (6 highest-binding regions shown: caudate [A], cingulate cortex [B], globus pallidus [C], putamen [D], nucleus accumbens [E], and substantia nigra [F]). Fits of 1- and 2-site models are statistically indistinguishable. Curves are comprised of BPND points for 11C-LY2975050 from multiple monkeys at multiple doses of LY2975050. Data points at mass doses below 1 μg/kg were from baseline scans. Mass doses for these studies were calculated as radioactivity dose divided by specific activity.
Binding Models for Derivation of LY2795050 ED50 at KOR
Fitted curves of the 2- and 1-site binding models are shown in Figure 6 for high-binding regions (caudate, cingulate cortex, globus pallidus, putamen, nucleus accumbens, and substantia nigra). On the basis of the F test, there was no significant difference between the fits (P = 0.94). Therefore, the 1-site binding model was deemed sufficient to describe SB of 11C-LY2795050. The shared ED50KOR estimated from the 1-site model was 15.6 ± 2.2 μg/kg. In the 2-site model, when ED50MOR was fixed at 119 μg/kg based on the 11C-carfentanil results, the shared ED50KOR was estimated at 12.9 ± 4.5 μg/kg. For a 10% change in ED50MOR, the change in estimated ED50KOR was less than 1%. Estimation of ED50KOR was therefore not sensitive to the choice of ED50MOR. Region-by-region estimates of BPND and associated shared estimates of ED50 are shown in a Supplemental Table 1.
In Vivo Selectivity
The selectivity of LY279505 for KOR over MOR (ratio of the ED50 values) was 7.6.
DISCUSSION
As reported previously, the in vitro binding affinities (Ki) of 11C-LY2795050 were 0.72 and 25.8 nM, respectively, for KOR and MOR (16) at room temperature (22°C). In our present work, in nonhuman primates, in vivo, ED50MOR was found to be 119 μg/kg based on occupancy studies with 11C-carfentanil PET and blocking doses of LY2795050. Further, ED50KOR was estimated to be 15.6 μg/kg based on fits of a 1-site binding model with data obtained from blocking studies with the tracer 11C-LY2795050 and varying doses of unlabeled LY2975050. Our present results suggest that the in vivo selectivity of the new KOR antagonist radioligand 11C-LY2795050 is 7.6:1 for KOR over MOR. Thus, the tracer has 4.7-fold-lower selectivity for KOR over MOR in vivo as compared with in vitro. This degree of discrepancy in tracer selectivity between in vitro and in vivo values is comparable to those for other tracers reported in the recent literature. Ekelund et al. (17) showed that the in vitro affinity ratio of 11C-NNC112 for the D1 versus 5-HT2A receptors was 100:1 whereas the in vivo selectivity was only 10:1.
In vivo and in vitro Kd (equilibrium dissociation constant) values are often quite different. Slifstein et al. (18) found a substantially lower affinity in vivo (Kd = 0.20 nM) of 18F-fallypride for D2 receptors than the in vitro estimates obtained at room temperature (Kd = 0.04 nM). On the other hand, at body temperature, the in vitro Kd value was found to be 2.03 nM (i.e., Kd(in vitro, room temperature) < Kd(in vivo) < Kd(in vitro, body temperature)). Thus, one possible source of the in vivo–in vitro discrepancy might be related to the temperature at which assays were performed.
One useful way to understand the in vivo selectivity is to recognize that the ratio of the SB of the tracer, 11C-LY2795050, at KOR versus MOR will be dependent on 3 factors: the ratio of the affinities (aka selectivity), the ratio of the receptor densities in each region (aka prevalence), and the occupancies of the respective sites by any cold drug or endogenous neurotransmitter that might be on-board. The SB ratio is the relative contribution of binding at KOR versus MOR that contributes to the total specific tracer binding, Eq. 5where
Eq. 5where  and
and 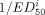 are the receptor density and affinity of the tracer, respectively, for site i. Occi is the occupancy of site i by a cold drug (e.g., naltrexone), tracer, or endogenous neurotransmitter.
are the receptor density and affinity of the tracer, respectively, for site i. Occi is the occupancy of site i by a cold drug (e.g., naltrexone), tracer, or endogenous neurotransmitter.
At baseline (no competition), in vivo selectivity is mainly determined by the ED50 ratio. Occupancy by drug at both sites is zero, so the occupancy ratio term in Equation 5 is approximately 1 (ignoring endogenous neurotransmitter). If ED50KOR is 16 μg/kg, then less than 5% occupancy by tracer would occur at approximately 1 μg/kg. In this study, injected mass doses of 11C-LY2795050 were 0.34 ± 0.23 μg/kg. Therefore, occupancy by tracer was negligible. If the prevalences of KOR and MOR are approximately equal in a given brain region (e.g., cingulate cortex (27)), then the SB ratio of 11C-LY2795050 will be ED50MOR/ED50KOR = 119/15.6 ≈ 7.6/1. Said another way, 88% of the SB signal from PET imaging will be attributable to binding at the KOR in a region with similar prevalence of KOR and MOR. Even in a region with a low prevalence of KOR (e.g., putamen, frontal cortex (27)), 72% of the SB detected will be due to binding at the KOR (Table 2).
Percentage of KOR Binding in Total SB in Regions
When a drug, such as naltrexone, is present in the brain, in vivo selectivity is determined by occupancies and ED50 ratio. That is, the occupancies of the drug at each receptor site in each region become important. It has been determined that clinical doses of naltrexone (50 mg), a nonselective opiate antagonist, lead to very high occupancy of MOR sites in the brain—an average of 94.9% according to Weerts et al. (10). If we consider a brain region with comparable prevalence of MOR and KOR and also use an average value of 91.5% occupancy of naltrexone at KOR (28), we can calculate: Eq. 6
Eq. 6
As the dose of naltrexone is increased from 0 to 50 mg, in vivo selectivity would rise gradually because the occupancy at KOR grows less rapidly than the occupancy at MOR. In other words, the SB ratio for KOR over MOR under naltrexone will actually be higher than at baseline because naltrexone has greater affinity for MOR than for KOR (29).
We determined in vivo selectivity, in part, by comparing model fits using 1- and 2-site binding models. A similar approach was taken by Searle et al. (30) in the analysis of 11C-PHNO data. They blocked 11C-PHNO binding with GSK598809, a selective D3 receptor antagonist, to gauge the selectivity for the D3 receptor as compared with the high-affinity D2 (D2high) receptor site. To check the assumption regarding negligible binding of GSK598809 at D2high sites, the authors compared model fits using a 2-site binding model that accounted for possible occupancy of D2high and D3 with a 1-site model that only accounted for D3 binding.
Similarly, we confirmed that a 2-site binding model was not necessary to describe the SB of 11C-LY2795050. The fits of our 2-site model to 11C-LY2795050 data were not significantly better than those of a 1-site model. Further, estimates of ED50KOR using the 2-site binding model were not different from those using a 1-site binding model (mean ± SE, 12.9 ± 4.5 μg/kg for the 2-site model and 15.6 ± 2.2 μg/kg for the 1-site model). In other words, we confirmed that the binding of 11C-LY2795050 at MOR is minimal. In Figure 6, all regions other than the putamen are well fitted using a 1-site binding model. The overall estimate of ED50KOR is not heavily dependent on the data in the putamen. If we eliminate the putamen, ED50KOR becomes 15 μg/kg.
In this study, we can obtain only the ED50 (dose of a compound [or drug] that gives 50% occupancy of the target receptor) values. Although the terms ED50 and Kd (or Ki; a concentration of compound [or drug] in the tissue) are not interchangeable, the ratios between in vitro Kd (or Ki) for KOR and MOR and between the in vivo ED50 for KOR and MOR can be compared to assess the selectivity difference between the in vitro and in vivo situations.
In comparing 1- and 2-site models, we chose to use BPND instead of VT, because BPND is more sensitive than VT to SB. If nonspecific VT is large, it is difficult to detect a change in SB via a change in VT, because regional VT is a sum of both nonspecific and specific volume of distribution. In our data, nonspecific VT values (low-binding regions: cerebellum or thalamus) have values that are approximately 50% of VT in the highest binding region (globus pallidus). Therefore, we assessed the validity of the cerebellum as the reference region for 11C-LY2795050 and noted that cerebellum VT did not decrease after any doses of LY2795050 (Fig. 3) whereas DVR (= BPND +1) showed dose-dependency in all other regions examined (Fig. 4).
Nucleus accumbens regions were excluded in the Lassen plot because that region has a much higher BPND value than others. Our finding is consistent with others’ findings showing that the nucleus accumbens has high MOR density (31). We chose to exclude the nucleus accumbens so that it would not drive the slope of the plot.
In our baseline data, the average koff (dissociation rate constant) value of 11C-LY2795050 in high-binding regions (globus pallidus, nucleus accumbens, and cingulate cortex) was 0.064 ± 0.02 (1/min). As a comparison, Pappata et al. (32) reported a koff of 0.1363 (1/min) for 11C-raclopride, which has proved to be ideal for displacement studies. We have not assessed the sensitivity of the new tracer’s binding to endogenous opiates, but this will be an important aspect to explore in future studies.
CONCLUSION
In this study, we measured ED50MOR (15.6 μg/kg) and ED50KOR (119 μg/kg) to calculate the in vivo selectivity of the novel antagonist tracer 11C-LY2795050 for KOR over MOR. In vivo selectivity of 11C-LY2795050 was approximately 7.6:1. This selectivity is 4.7-fold lower than the reported in vitro value. The same pattern has been found for other PET tracers in recent work. 11C-LY2795050 is sufficiently selective for KOR over MOR in vivo because, even in regions with equal KOR and MOR density, approximately 88% of the SB of 11C-LY2795050 that we measure with PET in a baseline scan will be due to binding of the tracer at the KOR.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This project was supported by the Yale PET Center. This publication was also made possible by CTSA grant UL1 RR024139 from the National Center for Research Resources (NCRR) and the National Center for Advancing Translational Science (NCATS), components of the National Institutes of Health (NIH), and NIH roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NIH. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Krista Fowles, Siobhan Ford, and Daniel Holden for animal experiments and the Yale PET Center chemistry team for synthesis of 11C-LY2795050.
Footnotes
Published online Aug. 5, 2013.
- © 2013 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication December 20, 2012.
- Accepted for publication April 8, 2013.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Impact of Pharmacological Manipulation of the {kappa}-Opioid Receptor System on Self-grooming and Anhedonic-like Behaviors in Male Mice
- Receptor Occupancy of the {kappa}-Opioid Antagonist LY2456302 Measured with Positron Emission Tomography and the Novel Radiotracer 11C-LY2795050
- Test-Retest Reproducibility of Binding Parameters in Humans with 11C-LY2795050, an Antagonist PET Radiotracer for the {kappa} Opioid Receptor