


Abstract
The folate receptor (FR) has proven a valuable target for nuclear imaging using folic acid radioconjugates. However, using folate-based radiopharmaceuticals for therapy has long been regarded as an unattainable goal because of their considerable renal accumulation. Herein, we present a novel strategy in which a DOTA–folate conjugate with an albumin-binding entity (cm09) was designed with the aim of prolonging circulation in the blood and therewith potentially improving tumor-to-kidney ratios. Methods: The folate conjugate cm09 was radiolabeled with 177LuCl3, and stability experiments were performed in plasma. Cell uptake studies were performed on FR-positive KB tumor cells, and an ultrafiltration assay was used to determine the plasma protein–binding properties of 177Lu-cm09. In vivo, 177Lu-cm09 was tested in KB tumor–bearing mice using SPECT/CT. The therapeutic anticancer effect of 177Lu-cm09 (20 MBq) applied as a single injection or as fractionated injections was investigated in different groups of mice (n = 5) by monitoring tumor size and the survival time of treated mice, compared with untreated controls. Results: Compound cm09 was radiolabeled at a specific activity of 40 MBq/nmol, a radiochemical yield of more than 98%, and a stability of more than 99% over 5 d in plasma. Ultrafiltration revealed significant binding of 177Lu-cm09 to serum proteins (∼91%) in plasma, compared with folate radioconjugate without an albumin-binding entity. Cell uptake and internalization of 177Lu-cm09 was FR-specific and comparable to other folate radioconjugates. In vivo studies resulted in high tumor uptake (17.56 percentage injected dose per gram [%ID/g] at 4 h after injection), which was almost completely retained for at least 72 h. Renal accumulation was significantly reduced (28 %ID/g at 4 h after injection), compared with folate conjugates that lack an albumin-binding entity (∼70 %ID/g at 4 h after injection). These circumstances enabled SPECT imaging of excellent quality. Radionuclide therapy (1 × 20 MBq) revealed complete remission of tumors in 4 of 5 cases and a significantly prolonged survival time, compared with untreated controls. Conclusion: The modification of a folate radioconjugate with an albumin-binding entity resulted in a significant increase of the tumor-to-kidney ratio of radioactivity, enabling for the first time, to our knowledge, the preclinical application of folic acid–targeted radionuclide therapy in mice.
The folate receptor (FR) is a relevant tumor-associated target because of its persistent expression in a variety of cancer types but limited expression in normal tissue and its ability to bind and internalize the vitamin folic acid and its conjugates with an affinity in the subnanomolar range (1–6). In recent years, several folic acid radioconjugates have been developed and evaluated for nuclear imaging of FR-positive cancer (7–9). The FR-targeting strategy has also been investigated with several nonradioactive therapeutic probes linked to folic acid (10,11). Among these, the most successful approach was the conjugation of folic acid to highly toxic low-molecular-weight chemotherapeutics (5,12). With several of these folate-based drug conjugates, respectable anticancer effects were achieved in preclinical (13–16) and clinical studies (17).
If β−-particle–emitting radioisotopes are used for targeted therapy, cancer cells are irradiated not only by decays taking place at or within the targeted cells but also by decays in neighboring or distant cells by the so-called cross-fire effect (18). In targeted radionuclide therapy, it is not necessary therefore to reach every cell within the tumor as is the case for targeted chemotherapy (19,20). For this reason, we are convinced that folic acid conjugates of therapeutic radioisotopes are a valuable tool to implement novel and effective anticancer therapies.
The results obtained with folate-based radiopharmaceuticals developed for SPECT and PET (7–9,21), however, do not hold much promise for successful therapeutic application of this class of compound. Despite the excellent tumor-targeting ability of radiofolates, a substantial fraction of radioactivity is always found in the kidneys because of significant FR expression in the proximal tubule cells (22–24). The risk of damage to the radiosensitive kidneys by particle-emitting radioisotopes has prevented the development of folate-targeted radionuclide therapy.
The fast clearance of folic acid conjugates from the blood circulation is usually seen as an advantage of the folic acid–targeting strategy because it minimizes the exposure of background organs to the therapeutic probe and improves the tumor-to-background contrast of imaging agents (12). However, it is precisely these pharmacokinetics that are responsible for the relatively low uptake of folate conjugates in tumor tissue and an extremely high accumulation of radioactivity in the kidneys. We reasoned that a prolonged blood circulation time could improve this dissatisfying situation.
It is well documented that serum protein binding of pharmaceuticals can be an effective means of improving the pharmacokinetic properties of otherwise rapidly cleared molecules by increasing their serum half-life (25). The conjugation of an albumin-binding peptide to an antibody fragment improved the tissue distribution of the fragment significantly in that it was rapidly deposited in the tumor tissue yet did not accumulate in the kidneys (26). On the basis of experiments that demonstrate the extension of the half-life of short-lived proteins by noncovalent association with albumin, it was suggested that the same strategy might also improve the unfavorable overall tissue distribution of folate radioconjugates.
Recently, a new class of low-molecular-weight albumin-binding entities based on the lead structure 4-(p-iodophenyl)butyric acid was identified from a DNA-encoded chemical library (27). These derivatives display a stable noncovalent binding interaction of variable affinity with both mouse and human serum albumin (27). The conjugation of the high-affinity albumin binder 2-(3-maleimidopropanamido)-6-(4-(4-iodophenyl)butanamido)hexanoate (dissociation constant ~ 3 μM) to antibody fragments resulted in a significantly increased tumor uptake (28).
Using this strategy, we synthesized a novel folic acid conjugate with both a DOTA chelator for coordination of a therapeutic radioisotope such as 177Lu (177Lu: half-life, 6.7 d; average β− energy, 134 keV; γ-energies, 113 keV, 208 keV) and the low-molecular-weight albumin-binding entity (27,28) as an additional functionality (Fig. 1).

Chemical structure of compound cm09 with 3 functionalities. Red = folic acid as a targeting molecule; green = DOTA chelator for coordination of 177Lu; blue = albumin-binding entity for modulation of the pharmacokinetic properties.
The goal of this study was to evaluate the in vitro characteristics of the 177Lu-DOTA-folate conjugate (177Lu-cm09) and to investigate its tissue distribution in mice bearing FR-positive KB tumor xenografts using SPECT/CT. The therapeutic anticancer efficacy was assessed by monitoring tumor growth inhibition in mice injected with 177Lu-cm09 using different application protocols.
MATERIALS AND METHODS
Organic Synthesis
The synthesis of the folate conjugate cm09 is described in the Supplemental Text (supplemental materials are available online only at http://jnm.snmjournals.org). The control compound EC0800 (Supplemental Text), which does not have albumin-binding properties, was a kind gift from Dr. Christopher P. Leamon (Endocyte Inc.) (29).
Radiofolate Synthesis
DOTA folates cm09 (10−3 M) and EC0800 (10−3 M) were mixed with HCl (0.05 M; 100-x μL) and sodium acetate (pH 8.0, 20 μL). 177Lu (177LuCl3 x μL [Isotope Technologies Garching, ITG GmbH], x μL) was added to obtain folate conjugate labeled at a specific activity of up to 40 MBq/nmol. The solution was heated for 10 min at 95°C. Sodium-diethylenetriaminepentaacetic acid solution (5 mM, pH 5, 10 μL) was added to complex any traces of unreacted 177Lu(III). Quality control was performed using high-performance liquid chromatography. The mobile phase consisted of an aqueous 0.05 M triethylammonium phosphate buffer (pH 2.25) (A) and methanol (B) with a linear gradient from 5% B to 80% B over 25 min (177Lu-cm09) or over 15 min (177Lu-EC0800) at a flow rate of 1 mL/min. The radiochemical yield was always more than 98% for 177Lu-cm09 (retention time, 19.7 min) and more than 96% for 177Lu-EC0800 (retention time, 11.5 min).
In Vitro Plasma Stability
177Lu-cm09 (50 μL, 10 MBq) was incubated in human plasma (250 μL) at 37°C. Aliquots of 50 μL were taken at different time points after incubation (0, 4, 24, 48, and 144 h). Plasma proteins were precipitated by the addition of 200 μL of methanol. After centrifugation, the supernatants were analyzed using high-performance liquid chromatography.
Cell Experiments
KB cells (human cervical carcinoma cell line, HeLa subclone; ACC-136) were purchased from the German Collection of Microorganisms and Cell Cultures (DSMZ). The cells were cultured as monolayers at 37°C in a humidified atmosphere containing 5% CO2. Importantly, the cells were cultured in a folate-free cell culture medium, FFRPMI (modified RPMI, without folic acid, vitamin B12, and phenol red; Cell Culture Technologies GmbH). FFRPMI was supplemented with 10% heat-inactivated fetal calf serum, l-glutamine, and antibiotics (penicillin–streptomycin–fungizone). For uptake studies, KB cells were seeded in 12-well plates to grow overnight (∼700,000 cells in 2 mL of FFRPMI medium per well). 177Lu-cm09 or 177Lu-EC0800 (∼38 kBq, 8 pmol) purified by high-performance liquid chromatography was added to each well. In some cases, cells were incubated with excess folic acid (100 μM) to block FRs on the surface of the KB cells. After incubation for 2 h at 37°C, the cells were washed 3 times with phosphate-buffered saline (PBS) to determine the total radiofolate uptake. To assess the internalized fraction, KB cells were washed with a pH 3 stripping buffer (30) to release FR-bound radiofolates from the cell surface. The lysis of cells was accomplished by the addition of NaOH, enabling transfer of the cell suspensions into tubes for measurement in a γ-counter. The concentration of proteins was determined for each sample using a Micro BCA Protein Assay kit (Pierce, Thermo Scientific) to standardize measured radioactivity to the average content of 0.3 mg of protein per well.
Protein Binding
An ultrafiltration assay was used to determine 177Lu-cm09 binding to plasma proteins in comparison to 177Lu-EC0800. Centrifree ultrafiltration devices ( 4104 centrifugal filter units [Millipore]; 30,000 Da nominal molecular weight limit, methylcellulose micropartition membranes) were used to separate the free fraction of the radiofolate from the plasma protein–bound fraction. 177Lu-cm09 or 177Lu-EC0800 in a volume of 25 μL was added to plasma samples of 250 μL and stirred in a vortex mixer for 10 s at room temperature. In addition, the radiotracers were mixed with PBS in equal volume ratios. Aliquots (250 μL) of the radioactive samples were loaded into the ultrafiltration devices and centrifuged at 2,500 rpm at 20°C for 40 min. Equal volumes of all ultrafiltrate solutions were taken and counted for radioactivity in a γ-counter. The counts of the filtered solutions were calculated as a fraction of the radioactivity in the corresponding loading solutions, which were set to 100%.
Biodistribution Studies
In vivo experiments were approved by the local veterinarian department and conducted in accordance with the Swiss law for animal protection. Six- to 8-wk-old female, athymic nude mice (CD-1 Foxn1nu) were purchased from Charles River Laboratories. The animals were fed with a folate-deficient rodent diet starting 5 d before tumor cell inoculation (31). Mice were inoculated with KB cells (5 × 106 cells in 100 μL of PBS) into the subcutis of each shoulder. Biodistribution studies were performed in triplicate approximately 14 d after cell inoculation when tumors reached a size of 200–450 mg. 177Lu-cm09 at a specific activity of 2–3 MBq/0.5 nmol was administered via a lateral tail vein. Blocking studies were performed by injection of excess folic acid (100 μg in 100 μL of PBS) immediately before radiofolate administration. The animals were sacrificed at predetermined time points after administration of the radiofolates. Selected tissues and organs were collected, weighed, and counted for radioactivity. The results were listed as percentage injected dose per gram of tissue weight (%ID/g), using reference counts from a defined volume of the original injectate counted at the same time. The biodistribution studies of 177Lu-EC0800, results of a variety of application protocols for 177Lu-cm09, and an estimation of the absorbed dose in tumors and kidneys are reported in the Supplemental Text.
SPECT/CT
SPECT/CT was performed with a 4-head multiplexing multipinhole camera (NanoSPECT/CT; Bioscan Inc.). Each head was outfitted with a tungsten-based collimator of nine 1.4-mm-diameter pinholes. SPECT/CT images were acquired at 1, 4, 24, and 72 h after injection of 177Lu-cm09 (35 MBq, 1.5 nmol) or 177Lu-EC0800 (35 MBq, 1.5 nmol) using Nucline software (version 1.02; Bioscan). In vivo whole-body SPECT images were obtained with a time per view of 40–70 s, resulting in a scan time of 30–60 min. CT scans were obtained with the integrated CT scanner using a tube voltage of 55 kVp and an exposure time of 1,000 ms per view. After acquisition, SPECT data were reconstructed iteratively with HiSPECT software (version 1.4.3049; Scivis GmbH) using 177Lu γ-energies of 56.1, 112.9, and 208.4 keV. The real-time CT reconstruction used a cone-beam filtered backprojection. SPECT and CT data were automatically coregistered because both modalities share the same axis of rotation. The fused datasets were analyzed with the InVivoScope postprocessing software (version 1.44; Bioscan, Inc.).
In Vivo Therapy
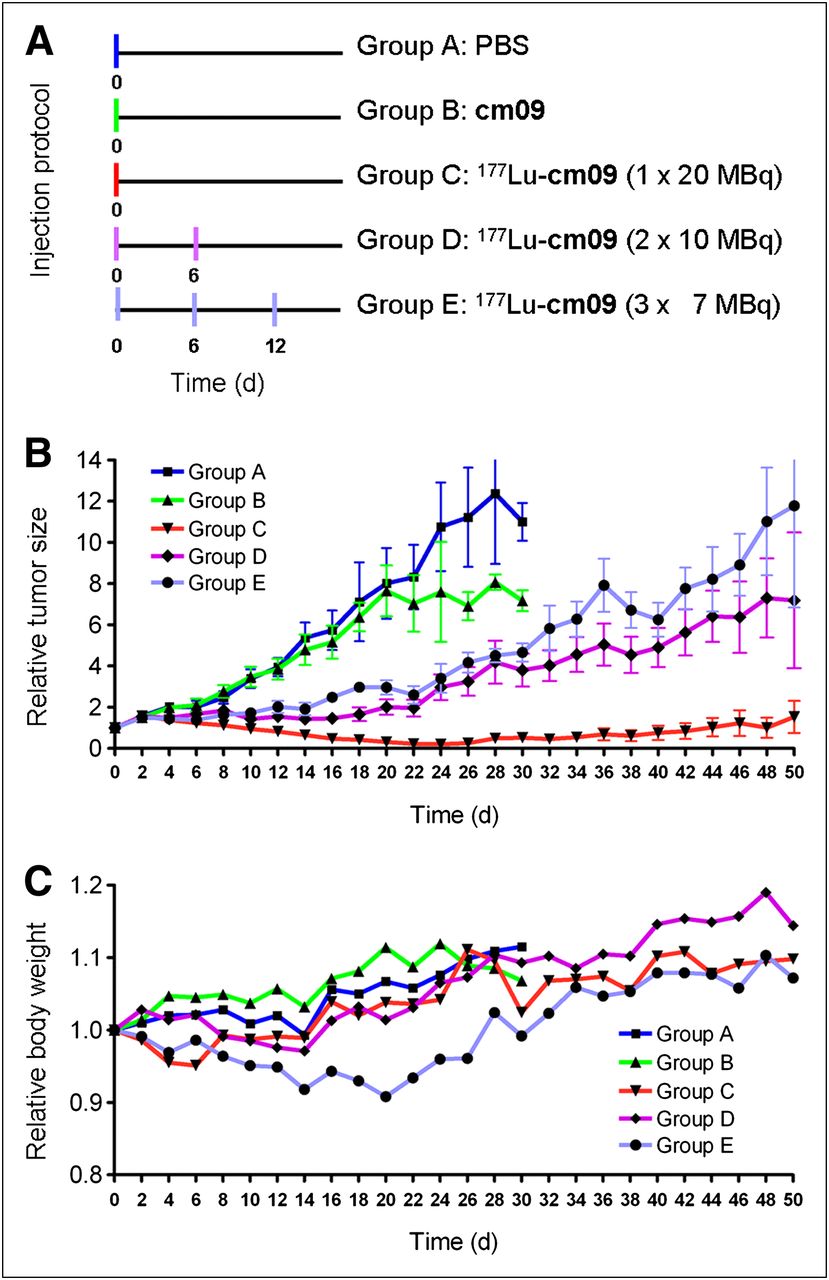
For the therapy study, mice were subcutaneously inoculated with KB cells (4.5 × 106 cells in 100 μL of PBS) at 4 d before therapy. Groups of 5 mice were injected with either PBS only (group A), the nonradiolabeled compound cm09 (group B), 1 × 20 MBq of 177Lu-cm09 (group C), 2 × 10 MBq of 177Lu-cm09 (group D), or 3 × 7 MBq of 177Lu-cm09 (group E) at a specific activity of 40 MBq/nmol when the average tumor volume had reached a value of 118–141 mm3. Human endpoint criteria were defined as weight loss of more than 15% of the initial body weight, a tumor volume of more than 1,500 mm3, active ulceration of the tumor, and abnormal behavior indicating pain or unease. Tumor volumes and body weights were determined at day 0 and then every other day until completion of the study after 12 wk. Tumors were measured using a digital caliper. Individual tumor size was calculated using the formula [0.5 × (L × W2)]. Mice were promptly removed from the study and euthanized on reaching one of the predefined endpoint criteria.
RESULTS
In Vitro Evaluation
177Lu-cm09 was stable (>99%) in human plasma for at least 6 d. These findings were in agreement with the results obtained with the control compound 177Lu-EC0800. Radiolysis of 177Lu-cm09 at a high radioactivity concentration (80 MBq in 400 μL of PBS) was not observed during the first 4 h of investigation. After 24 and 48 h of incubation at room temperature, marginal amounts of a radiolytic product were detected. These observations were in contrast to those obtained with 177Lu-EC0800, which was almost completely decomposed after 48 h (Supplemental Text). The logD value of 177Lu-cm09 was low (−4.25 ± 0.41) and only slightly higher than the logD value of the control compound 177Lu-EC0800 (−4.81 ± 0.36, Supplemental Text). Cell uptake and internalization experiments performed with 177Lu-cm09 showed almost identical results to those obtained with 177Lu-EC0800 and other folic acid radioconjugates previously evaluated by our group (32–34). Approximately 30% of FR-bound 177Lu-cm09 was internalized, whereas the uptake was reduced to background levels if cells were coincubated with excess folic acid (Fig. 2). The comparably high FR affinity of 177Lu-cm09 and 177Lu-EC0800 was demonstrated in vitro using KB cells (Supplemental Text).
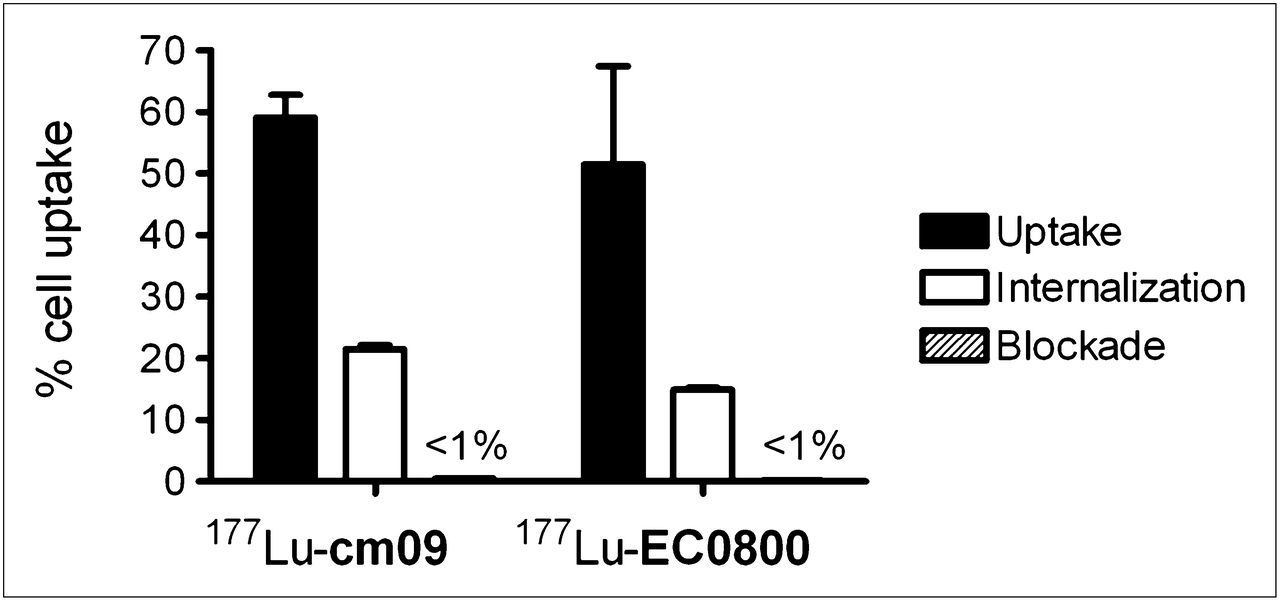
Results of cell experiments performed with 177Lu-cm09 and 177Lu-EC0800. FR-specific uptake, internalization, and blockade with excess folic acid were comparable for both radiotracers.
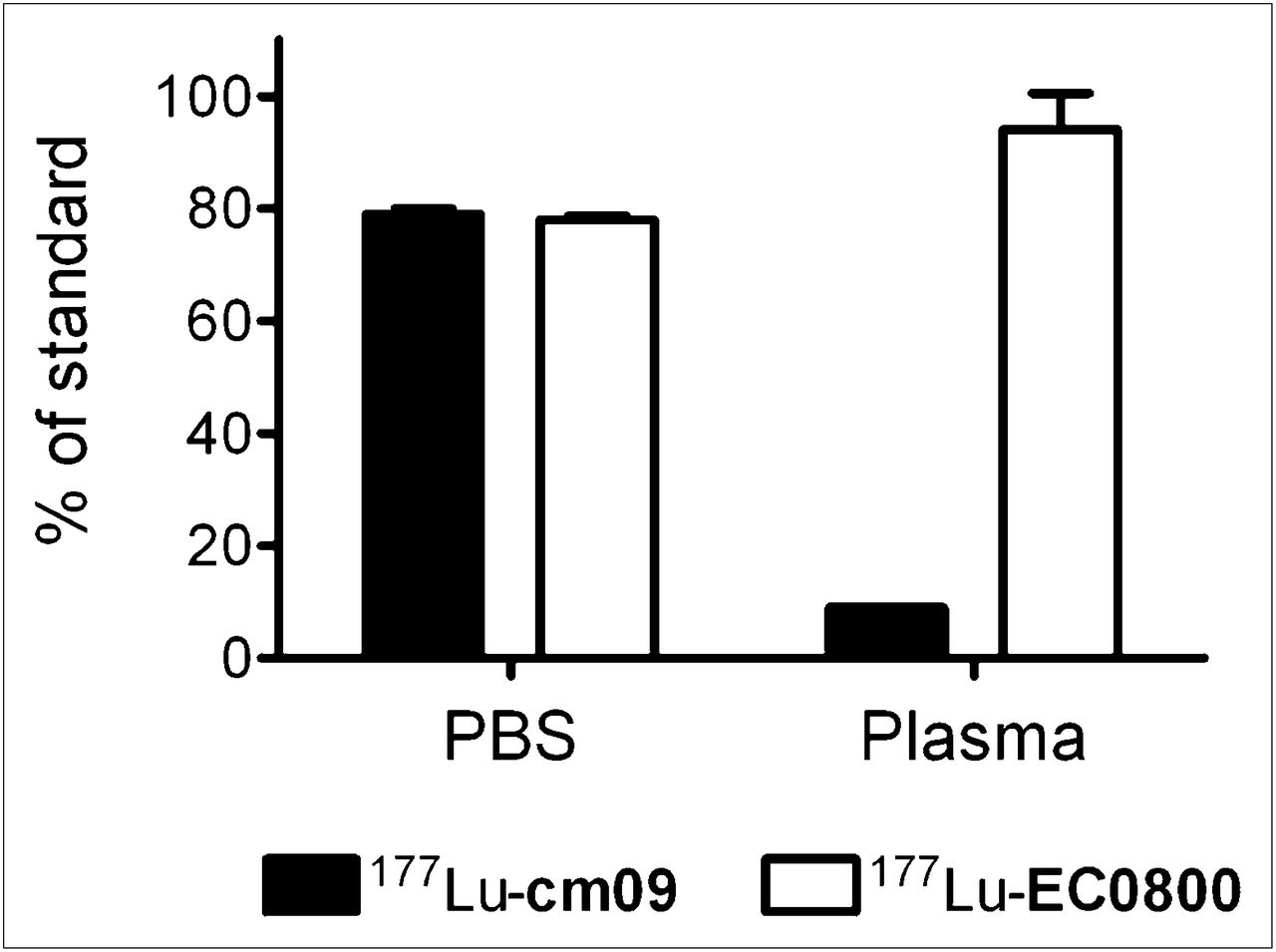
The ultrafiltration experiments showed significant differences between 177Lu-cm09 and 177Lu-EC0800 (Fig. 3). In contrast to 177Lu-EC0800, which was readily filtered, the filtered fraction of a plasma sample incubated with 177Lu-cm09 contained little radioactivity, indicating that most 177Lu-cm09 was bound to plasma proteins and thus not able to penetrate the filter membrane. A control experiment using protein-free PBS instead of plasma showed radioactivity in the filtrate for both 177Lu-cm09 and 177Lu-EC0800.

Results of ultrafiltration assay. In PBS, 177Lu-cm09 and 177Lu-EC0800 penetrated the filter with equal efficiency. After mixing with plasma, most 177Lu-cm09 is bound to serum albumin and therefore only a small fraction was filtered. 177Lu-EC0800 does not have an albumin-binding entity and was therefore filtered from both PBS and plasma samples.
Biodistribution Studies
The results of the biodistribution studies are shown in Table 1. The albumin-binding properties of 177Lu-cm09 are reflected in the fact that a significant fraction of radioactivity, which steadily decreased to background levels over time, was found in the blood at early time points after injection (8.15 ± 1.21 %ID/g at 1 h after injection). As a consequence, 177Lu-cm09 uptake in the tumor tissue was already high shortly after injection (10.84 ± 1.32 %ID/g at 1 h after injection) and reached a maximum accumulation at 24 h after injection (19.46 ± 3.13 %ID/g). Five days after administration of 177Lu-cm09, there was still a high level of radioactivity in tumor xenografts (6.72 ± 1.37 %ID/g), indicating slow washout from the tumor tissue. The dilution of accumulated radioactivity in growing tumors over time may also have contributed to the reduction of uptake at later time points. Significant and FR-specific uptake of 177Lu-cm09 was also found in the kidneys, with a maximum value of 30.09 ± 4.04 %ID/g determined at 24 h after injection. Shortly after injection of 177Lu-cm09, low levels of radioactivity were also found in the lung, liver, intestinal tract, muscle, and bones.
Biodistribution of 177Lu-cm09 in KB Tumor–Bearing Female Nude Mice
To investigate the influence of the molar amount of folate conjugate on the overall tissue distribution, biodistribution studies were performed with different molar amounts (0.25–1.00 nmol) of cm09, that is, with 177Lu-cm09 at different specific activities. There was a slight trend toward increasing accumulation of radioactivity in the blood and in the tumors and kidneys as the amount of cm09 injected was increased (Supplemental Text and Supplemental Table 2). Uptake in FR-negative tissues was not influenced by the amount of folate conjugate cm09, and tumor-to-background ratios also remained largely unchanged.
In a separate experiment, 177Lu-cm09 was mixed with 2% or 10% serum albumin or human or murine serum before administration. It was hypothesized that the free fraction of 177Lu-cm09, which is filtered through the kidneys, would be decreased if 177Lu-cm09 was mixed with an albumin solution before injection; however, these interventions did not influence the biodistribution data (Supplemental Text and Supplemental Table 3). Variation of the injection route (intravenous vs. subcutaneous vs. intraperitoneal) did not significantly affect the overall tissue distribution of 177Lu-cm09 (Supplemental Text and Supplemental Table 4). The only exception was tumor uptake, which was slightly lower at 4 h after subcutaneous injection than at 4 h after intravenous or intraperitoneal administration of 177Lu-cm09.
The effect of preinjected pemetrexed was also investigated (Supplemental text and Supplemental Table 5). The favorable effect of reducing kidney uptake without affecting tumor accumulation of radioactivity, which has been observed with other folate conjugates (32,35), was only observed at 4 h after injection. At that time point, the tumor-to-kidney ratio was increased (1.07 ± 0.25), compared with the ratio (0.65 ± 0.07) obtained in mice that did not receive pemetrexed. Repeated injection of pemetrexed would probably have improved the tumor-to-kidney ratios also at later time points. However, this experimental setting was not investigated because of the potential toxic side effects of high-dose pemetrexed.
The absorbed dose was estimated for KB tumor xenografts (1.80 Gy/MBq) and kidneys (3.44 Gy/MBq) based on biodistribution data (Supplemental Text).
SPECT/CT
In vivo SPECT/CT scans of KB tumor–bearing mice are shown in Figure 4. In the case of 177Lu-cm09, tumor-to-background contrast was low immediately after injection but increased quickly over time. In experiments performed with 177Lu-EC0800, comparatively little radioactivity was found in tumors, whereas uptake in the kidneys was high at all time points investigated.
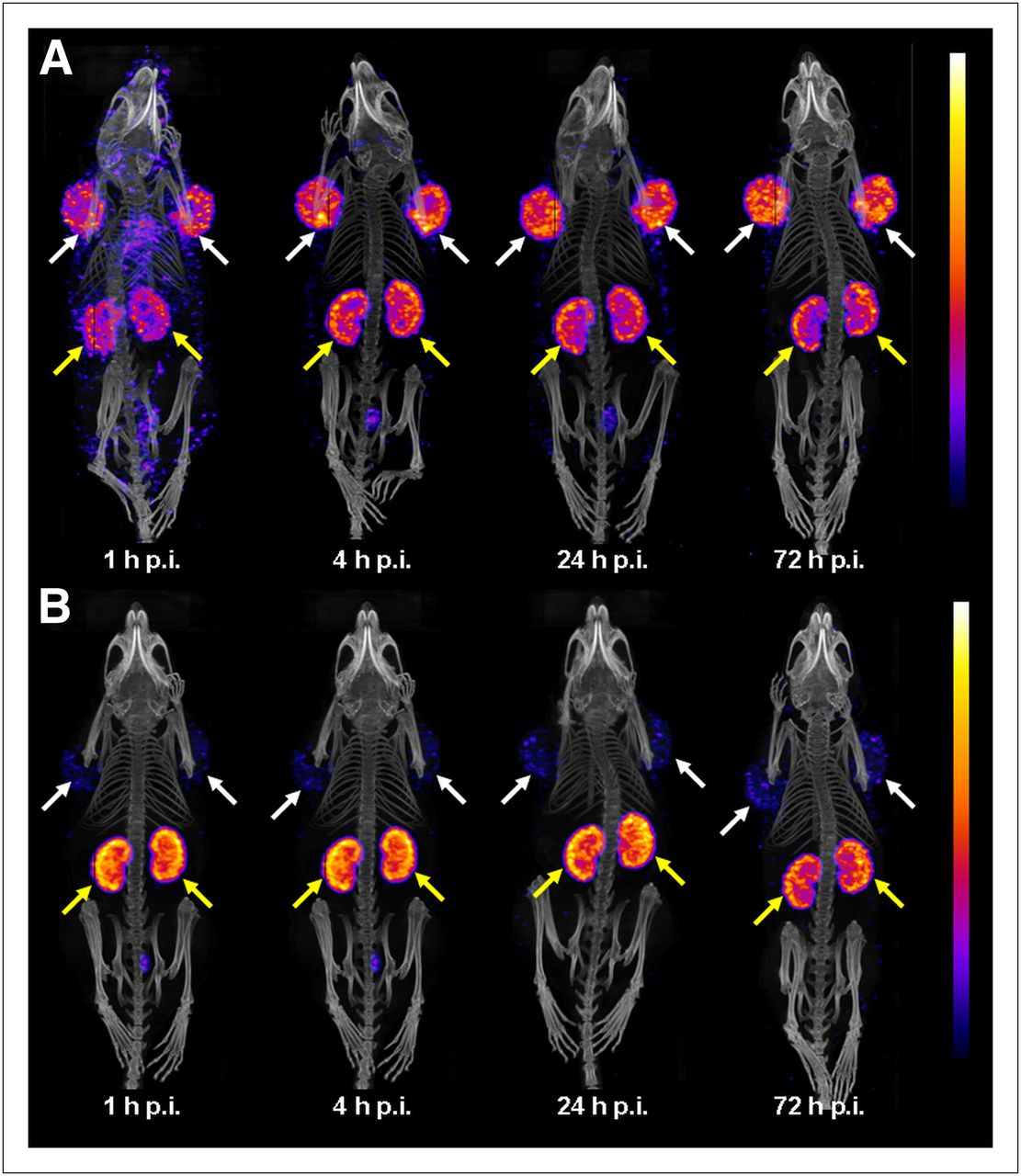
SPECT/CT images of KB tumor–bearing mice injected with 177Lu-cm09 (A) and 177Lu-EC0800 (B). Accumulation of radioactivity was found in FR-positive tumors (white arrows) and kidneys (yellow arrows). Images show a significantly improved tumor-to-kidney ratio (∼1.0 vs. ∼0.2) at 1, 4, 24, and 72 h after injection in mice that received 177Lu-cm09, compared with mice that received 177Lu-EC0800. p.i. = after injection.
Therapy Study
After successful performance of an in vivo pilot study with a therapeutic dose of 177Lu-cm09 (Supplemental Text), a therapy study was performed with 5 groups (A–E) of 5 mice each, over a period of 3 months. The injection protocol and the relative tumor size and body weights of mice from each group are shown in Figure 5. Tumor growth was comparable for mice in control groups A and B (group A received only PBS and group B unlabeled folate compound cm09) (Fig. 5A). In comparison, tumor growth in mice that received 177Lu-cm09 was clearly reduced. In terms of tumor response, the best results were observed in group C (1 × 20 MBq of 177Lu-cm09, Fig. 5B), in which tumor xenografts disappeared completely in 4 of the 5 mice. Among the mice in groups D and E, which received 177Lu-cm09 in fractions (2 × 10 MBq or 3 × 7 MBq, respectively), the relative tumor size was only about 30% of the relative tumor size of control mice at day 28 but 10-fold larger than in mice of group C, which received the whole amount of 177Lu-cm09 in a single injection (Fig. 5B). Body weight loss was most significant in mice that received 3 injections of 177Lu-cm09 (group E, Fig. 5C). However, none of the animals had to be euthanized because of excessive loss of body weight.
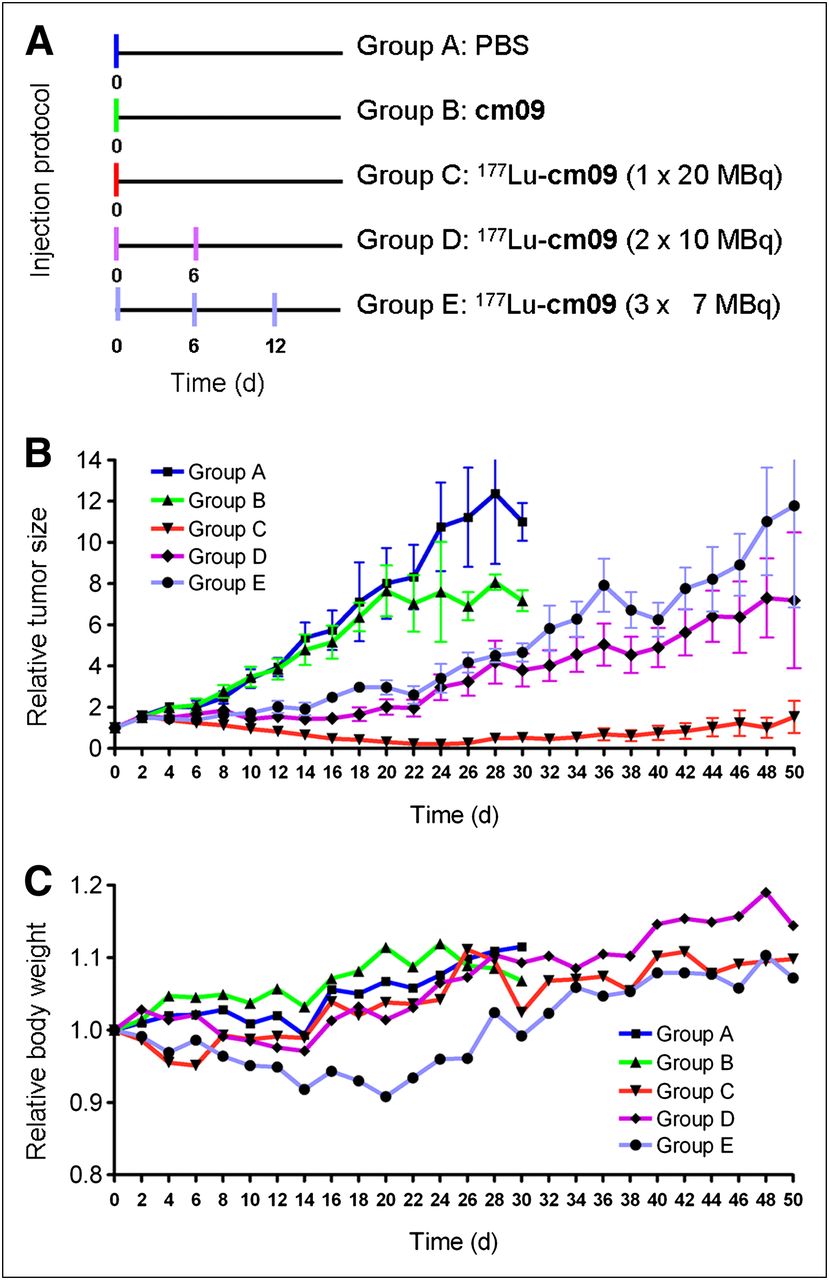
Design and results of the therapy study: application protocol (A), average relative tumor size (B), and average relative body weight (C) for each group of mice (A–E) included in this study.
Survival curves of mice from each group are shown in Figure 6. The average survival times of control mice were 27 d (group A) and 24 d (group B). In the case of groups D and E, the average survival times were almost double (48 and 46 d, respectively). For mice in group C, the average survival time was undefined because only 1 mouse reached an endpoint criterion whereas the other 4 mice in group C survived with complete tumor response until the end of the study at day 84.
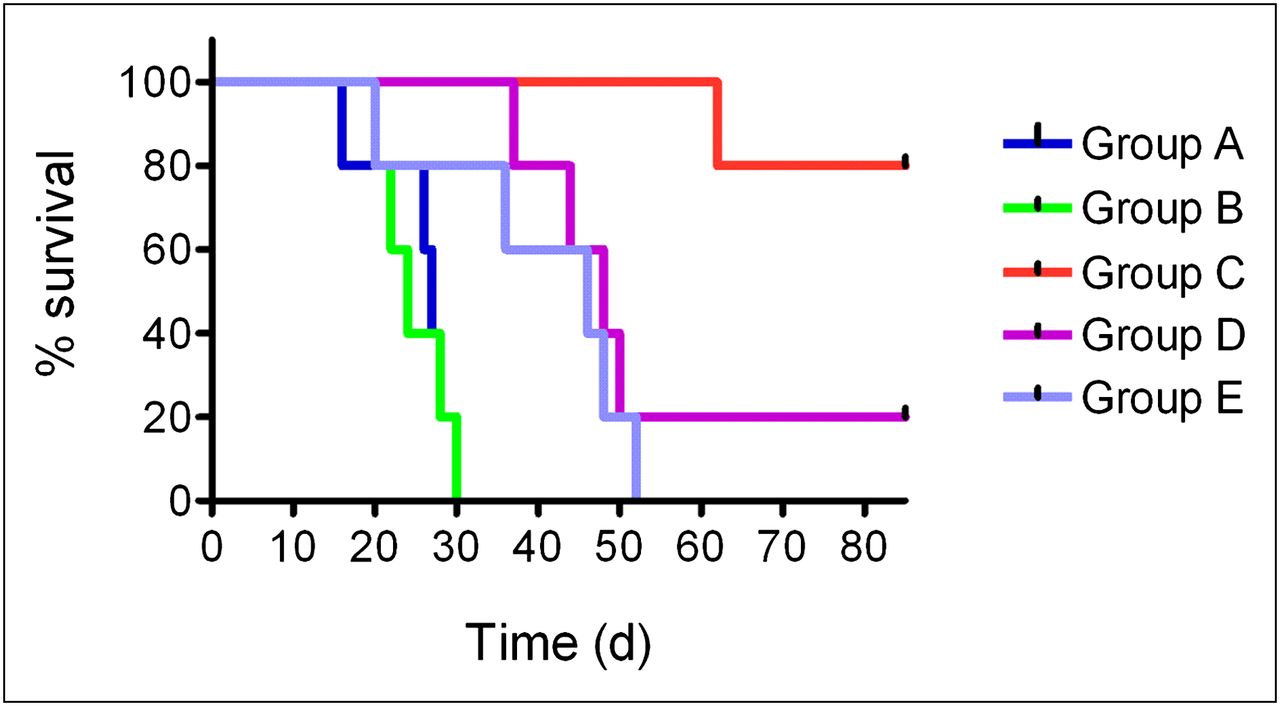
Survival curves of mice from groups A–E. (A, dark blue) control group. (B, green) unlabeled cm09. (C, red) 1 × 20 MBq of 177Lu-cm09. (D, violet) 2 × 10 MBq of 177Lu-cm09. (E, light blue) 3 × 7 MBq of 177Lu-cm09.
Pictures of mice, results of autoradiography studies performed with tumor sections of mice from each group, and values of the analysis of plasma chemistry are reported in the Supplemental Text.
DISCUSSION
The development of strategies to overcome the unfavorable accumulation of folate-based radiopharmaceuticals in the renal tissue has been the focus of intense research in our group (29). In recent work, we addressed the low tumor-to-kidney ratios of folate radioconjugates by pharmacologic intervention. We found that administration of the antifolate pemetrexed before the folate radioconjugate significantly reduced the undesired renal accumulation, whereas uptake in tumor xenografts was unaffected (35–37). Despite the significantly improved tumor-to-background contrast, which can be achieved by the preinjection of pemetrexed, potentially toxic side effects, particularly under folate-deficient conditions, make it difficult to justify using a chemotherapeutic agent solely to modulate the tissue distribution of a folate radioconjugate.
In this study, we pursued a different strategy, this time using chemical modification of a folate radioconjugate, with the aim of improving the overall tissue distribution of radioactivity, thus avoiding additional pharmacotherapy. In vitro investigations using an ultrafiltration assay showed clearly that the new radioconjugate 177Lu-cm09 had significant plasma-binding properties, compared with 177Lu-EC0800, which does not incorporate an albumin-binding entity, and was investigated in parallel as a control compound. The investigation of the uptake and internalization into FR-positive tumor cells showed that 177Lu-cm09 had the same excellent FR specificity as 177Lu-EC0800. 177Lu-cm09 (log D, −4.25 ± 0.41) retained the favorable hydrophilic character associated with DOTA folate conjugates (29,32,38) such as 177Lu-EC0800.
Using 177Lu-cm09 in vivo, we obtained exceptional results that were significantly better than with any other folate radiotracer previously tested by our group (29,32) or by others (38,39). As a consequence of serum protein binding and hence an extended blood circulation time, accumulation of 177Lu-cm09 in tumor xenografts reached the remarkable value of 19.46 ± 3.13 %ID/g (24 h after injection), almost 3-fold higher than tumor uptake of 177Lu-EC0800 (7.00 ± 1.22 %ID/g at 24 h after injection). In addition, the adversely high kidney uptake usually associated with folate radiotracers including 177Lu-EC0800 (>70 %ID/g between 1 and 4 h after injection) was significantly reduced for 177Lu-cm09 (∼15, ∼23, and ∼30 %ID/g at 1, 4, and 24 h after injection, respectively). This reduction in uptake resulted in tumor-to-kidney ratios that were 5- to 6-fold higher than values obtained with 177Lu-EC0800 and other DOTA folate conjugates (Supplemental Text) (29,38). These results unambiguously proved our hypothesis that the in vivo tissue distribution of folate radioconjugates would be improved by increasing their blood circulation time and facilitated the development of a FR-targeted radionuclide therapy. The accumulation of folate radioconjugates in the choroid plexus of the brain, which also expresses the FR (2), was not visible on SPECT images but may be a critical point in view of a therapeutic application of 177Lu-cm09 in patients (Supplemental Text).
The therapeutic application of 177Lu-cm09 showed excellent results in terms of tumor growth inhibition, particularly if the whole dose of 20 MBq was administered in a single injection. The results of a fractionated injection (2 × 10 MBq or 3 × 7 MBq) were inferior in terms of tumor growth inhibition and body weight loss for the test animals. The fact that fractionated therapy application was less successful could be attributed to the increasing tumor size. Compared with the average tumor size at day 0 (∼118 cm3), tumors were 1.4- to 1.6-fold larger at day 6 (groups D and E) when the second dose was administered and about 2-fold larger at day 12 (group E) when the third dose was administered. Weak vascularization of xenografts or intratumoral pressure could account for the reduced therapy response of larger tumors.
To exclude the possibility that repeated therapy application failed because of the loss of FR expression in tumor cells after the first dose of 177Lu-cm09, in vitro autoradiography studies were performed on tumor sections from mice from each group. The results confirmed that FR expression levels were retained in both tumor tissue from control animals and escape tumors from treated animals.
Normal behavior was observed in the animals throughout the study, and body weight loss shortly after therapy application was marginal. Analysis of plasma samples from the mice showed creatinine and blood urea nitrogen levels in the reference range. Plasma levels of alkaline phosphatase increased with the tumor burden and were therefore lowest in group C, where the treatment reduced tumor growth most effectively. Although there were no signs of toxicity, the dose absorbed by the kidneys is still high. For clinical translation, it will be necessary to find methods to further reduce renal accumulation of radioactivity and hence reduce the dose to the kidneys (40). Investigations of the potential long-term damage to the kidneys after therapy and application of radioprotective agents are subjects of current investigations in our laboratories.
CONCLUSION
Installation of an albumin-binding entity into the structure of a folate-based radioconjugate improved the overall tissue distribution significantly. Tumor uptake was doubled, and kidney retention was reduced to 30% of the value obtained with folate conjugates without an albumin-binding entity. These characteristics enabled the first folic acid–targeted radionuclide therapy study in mice. Excellent results were achieved in terms of tumor growth inhibition, and radiotoxic side effects were not observed. The results are a milestone in the field of FR-targeted anticancer therapy.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This project was supported by the Swiss National Science Foundation (Ambizione Grant PZ00P3_121772) and COST (Action BM0607). No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Dr. Peter Bläuenstein, Nadja Romano, and Alain Blanc for technical assistance.
Footnotes
Published online Dec. 12, 2012.
- © 2013 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication April 11, 2012.
- Accepted for publication August 7, 2012.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Dansylated Amino Acid-Modified Long-Acting PSMA Derivatives 68Ga/177Lu-LNC1011 as Prostate Cancer Theranostics
- Design of a Fibroblast Activation Protein-Targeted Radiopharmaceutical Therapy with High Tumor-to-Healthy-Tissue Ratios
- Fibroblast Activation Protein-Targeted Radioligand Therapy for Treatment of Solid Tumors
- Albumin-Binding PSMA Ligands: Implications for Expanding the Therapeutic Window
- Safety, Pharmacokinetics, and Dosimetry of a Long-Acting Radiolabeled Somatostatin Analog 177Lu-DOTA-EB-TATE in Patients with Advanced Metastatic Neuroendocrine Tumors
- Dual-Target Binding Ligands with Modulated Pharmacokinetics for Endoradiotherapy of Prostate Cancer
- Novel "Add-On" Molecule Based on Evans Blue Confers Superior Pharmacokinetics and Transforms Drugs to Theranostic Agents
- Promising Prospects for 44Sc-/47Sc-Based Theragnostics: Application of 47Sc for Radionuclide Tumor Therapy in Mice
- Promises of Cyclotron-Produced 44Sc as a Diagnostic Match for Trivalent {beta}--Emitters: In Vitro and In Vivo Study of a 44Sc-DOTA-Folate Conjugate
- 177Lu-EC0800 Combined with the Antifolate Pemetrexed: Preclinical Pilot Study of Folate Receptor Targeted Radionuclide Tumor Therapy