


Abstract
The objective of this study was to examine 18F-FDG uptake in microscopic tumors grown intraperitoneally in nude mice and to relate this to physiologic hypoxia and glucose transporter-1 (GLUT-1) expression. Methods: Human colon cancer HT29 and HCT-8 cells were injected intraperitoneally into nude mice to generate disseminated tumors of varying sizes. After overnight fasting, animals, breathing either air or carbogen (95% O2 + 5% CO2), were intravenously administered 18F-FDG together with the hypoxia marker pimonidazole and cellular proliferation marker bromodeoxyuridine 1 h before sacrifice. Hoechst 33342, a perfusion marker, was administered 1 min before sacrifice. After sacrifice, the intratumoral distribution of 18F-FDG was assessed by digital autoradiography of frozen tissue sections. Intratumoral distribution was compared with the distributions of pimonidazole, GLUT-1 expression, bromodeoxyuridine, and Hoechst 33342 as visualized by immunofluorescent microscopy. Results: Small tumors (diameter, <1 mm) had high 18F-FDG accumulation and were severely hypoxic, with high GLUT-1 expression. Larger tumors (diameter, 1–4 mm) generally had low 18F-FDG accumulation and were not significantly hypoxic, with low GLUT-1 expression. Carbogen breathing significantly decreased 18F-FDG accumulation and tumor hypoxia in microscopic tumors but had little effect on GLUT-1 expression. Conclusion: There was high 18F-FDG uptake in microscopic tumors that was spatially associated with physiologic hypoxia and high GLUT-1 expression. This enhanced uptake was abrogated by carbogen breathing, indicating that in the absence of physiologic hypoxia, high GLUT-1 expression, by itself, was insufficient to ensure high 18F-FDG uptake.
PET with 18F-FDG has emerged as an important clinical tool for cancer detection, staging, and monitoring of response and is routinely used in the clinical management of several cancer types (1). The uptake of 18F-FDG, an analog of glucose, is largely proportional to the rate of glucose metabolism, enabling this parameter to be quantified (2). In hypoxic conditions, cancer cells may undergo a switch from aerobic to anaerobic glucose metabolism. This adaptive response involves the coordinated expression of many hypoxia-inducible factor (HIF)–regulated proteins, such as glucose transporter-1 (GLUT-1), and various glycolytic enzymes (3). Glucose metabolism in hypoxic cancer cells has been studied in cell culture and in animal models of macroscopic tumors (2); however, results have been mixed and controversial (4–14).
We have previously shown in an animal model of disseminated peritoneal disease derived from HT29 and HCT-8 colorectal cancer cells (15,16) that microscopic tumors (diameter, <1 mm) were poorly perfused and intensely hypoxic. In contrast, larger tumors (diameter, ∼1–4 mm) were well perfused and not significantly hypoxic. We also found that hypoxia in microscopic peritoneal tumors could be substantially reduced by having animals breathe carbogen (95% O2 +5% CO2) (15,16). Glucose metabolism in microscopic tumors and its relationship to hypoxia has not yet been investigated.
Noninvasive PET is impractical for directly assessing 18F-FDG uptake in individual microscopic tumors, which are too small to be seen (16). However, such models may still provide a useful means of examining the relationship between radiotracer distribution and the features of microscopic tumors, including their hypoxic status. Digital autoradiographic detection systems can quantitatively determine the spatial distribution of radioactivity with a pixel resolution of 25–50 μm. The spatial distributions of microenvironmental variables such as physiologic hypoxia, upregulated protein expression, blood perfusion, and cellular proliferation can be visualized on tumor sections by immunofluorescence methods (17). In addition, the disseminated peritoneal disease model provides a diversity of tumors of differing size and hypoxic status growing in the same animal, thereby reducing or eliminating issues associated with interanimal variability.
In the current study, we report for the first time the use of correlative imaging methodologies to examine the uptake of 18F-FDG in microscopic tumors and relate this to hypoxic status. 18F-FDG uptake, visualized by digital autoradiography (DAR) at 50-μm pixel resolution, was compared with immunofluorescent visualization of pimonidazole binding and GLUT-1 expression. We also report on how 18F-FDG uptake in microscopic tumors is altered by carbogen breathing.
MATERIALS AND METHODS
Tumor Cell Lines and Animals
Two human cancer cell lines were used in experiments: HT29 and HCT-8, both originally derived from colorectal adenocarcinomas. Cancer cell lines were purchased from American Type Cell Collection. HT29 and HCT-8 cells were maintained in McCoy's 5A modified medium (Gibco) and RPMI 1640 medium (Cellgro), respectively. All media were supplemented with 10% fetal bovine serum (Gemini), 1% glutamine, and 1% antibiotic mixture (Cellgro). Cells were grown at 37°C in a humidified 5% CO2 incubator. Exponentially growing cells were harvested with 0.05% trypsin plus ethylenediaminetetraacetic acid for HT29 and 0.25% trypsin plus ethylenediaminetetraacetic acid (Cellgro) for HCT-8, washed and suspended in phosphate-buffered saline (PBS). The number of cells was counted using a Coulter counter (Beckman-Coulter).
All experiments were performed using 6- to 8-wk-old female athymic NCr-nu/nu mice purchased from NCI-Frederick Cancer Research Institute. Nude mice were maintained and used according to institutional guidelines. The experimental protocols were approved by the Institutional Animal Care and Use Committee. Animals were housed 5 per cage and kept in the institutional small-animal facility at a constant temperature and humidity. Food pellets and water were provided ad libitum.
Establishment of Microscopic Tumors in Animals
Microscopic Peritoneal Tumors
Disseminated microscopic tumors were generated in the peritoneum as previously described (15,16). Briefly, 5–10 × 106 cells/0.1–0.2 mL of cancer cell suspensions were injected intraperitoneally into unanesthetized mice, and experiments were performed typically 6–7 wk (HT29) or 3–4 wk (HCT-8) later. At these times, distributions of tumors ranging from a few hundred micrometers up to several millimeters in diameter were observed to be present on or in the intestinal serosa.
HT29 Ascites Tumors
At the time of sacrifice, ascites was evident in most animals injected intraperitoneally with HT29 cells. The ascites fluid was observed to be bloody and contained a distribution of free-floating tumor cell aggregates of sizes up to 1 mm in diameter, denoted here as ascites tumors. HT29 ascites tumors were typically ellipsoidal, with ductlike structures in the interior.
Experimental Procedures
The hypoxia marker pimonidazole hydrochloride (1-[(2-hydroxy-3-piperidinyl)propyl]-2-nitroimidazole hydrochloride) (Chemicon International) was dissolved in physiologic saline at a concentration of 20 mg/mL. The proliferation marker bromodeoxyuridine (Roche Diagnostics) was first dissolved in dimethyl sulfoxide and further diluted in physiologic saline to a final concentration of 20 mg/mL. The blood perfusion marker Hoechst 33342 (Sigma-Aldrich) was dissolved in physiologic saline at a concentration of 5 mg/mL. In all cases, fresh drug solutions were prepared on the day of injection.
All animals were kept fasting overnight before experiments, which were performed without anesthesia. Mice were injected via the tail vein with a mixture of 18F-FDG (7.4–13.3 MBq), pimonidazole (2 mg), and bromodeoxyuridine (4 mg) 1 h before sacrifice (total injection volume, 0.4 mL). Hoechst 33342 (0.5 mg, 0.1 mL) was injected via the tail vein 1 min before sacrifice. For experiments featuring carbogen breathing, mice were placed in a plastic chamber (20 × 10 × 10 cm) into which carbogen (95% O2; 5% CO2) was delivered at a flow rate of 5 L/min (15). After 1 h of breathing carbogen, animals were injected with the 18F-FDG–pimonidazole–bromodeoxyuridine mixture and then were returned to the carbogen chamber for another hour before Hoechst 33342 administration and sacrifice.
In total, 18F-FDG uptake in disseminated HT29 peritoneal tumors was examined in 7 air-breathing and 4 carbogen-breathing animals and in 2 air-breathing and 2 carbogen-breathing animals with disseminated HCT-8 peritoneal tumors.
Preparation of Frozen Tumor Sections
Immediately after animal sacrifice, tumor tissues or ascites fluid were removed for subsequent processing. Peritoneal tumors (adhering to the intestinal serosa) were washed with cold PBS to remove any attached ascites tumors before freezing and embedding in optimal-cutting-temperature medium (4583; Sakura Finetek). Ascites tumors were harvested, washed with cold PBS to remove red blood cells, frozen, and embedded in optimal-cutting-temperature medium. Immediately thereafter, 5 contiguous 8-μm-thick tissue sections were cut using an HM500 cryostat microtome (Microm International GmbH) and adhered to poly-l-lysine–coated glass microscope slides (Polysciences, Inc.).
18F-FDG DAR
As described previously (18), autoradiograms were obtained by placing the tumor sections in a film cassette against a Fujifilm BAS-MS2325 imaging plate (Fuji Photo Film Co.). Plates were exposed overnight and read by a Fujifilm BAS-1800II bioimaging analyzer (Fuji Photo Film Co.), which generated digital images with pixel dimensions of 50 × 50 μm. DAR image intensity was expressed in the machine readout parameter of photostimulable luminescence per square millimeter (PSL/mm2) and was quantified using Multi Gauge software (version 2.2; Fujifilm). Subsequently, the derived PSL/mm2 values were converted to MBq/g based on the known start and end times of the DAR exposure and a previously measured system calibration factor of 103 PSL/mm2 per MBq h/g (18). Finally, tumor uptakes were expressed in terms of percentage injected dose per gram based on the known administered activities.
Visualization of Pimonidazole, GLUT-1, Bromodeoxyuridine, Hoechst 33342, and Hematoxylin and Eosin on Tumor Sections
Images of the distributions of pimonidazole, GLUT-1, bromodeoxyuridine, and Hoechst 33342 were obtained after completion of 18F-FDG DAR exposures as described previously (15). To minimize issues associated with section alignment and registration, the same tumor section used for DAR or contiguous adjacent sections were used for all images. Briefly, slides were air-dried, fixed in cold acetone (4°C) for 20 min, and incubated with SuperBlock (37515; Pierce Biotechnology) at room temperature for 30 min. All antibodies were also applied in SuperBlock. Sections were then incubated with fluorescein isothiocyanate–conjugated antipimonidazole monoclonal antibody (Chemicon International), diluted 1:25, for 1 h at room temperature. GLUT-1 staining was performed either on the same section as that stained for pimonidazole or on the adjacent section by incubating for 1 h at room temperature with rabbit anti-GLUT-1 polyclonal antibody (Millipore) diluted 1:50. Sections were washed 3 times in PBS, each wash lasting 5 min, and incubated for 1 h at room temperature with either AlexaFluor568- (for sections costained with pimonidazole) or AlexaFluor488-conjugated goat antirabbit antibody (1:100, Molecular Probes) and washed again. For bromodeoxyuridine staining, sections adjacent to those used for pimonidazole were treated with 2N HCl for 10 min at room temperature, followed by 0.1 M Borax for 10 min at room temperature. Sections were then exposed to AlexaFluor594-conjugated antibromodeoxyuridine antibody (1:20 dilution, Molecular Probes) for 1 h at room temperature and washed. To control for nonspecific binding of antibodies, stained sections were processed from similar tumors that had not been exposed to pimonidazole or bromodeoxyuridine. Controls for GLUT-1 staining consisted of sections in which primary antibody was omitted.
Images were acquired at ×100 magnification using an Olympus BX40 fluorescence microscope (Olympus America Inc.) equipped with a motorized stage (Prior Scientific Instruments Ltd.). Hoechst 33342 and pimonidazole were imaged using blue and green filters, respectively. GLUT-1 was imaged using a red or green filter, depending on the fluorophore used, and bromodeoxyuridine was imaged using a red filter.
After acquisition of fluorescence images, tumor sections were stained with hematoxylin and eosin and imaged by light microscopy. Microscopic images were coregistered and analyzed using Photoshop 7.0 (Adobe).
Statistical Analysis
Statistical significance was examined by 2-tailed Student t test. A P value less than 0.05 was considered a statistically significant difference.
RESULTS
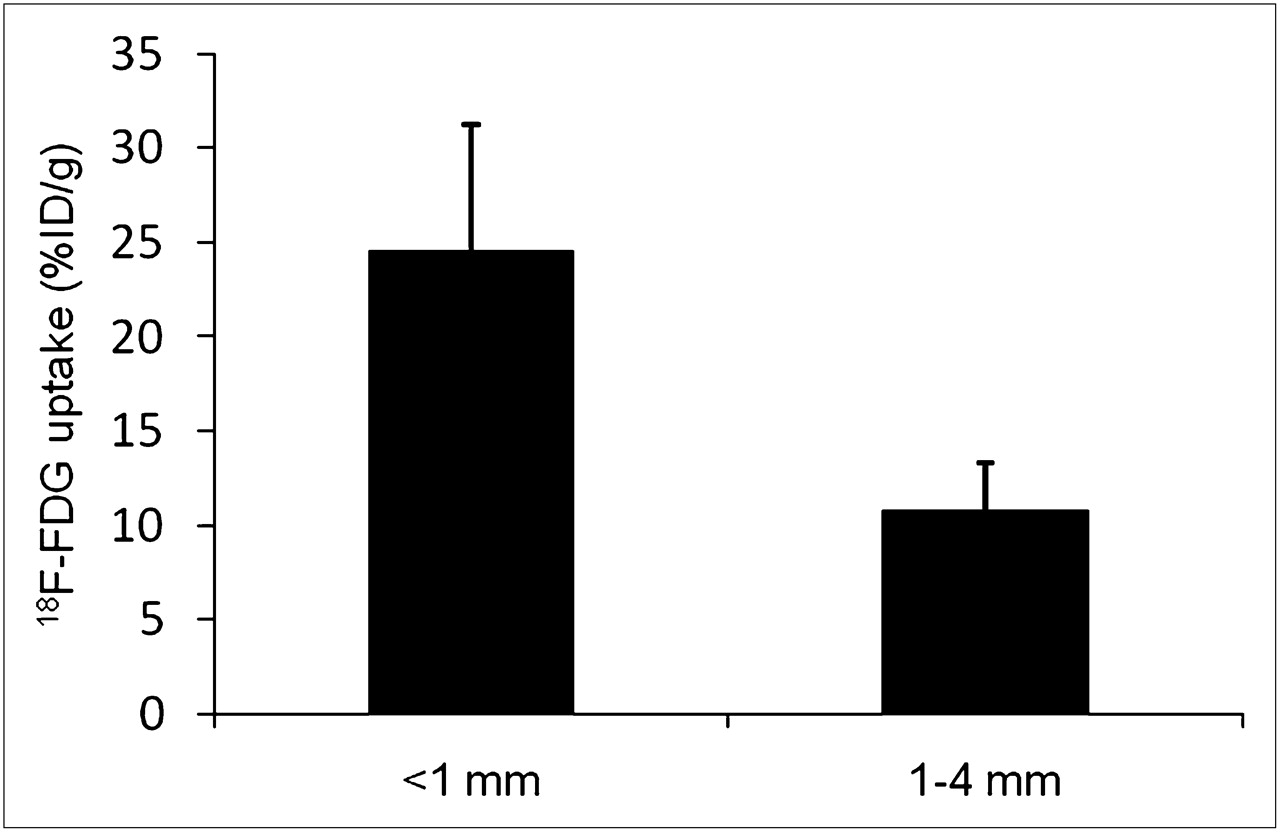
18F-FDG uptake in disseminated peritoneal disease arising from HT29 tumor cells was studied in 7 air-breathing animals. In all cases, results were broadly similar. Figure 1 shows a representative example of the relationship between 18F-FDG uptake and pimonidazole binding, GLUT-1 expression, cellular proliferation (as visualized by bromodeoxyuridine incorporation), and blood perfusion (as visualized by Hoechst 33342). In general, there was spatial colocalization between high levels of 18F-FDG uptake, pimonidazole binding, and GLUT-1 expression. Such regions tended to correspond to low levels of cellular proliferation and blood perfusion. In particular, the smallest tumor deposits (diameter, <∼1 mm) were hypoxic (as evidenced by high pimonidazole binding) and had high 18F-FDG uptake. In these tumors, GLUT-1 expression was high, bromodeoxyuridine staining was confined to the rim, and blood perfusion was minimal. Larger tumors (diameter, ∼1–4 mm) were not hypoxic (low pimonidazole binding) and displayed relatively low 18F-FDG uptake and GLUT-1 expression. Additionally, bromodeoxyuridine-positive cells were distributed throughout the larger tumors, and blood perfusion was relatively high. Figure 2 shows quantitative 18F-FDG uptake in a collection of peritoneal HT29 tumors from a single air-breathing mouse (5 tumors [<1 mm] and 4 tumors [1–4 mm]). 18F-FDG uptake was significantly greater in the submillimeter tumors than in the larger ones.
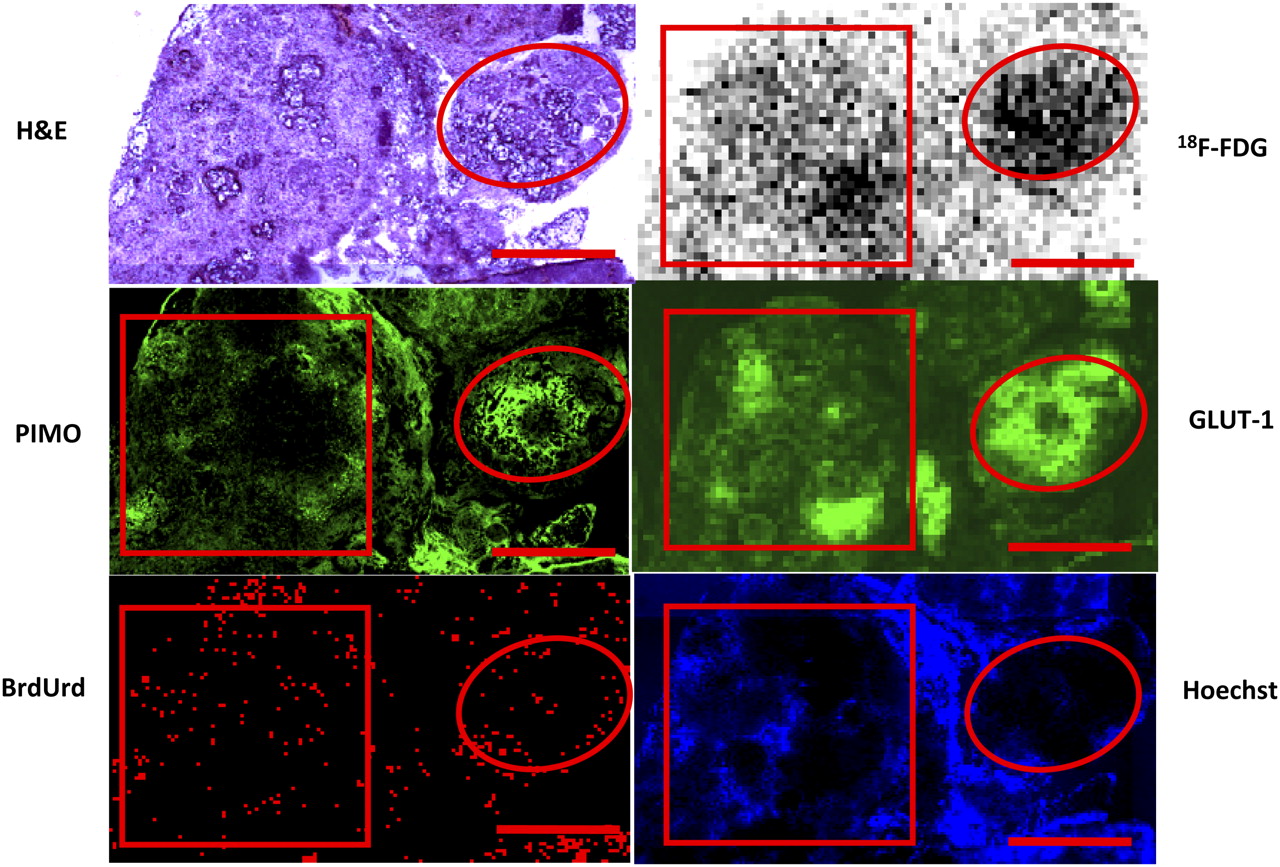
18F-FDG uptake in HT29 peritoneal tumors in air-breathing condition. Part of larger tumor (square) has relatively low levels of 18F-FDG uptake, pimonidazole binding, and GLUT-1 expression, with relatively high levels of cell proliferation and blood perfusion. Microscopic tumor (circle) has relatively high 18F-FDG uptake, pimonidazole binding, and GLUT-1, with lower cell proliferation and little perfusion. Similar results were seen in 7 animals. Scale bar is 1 mm. PIMO = pimonidazole; BrdUrd = bromodeoxyuridine.

Quantitative 18F-FDG uptake based on collection of intraperitoneal HT29 tumors derived from 5 tumors (diameter, <1 mm) and 4 tumors (diameter, 1–4 mm), all from a single air-breathing animal. 18F-FDG uptake was significantly higher in smaller tumors than in larger ones, P < 0.001. %ID/g = percentage injected dose per gram.
Two air-breathing animals with disseminated peritoneal disease arising from HCT-8 tumor cells were also studied. The results in this case were similar to those for HT29 tumors. An example is shown in Supplemental Figure 1 (supplemental materials are available online only at http://jnm.snmjournals.org).
Figure 3 shows an example of HT29 ascites tumors. These resembled the microscopic peritoneal tumors in that they had high 18F-FDG uptake, pimonidazole binding, and GLUT-1 expression throughout, with bromodeoxyuridine positivity seen only in the rim.

Comparison of 18F-FDG uptake with tumor hypoxia, GLUT-1 expression, and cellular proliferation in 2 HT29 ascites tumors from an air-breathing animal. Ascites tumors had high 18F-FDG uptake, pimonidazole binding, and GLUT-1 expression, with proliferation (bromodeoxyuridine) confined to rim. Scale bar is 500 μm. PIMO = pimonidazole; BrdUrd = bromodeoxyuridine.
Figure 4 illustrates the differences in 18F-FDG uptake in disseminated peritoneal HT29 microscopic tumors (diameter, <1 mm) between air- and carbogen-breathing conditions. A total of 4 animals from 2 independent experiments were studied with carbogen breathing, and in all cases results were broadly similar. For example, in an experiment that included 2 carbogen-breathing mice and 2 air-breathing mice, after 2 h of carbogen breathing (1 h before and 1 h after 18F-FDG administration), there was a major reduction in 18F-FDG uptake in microscopic tumors, compared with that for air-breathing animals. The histogram of Figure 4D is based on data from a total of 9 tumors (<1 mm) from 2 air-breathing animals and 11 tumors (<1 mm) from 2 carbogen-breathing animals. 18F-FDG tumor uptake was significantly reduced for the carbogen-breathing animals.

(A) Comparison of HT29 peritoneal tumors from animals breathing air or carbogen (95% O2; 5% CO2). 18F-FDG uptake and pimonidazole binding were markedly reduced for carbogen breathing whereas GLUT-1 expression was unaffected. Scale bar is 500 μm. (B) For air-breathing conditions, overall 18F-FDG uptake was higher, and several hot spots were observed. In contrast, carbogen breathing (C) resulted in significantly less 18F-FDG uptake in microscopic tumors. Similar results were seen in 4 carbogen-breathing animals. Scale bars are 4 mm. (D) Difference in 18F-FDG uptake between submillimeter HT29 tumors in air-breathing (9 tumors from 2 animals) and carbogen-breathing (11 tumors from 2 animals) animals was significant P < 0.001. PIMO = pimonidazole; H&E = hematoxylin and eosin.
We also attempted to quantify the degree of immunohistochemical staining based on mean fluorescence intensity. For pimonidazole, there was also an apparent decrease in binding for carbogen breathing (mean fluorescence intensity, 82 ± 9; n = 11 microscopic tumors from 2 mice) in comparison to air breathing (mean fluorescence intensity, 186 ± 16; n = 9 microscopic tumors from 2 mice). Taking these numeric values at face value, the difference was statistically significant (P < 0.001). In contrast, the level of GLUT-1 expression quantified by fluorescence intensity in microscopic tumors was not significantly different between carbogen-breathing (153 ± 24, n = 11) and air-breathing conditions (151 ± 22, n = 9) (P = 0.84). However, the linearity of the relationship between fluorescence intensity and antigen concentration is not established.
Two carbogen-breathing animals with disseminated peritoneal disease arising from HCT-8 cancer cells were also studied. Results were generally similar to those of the HT29 tumors, featuring reduced 18F-FDG uptake and pimonidazole binding coupled with unchanged GLUT-1 expression (compare Supplemental Figs. 1 and 2).
For submillimeter-sized tumors, quantitative analysis showed that 18F-FDG uptake was significantly lower for carbogen breathing than for air breathing for both tumor cell lines (Fig. 4D; Supplemental Fig. 3). The ratio of percentage injected dose per gram of 18F-FDG uptake between air and carbogen conditions was 2.5 ± 0.5 for HT29 (9 tumors from 2 air-breathing animals; 11 tumors from 2 carbogen-breathing animals) and 2.6 ± 0.2 for HCT-8 (3 tumors from 1 air-breathing animal; 6 tumors from 1 carbogen-breathing animal).
DISCUSSION
We have previously reported the existence of severe hypoxia in microscopic tumors derived from HT29 and HCT-8 colorectal cancer cells grown intraperitoneally in nude mice (15,16). The current study confirms these findings and extends them to include the use of the PET tracer 18F-FDG. In all cases, there was a clear qualitative agreement between the level of 18F-FDG uptake and the degree of binding of the hypoxia marker pimonidazole, suggesting a strong association between 18F-FDG uptake and physiologic hypoxia. In air-breathing animals, enhanced 18F-FDG uptake was also associated with high expression of GLUT-1, but this was not maintained in the case of carbogen breathing. Relatively short-term carbogen breathing was effective in reducing physiologic hypoxia in microscopic tumors, as evidenced by reduced pimonidazole binding, but had no obvious effect on the level of GLUT-1 expression. The observation that carbogen breathing also significantly reduced the uptake of 18F-FDG is strong evidence that although high GLUT-1 expression may be necessary, it was not sufficient to ensure high 18F-FDG uptake. In the tumor models used in this study, physiologic hypoxia was also necessary for high 18F-FDG uptake.
These results are consistent with previously published work on the relationship between hypoxia, glucose demand, and 18F-FDG uptake in vitro (2). Clavo et al. (7) showed that 3H-FDG uptake in HTB 63 melanoma and HTB 77 IP3 ovarian carcinoma cell lines was increased in hypoxic conditions in a time- and O2 concentration–dependent manner. This appeared to be partly due to increased membrane expression of GLUT-1. In contrast, Burgman et al. (5) found that hypoxia increased 3H-FDG uptake in MCF7 breast carcinoma cells without any increase in either glucose transporter protein or hexokinase. In this case, the increase in 3H-FDG uptake was attributed to an increase in glucose transporter activity due to hypoxia-induced alterations in cellular redox state. Our observation of significant changes in 18F-FDG uptake between air and carbogen breathing coupled with no apparent changes in GLUT-1 expression is more supportive of the mechanism proposed by Burgman et al. (5). Although it is possible that changes in hexokinase activity may also be involved, Waki et al. found no correlation between hexokinase activity and 3H-2-deoxyglucose uptake for 16 tumor cell lines (19).
Our results also agree with some in vivo studies using macroscopic tumors that showed a spatial correlation between the intratumoral distribution of 18F-FDG and those of the hypoxic marker pimonidazole (9,13) and the expression of GLUT-1 (also GLUT-3 and hexokinase-II) (4). It has been noted (13) that differences between 18F-FDG uptake in oxic and hypoxic regions would be reduced for tumors with high levels of aerobic glycolysis, a characteristic feature of cancer (20). This is because both anaerobic and aerobic glycolysis are inefficient sources of cellular energy (in comparison to oxidative phosphorylation) and both have a high demand for glucose. In the current study, a carbogen-induced decrease in tumor hypoxia led to a significant reduction in 18F-FDG accumulation using the human colorectal cancer cell lines HT29 and HCT-8. This reduction suggests that these cells were able to downregulate glycolysis in the newly oxygenated environment and that this change occurred rapidly, before noticeable alteration of GLUT-1 expression.
The finding that carbogen breathing–mediated reoxygenation produced a rapid decrease in tumor 18F-FDG uptake raises some issues of potential clinical significance. In particular, the use of changes in 18F-FDG tumor uptake to monitor response to cancer therapy may be subject to complications of interpretation due to treatment-induced changes in tumor hypoxic status. Additionally, oxygen breathing at the time of 18F-FDG administration may decrease uptake in patient tumors and should probably be avoided if possible. Finally, although we found that microscopic peritoneal and ascites tumors were hypoxic, with high 18F-FDG uptake, it is unlikely that these could be visualized individually because of their small size. However, it is possible that ensembles of microscopic tumors could be visualized. This may partly explain why malignant ascites may be evaluated by 18F-FDG PET/CT, as has recently been reported (21).
At a practical level, the disseminated peritoneal disease model used in this study has advantages over macroscopic subcutaneous xenografts as a system for examining mechanisms of radiotracer uptake. Macroscopic xenografts have a complex internal structure, with microscopic regions of aerobic, hypoxic, and necrotic tumor together with normal stroma, all in close proximity. Consequently, when comparing digital autoradiograms with immunohistochemical images, problems may arise due to image mismatch. In contrast, individual microscopic tumors have a close-to-homogeneous internal structure, for example, either uniformly hypoxic and unperfused or oxygenated and well perfused. It is thus much easier to demonstrate that radiotracer autoradiograms match images of pimonidazole binding or hypoxia-regulated protein expression. A second advantage is that because many peritoneal tumors (of differing size, hypoxic status, protein expression, etc.) can grow in an individual animal, it is possible to administer a radiotracer without confounding variations due to injected dose or interanimal pharmacokinetics. Therefore, the disseminated peritoneal disease model is, at least, a valuable supplement to macroscopic xenograft models as a test system for radiotracer development.
We would also contend that the microscopic tumor model is of potential use for studying metastatic cancer, because microscopic tumors growing in animals may mimic some aspects of microscopic disease in patients. As most cancer-related deaths are due to the development of metastatic disease rather than the growth of primary tumors, the prevention or elimination of metastases before they become clinically detectable would be expected to reduce cancer mortality rates. However, if the preangiogenic phase of micrometastatic development involves an episode of severe hypoxia, the efficacy of adjuvant or neoadjuvant treatments in the form of chemotherapy or radiotherapy may be compromised by hypoxic resistance (15,16). If this is true, then new strategies will be required to meet the challenge, possibly with the aim of converting hypoxia into a target for systemic therapies. The high demand for glucose displayed by hypoxic microtumors suggests that glucose metabolism may be a suitable target for developing novel therapies for micrometastatic disease. Several therapeutic strategies are under investigation to exploit or interrupt tumor glycolytic metabolism (22,23). Future studies to test the therapeutic efficacy of targeting glucose metabolism in micrometastatic model are warranted.
CONCLUSION
In a peritoneal model of disseminated microscopic disease, 18F-FDG uptake was significantly increased in hypoxic microscopic tumors. This enhanced uptake could be abrogated by carbogen breathing. Physiologic hypoxia was a necessary condition for increased 18F-FDG uptake in microscopic tumors.
Acknowledgments
This work was supported in part by NIH grants R01 CA84596 and P01 CA115675, and NCI grant P30-CA 08748.
Footnotes
-
COPYRIGHT © 2010 by the Society of Nuclear Medicine, Inc.
References
- Received for publication September 30, 2009.
- Accepted for publication December 21, 2009.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- The Evolving Role of Succinate in Tumor Metabolism: An 18F-FDG-Based Study
- The Reverse Warburg Effect and 18F-FDG Uptake in Non-Small Cell Lung Cancer A549 in Mice: A Pilot Study
- Tumor Microenvironment-Dependent 18F-FDG, 18F-Fluorothymidine, and 18F-Misonidazole Uptake: A Pilot Study in Mouse Models of Human Non-Small Cell Lung Cancer