


Abstract
Studies in rodents indicate that the disruption of P-glycoprotein (P-gp) function increases drug distribution into the developing fetus and organs such as the brain. To simultaneously and serially evaluate the effect of P-gp activity and inhibition on the tissue distribution of drugs in a more representative animal model, we tested the feasibility of conducting whole-body PET of the pregnant nonhuman primate (Macaca nemestrina). We used 11C-verapamil as the prototypic P-gp substrate and cyclosporine A (CsA) as the prototypic inhibitor. Methods: Four pregnant macaques (gestational age, 145–159 d; gestational term, 172 d) were imaged after the intravenous administration of 11C-verapamil (30–72 MBq/kg) before and during intravenous infusion of CsA (12 or 24 mg/kg/h, n = 2 each). The content of verapamil and its metabolites in plasma samples was determined using a rapid solid-phase extraction method. The plasma and tissue time–radioactivity concentration curves of 11C were integrated over 0–9 min after each verapamil injection. The tissue or arterial plasma area under the time–concentration curve (AUCtissue/AUCplasma) served as a measure of the tissue distribution of 11C radioactivity. CsA effect on 11C radioactivity distribution was interpreted as P-gp inhibition. The change in the fetal liver AUC ratio served as a reporter of placental P-gp inhibition. Results: CsA effect on tissue distribution of 11C radioactivity (AUC ratios) did not increase with the mean blood concentration of CsA, indicating a near-maximal P-gp inhibition. CsA increased maternal brain and fetal liver distribution of 11C radioactivity by 276% ± 88% (P < 0.05) and 122% ± 75% (P < 0.05), respectively. Changes in other measured tissues were not statistically significant. Conclusion: These data demonstrate for the first time, to our knowledge, the feasibility of simultaneous, serial, noninvasive imaging of P-gp activity and inhibition in multiple maternal organs and the placenta in the nonhuman primate. Our findings, consistent with previous data in rodents, indicate that the activity of P-gp in the placenta and the blood–brain barrier is high and that the inhibition of P-gp facilitates drug distribution across these barriers.
The multidrug resistance transporter P-glycoprotein (P-gp) affects drug pharmacokinetics and pharmacodynamics. The importance of P-gp results from its broad substrate selectivity and its strategic localization at critical sites for drug pharmacokinetics, including the blood–brain barrier (BBB), placenta, intestine, liver, and kidneys (1–3). Studies in rodents with genetically or chemically disrupted P-gp function provide evidence for the role of P-gp in the pharmacokinetics of many drugs, including chemotherapeutic, antiretroviral, and cardiac agents (1,3). In each case, the greatest impact of P-gp on drug distribution is at the BBB and the placenta. For example, the chemical inhibition of P-gp function in pregnant mice by the cyclosporine A (CsA) analog PSC833 increases the accumulation of digoxin in the maternal brain 8-fold and in the fetus up to 2.8-fold, but the magnitude of change in the maternal liver, kidneys, and spleen is much less (4).
It has been widely assumed that P-gp in the human BBB and the placental barrier is as important as that in rodents. In addition, it is commonly accepted that inhibition of P-gp may result in a significant increase in tissue distribution of P-gp substrate drugs, thereby increasing the efficacy or toxicity of drugs such as the anti-HIV protease inhibitors and cancer chemotherapeutic agents. However, because species can differ in the level of expression and activity of transporters, it is currently unknown whether this assumption is correct. We have begun to investigate this issue using PET as a noninvasive method to asses P-gp function at the human BBB. Using 11C-verapamil as the P-gp substrate and CsA as the P-gp inhibitor, we found that the brain distribution of 11C radioactivity increases by 88% with average CsA blood concentrations of 2.8 μM (5). This increase is less than that predicted by genetic ablation or CsA inhibition of P-gp in rodents (2,6,7). When rats are studied at the same blood concentration of CsA as that achieved in our human study, the inhibition of P-gp–mediated transport at the BBB is similar (7). However, the maximal magnitude of P-gp inhibition by CsA at the human BBB cannot be assessed because of concerns about CsA toxicity.
Placental P-gp can limit drug delivery to the fetus, such as in the case of the anti-HIV protease inhibitors. Alternatively, the barrier may protect the fetus against the effect of maternal cancer chemotherapy, such as doxorubicin, which has been safely used in pregnancy (8). Thus, more detailed knowledge of P-gp function at the blood–placental barrier is important in drug therapy for pregnant women. However, because of differences in placental physiology, rodents may not be a good model for drug transfer across the human placenta (9). Moreover, because of fetal radiation exposure, the noninvasive measurement by PET of placental P-gp function in humans is not possible. Although we and others have previously shown that the level of P-gp expression in the human placenta decreases with gestational age (10,11), assessment of its function at various gestational ages is not possible in humans. When compared with rodent models, the pregnant macaque provides a more representative model for the study of the maternal–fetal transfer of drugs (12–14). Therefore, using 11C-verapamil as the model P-gp substrate and CsA as the inhibitor, we tested the feasibility of noninvasive, simultaneous PET of P-gp activity and its inhibition in maternal organs and in the placenta in the pregnant macaque, Macaca nemestrina. Our hypothesis was that P-gp inhibition by CsA would result in a significant increase in distribution of 11C radioactivity into the tissues most protected by P-gp, that is, the maternal brain and fetus.
MATERIALS AND METHODS
Chemicals and Reagents
Racemic verapamil was purchased from Sigma-Aldrich. D-617 (3,4-dimethoxy-α-[3-(methylamino)propyl]-α-(1-methylethyl)-benzeneacetonitrile) and norverapamil were kindly supplied by Knoll AG. Palladium on alumina (0.5%), anhydrous acetonitrile, and zinc (99%) was purchased from Sigma-Aldrich. Ethanol, anhydrous sodium sulfate, and phosphate-buffered saline (PBS) solution were United States Pharmacopeia products. All other chemicals were purchased from multiple vendors and were of the highest chemical purity available.
Radiopharmaceuticals
15O-water, 11C-CO, and racemic 11C-verapamil were synthesized as previously reported (15). The radiochemical purity of the radiopharmaceuticals was greater than 95%, with the specific activity of 11C-verapamil greater than 57 GBq/μmol.
Animal Preparation
All experimental procedures were approved by the University of Washington Animal Care Committee. Four pregnant macaques were included in the study (Table 1). After an overnight fast, the animal was sedated with an intramuscular injection of ketamine (10 mg/kg) and atropine (0.04 mg/kg). After the animal was placed on a custom-built platform to ensure immobilization in the PET scanner (and also later during the MRI study), it was administered intravenous propofol (5 mg/kg) and intubated. Then, under isoflurane (0.5%−2.0%, v/v) anesthesia, the femoral artery and both femoral veins of the macaques were cannulated for blood sampling, radioactive tracer administration, and CsA infusion, respectively. To reduce spillover of radioactivity from the urinary excretion of the tracers, the animal was equipped with a urinary catheter and well hydrated. Hydration was provided by an intravenous infusion of Normosol-R (a nonpyrogenic, isotonic electrolyte solution) (Hospira, Inc.). Before the animal was positioned in the scanner, a bedside ultrasound was conducted to determine fetal position and heart rate. During the entire study, the animal was monitored noninvasively for blood oxygen saturation, body temperature, and blood pressure.
Characteristics of Animals at Time of PET Study
PET Protocol
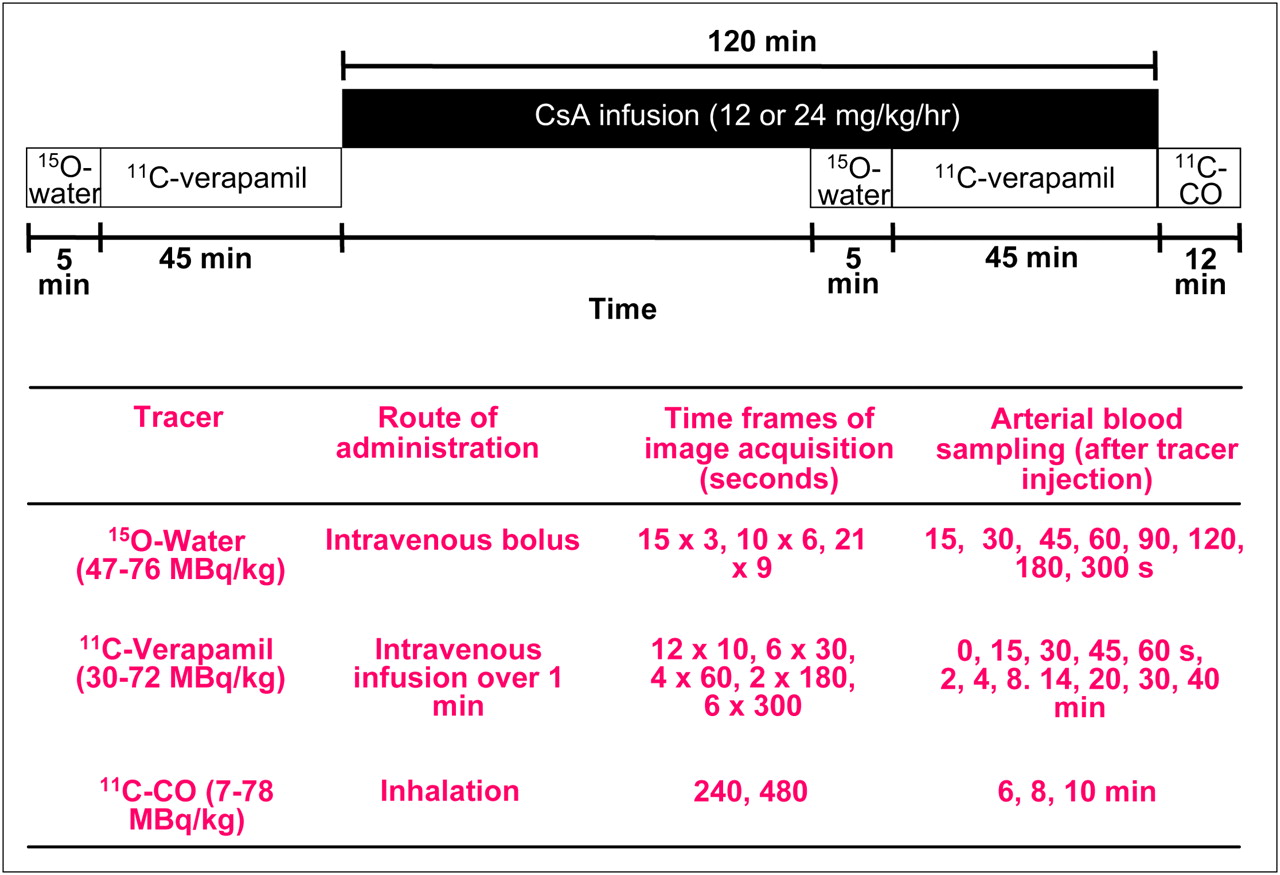
All PET studies were performed in a 2-dimensional acquisition mode on a PET scanner (Advance; GE Healthcare) providing 35 image planes over a 15-cm axial field of view (16). To include both the maternal brain and the fetal compartment in the field of view, the animal was positioned on its back, transverse to the scanner axis. This allowed dynamic imaging of the maternal brain, fetus, and all major maternal organs. At the beginning of each study, a 15-min attenuation scan was acquired with a rotating 68Ge source. The attenuation scan was repeated if any significant change in the position of the animal was noted between the substudies. Each substudy is described in detail in Figure 1. The animals were administered 47–73 MBq/kg of 15O-water (5 mL; intravenously, to measure tissue blood flow) as saline; immediately afterward, images were acquired and arterial blood samples (0.5 mL) were obtained (Fig. 1). Blood radioactivity was counted using a γ-counter (Cobra II Auto Gamma; Packard Corp.). At 5–15 min after the 15O-water study, 11C-verapamil (34−72 MBq/kg) was infused intravenously over a period of 1 min, in a total volume of 5–10 mL of isotonic saline containing less than 13% (v/v) (<1.3 mL) ethanol. At the onset of tracer infusion, a 45-min dynamic sequence was started and blood samples (0.5 mL) were taken (Fig. 1). From these samples, aliquots of plasma (100 μL) were counted. A larger volume of blood (3–5 mL) was collected at 1, 4, 8, 14, 20, and 40 min to determine plasma verapamil and metabolite concentrations by solid-phase extraction (SPE). At the completion of the 11C-verapamil scanning, CsA was administered as an infusion (12 or 24 mg/kg/h), for a maximum of 2 h. A second 15O-water imaging study (injected dose, 48–76 MBq/kg) was conducted at approximately 45 min after the initiation of CsA infusion, to measure any changes in the tissue blood flow induced by CsA. After 1 h of CsA infusion, the 11C-verapamil administration was repeated (30–66 MBq/kg), followed by an imaging and blood-sampling sequence identical to the one described above. To measure blood CsA concentrations, blood samples (1 mL) were taken at 1, 4, 8, 14, 20, and 40 min after the start of the second 11C-verapamil infusion. CsA blood concentrations were determined by liquid chromatography–mass spectrometry by the Department of Laboratory Medicine, University of Washington. At the end of the second 11C-verapamil substudy, 11C-CO was administered by inhalation to determine tissue blood volumes (Fig. 1).
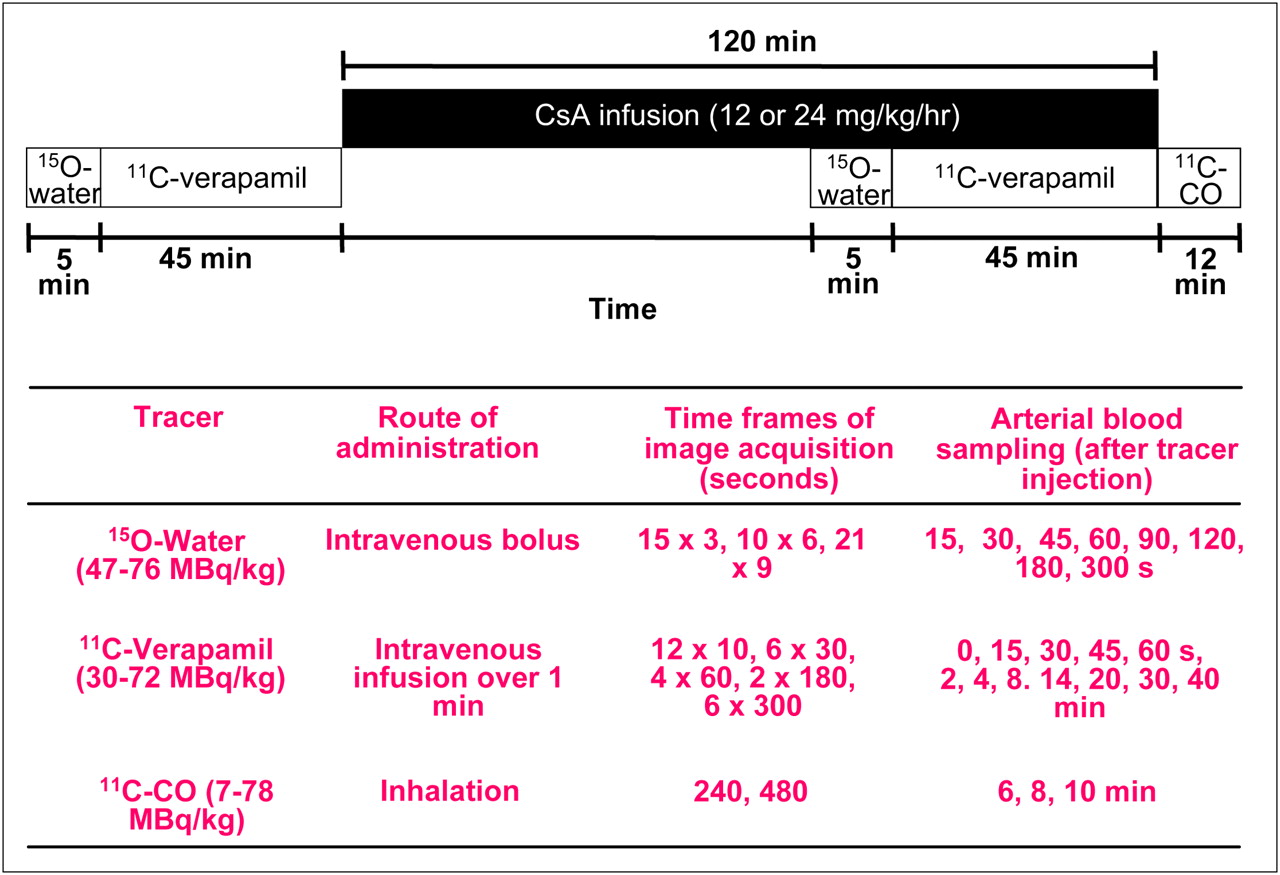
Schematic of PET protocol. Protocol for 1 animal was modified and shortened.
This protocol was used in 3 animals. The protocol was modified in the fourth animal, to reduce the compression of the central vessels by the pregnant uterus, as follows: The animal was placed in a lateral decubitus position, the duration of the second 11C-verapamil substudy was shortened to 25 min, 11C-verapamil administration was initiated 30 min into CsA infusion (12 mg/kg/h CsA), and the 11C-CO study was not performed.
Reconstruction of PET Images
After correction for random coincidences, scatter, and attenuation, images were reconstructed onto a 128 × 128 matrix of 35-slice volumes using a 2-dimensional filtered backprojection method with a 12-mm Hanning filter. Standardized uptake value image sets were created by summing the dynamic data from 0 to 5 min for 15O-water, from 1 to 9 min for 11C-verapamil, and from 4 to 12 min for 11C-CO to facilitate region-of-interest (ROI) placement for image analysis. The tomograph, dose calibrator, and γ-counter were cross-calibrated to express all measurements in common units of radioactivity (μCi or MBq).
Image Analysis
Within 2 wk of the PET study, the animal was imaged (in the same position as in the PET scanner) with a 1.5-T MRI instrument (Singa; GE Healthcare). ROIs were identified on T1- and T2-weighted MR images and summed 11C-verapamil images. We used the MR images and PET transmission scans as guides to generate ROIs from contiguous slices to create volumes of interest for each tissue type using conventional image-processing software (Alice [HIPG]; PMOD, Version 2.9 [PMOD Technologies]). Regions smaller than three times the machine resolution (1.2 cm) were corrected for partial volume (17). Volumes of interest were applied to both the dynamic image sets and the static summed standardized uptake value images for data extraction.
To better facilitate the visualization of the effect of CsA, images from the first-study (without CsA) scans were subtracted from those from the second-study (with CsA) (PMOD) scans, after normalization to injected dose and coregistration, to compensate for the effect of any animal motion between scans. This algorithm allowed visualization on a pixel-by-pixel basis of the net effect of CsA on tissue distribution of 11C radioactivity.
Estimation of Tissue Blood Flow
Image-based arterial-input functions from dynamic 15O-water studies were created from cardiac ROIs over the maternal heart derived by segmentation (18) and scaled by radioactive measurements of late arterial blood samples. A 1-compartment model (19) using the input function and the dynamic image sequence was used to generate parametric image volumes of blood flow (20). The manual placement of specific tissue ROIs was used to recover the average blood flow for each region.
Quantification of Verapamil and Its Metabolites in Plasma
The amount of radioactive verapamil and its metabolites in the plasma was determined after SPE and high-performance liquid chromatography, as previously described (5,21).
Data and Statistical Analysis
For tissue or arterial plasma area-under-the-curve (AUCtissue and AUCplasma, respectively) analysis, image and plasma data were decay-corrected to the injection time before curve integration. Then the ratio of AUCtissue to AUCplasma was computed. This AUC ratio served as a measure of tissue distribution of 11C radioactivity. The effect of CsA on this distribution was interpreted as P-gp inhibition. Data were expressed as mean ± SD. The difference between studies performed with or without CsA was analyzed by the Mann–Whitney test. Significance was set at a P value of less than 0.05.
RESULTS
The first 2 animals tolerated the imaging protocol with no adverse events and delivered full-term, viable babies. In these animals, 11C-verapamil injection and CsA infusion did not affect blood pressure or heart rate. The third animal, which had been positioned on its back and treated with 24 mg/kg/h of CsA, experienced significant blood pressure fluctuations at the end of the study, after imaging had been completed. Eight days after the PET study, this animal underwent a C-section to deliver a fetus that was diagnosed as nonviable by ultrasound. The fourth animal was positioned on its left side to avoid central vessel compression, and the imaging protocol was modified. The fourth study was completed successfully, without any adverse events experienced by the mother or her fetus.
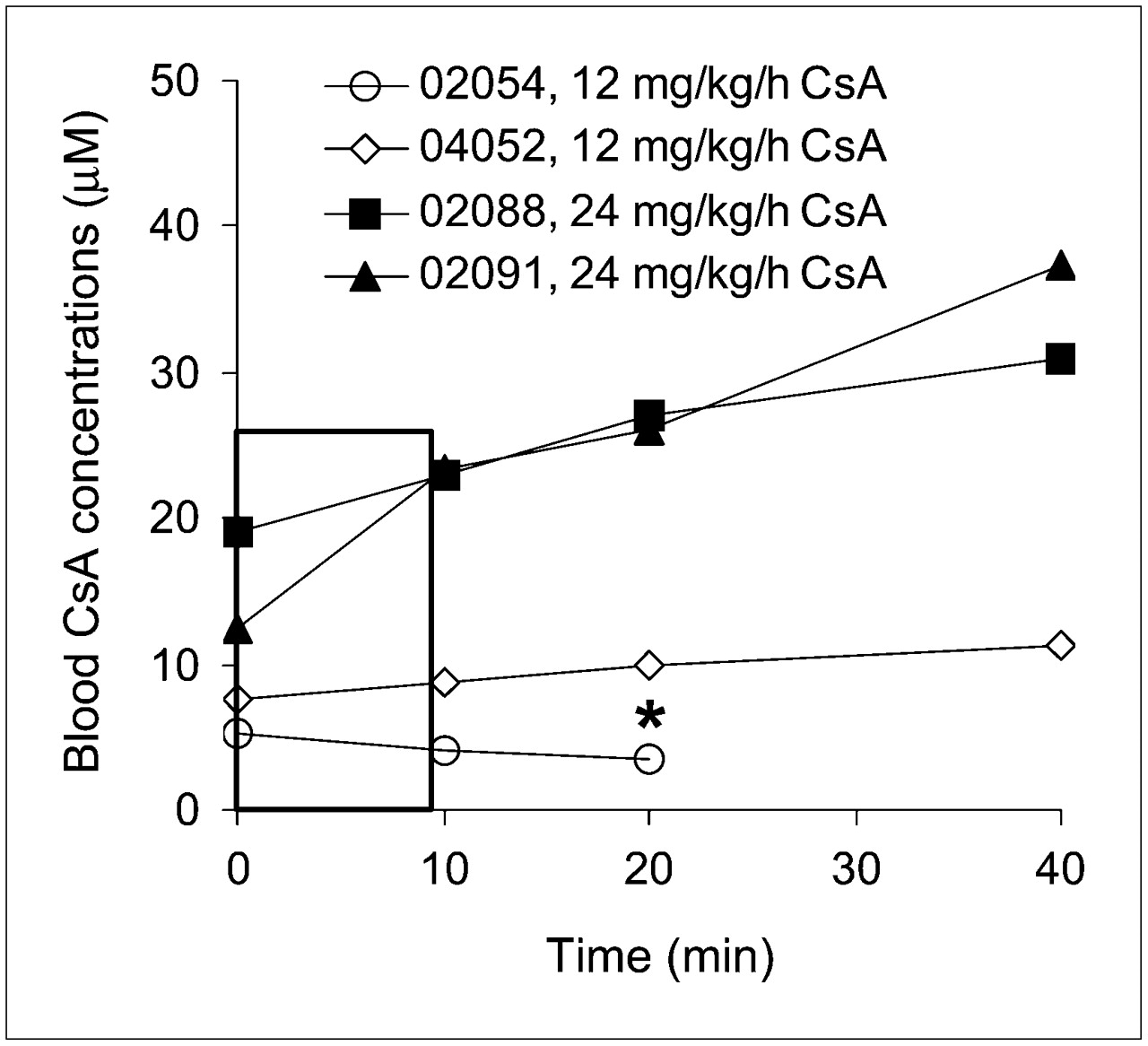
The blood concentrations of CsA (Fig. 2) were relatively constant at the 12 mg/kg/h rate but not at the 24 mg/kg/h rate. Although the number of animals was too small to statistically compare the effect of CsA dose on percentage of plasma radioactivity consisting of verapamil or its metabolite, these percentages appeared to be unaffected by CsA dose (Supplemental Table 1; supplemental materials are available online only at http://jnm.snmjournals.org); thus, we averaged the results across all 4 animals (Fig. 3). Because of the shortened protocol of the fourth study, results at 40 min represent 3 animals and have not been analyzed statistically.
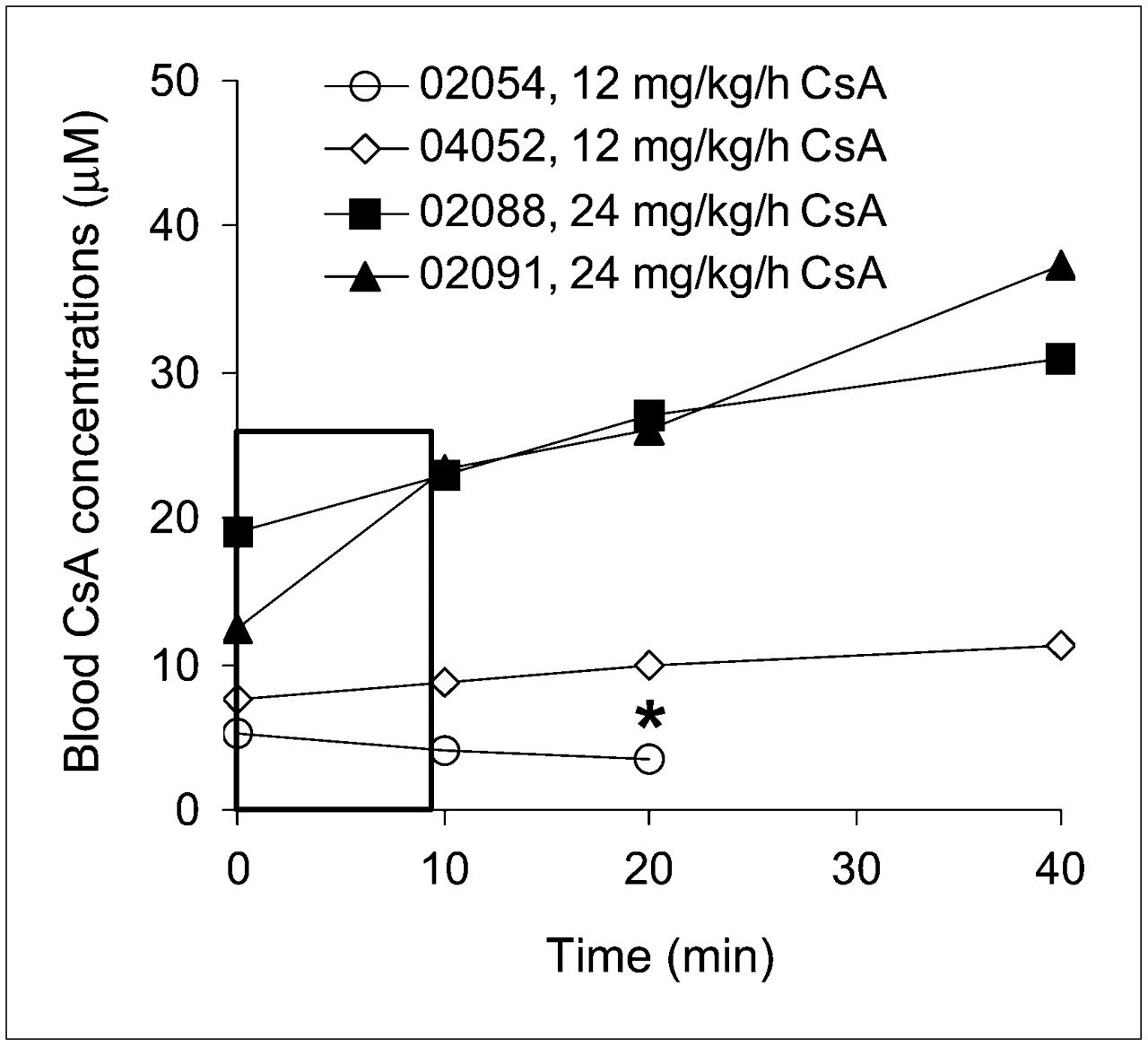
Blood CsA concentration time profiles from time of 11C-verapamil injection (t = 0). Legend indicates animal and CsA dose. Rectangle highlights modest change in CsA blood concentration over 0–9 min, the focus of our data analysis. *Animal was studied for only 25 min.
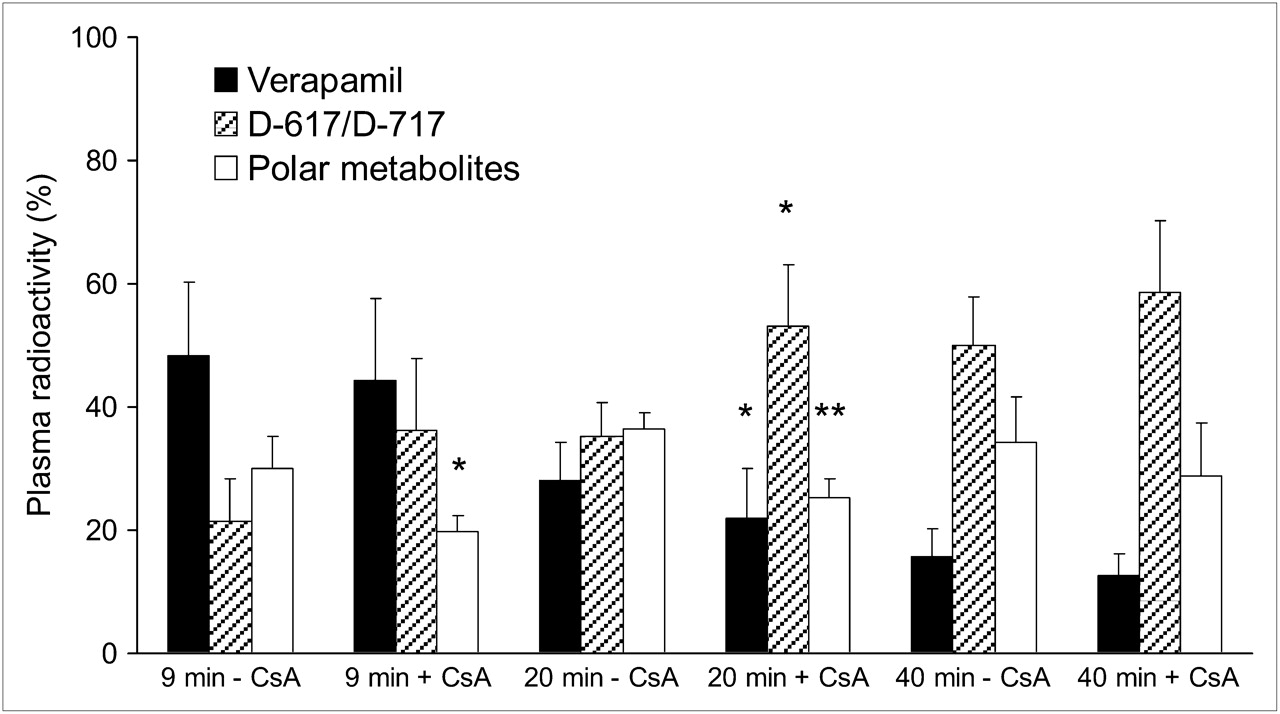
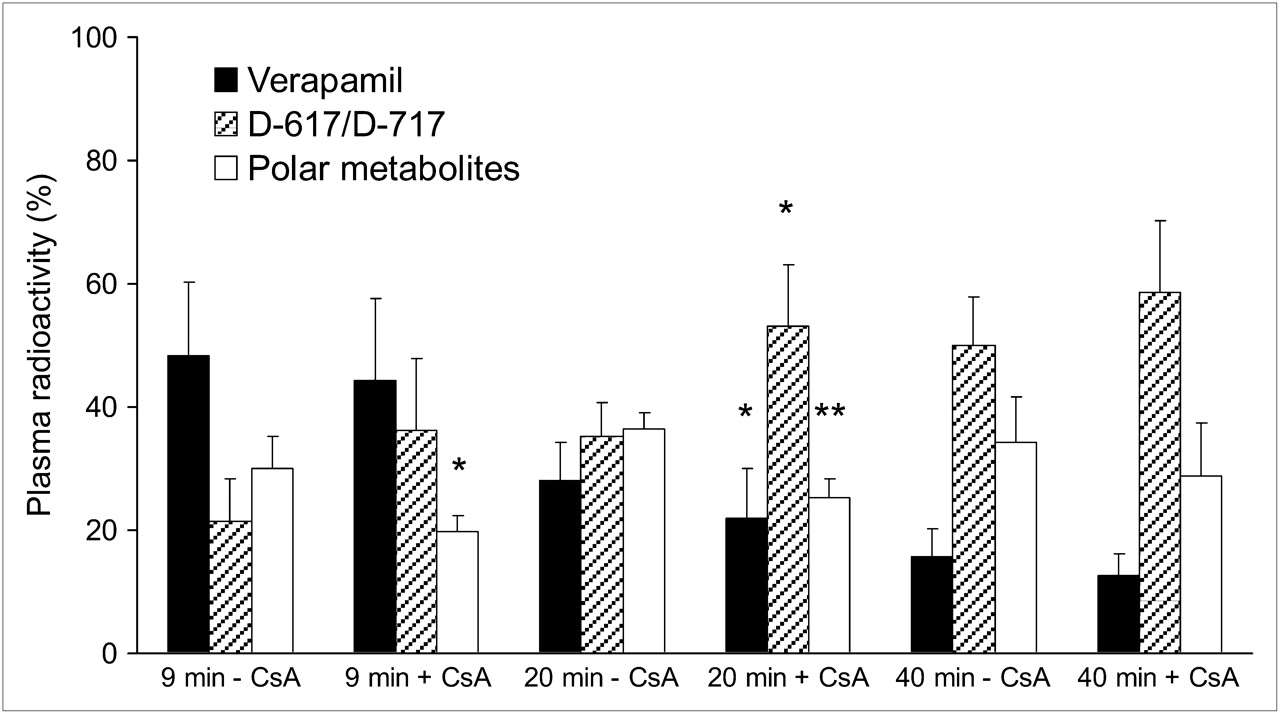
Percentage of 11C radioactivity in plasma before and during CsA infusion show that verapamil is rapidly metabolized in pregnant macaques. At 9 min, CsA did not significantly affect plasma radioactivity of verapamil and D-617/D-717 but decreased radioactivity of polar metabolites. At 40 min, less than 20% of total radioactivity was verapamil with or without CsA. Data are expressed as mean ± SD (n = 4). Data without and with CsA (at given time point) were compared using Mann–Whitney test. *P < 0.05.
Our rapid SPE method did not separate 3,4-dimethoxy-α-[3-(methylamino)propyl]-α-(1-methylethyl)-benzeneacetonitrile (D-617) from its O-demethylated metabolite, D-717. As in humans, only the dealkylated metabolites, D-617/D-717, and unknown polar metabolites were observed in the plasma of the macaques (Fig. 3). At 40 min after each injection, irrespective of CsA presence or its dose, verapamil accounted for less than 20% of plasma radioactivity. However, at 9 min, verapamil accounted for 48% ± 12% and 44% ± 13% of total plasma radioactivity in the studies without and with CsA, respectively (n = 4, Fig. 3). At the same time, D-617/D-717 accounted for 21% ± 7% and 36% ± 11% of total plasma radioactivity, respectively (n = 4). To minimize the influence of metabolism in the analysis of tissue verapamil radioactivity, we chose to integrate the plasma and the tissue concentration time curves over the first 9 min (AUC0–9). At 5 min, the midinterval of this period, mean blood CsA concentrations were 4.7 and 8.2 μM for the 12 mg/kg/h and 17.9 and 21.0 μM for 24 mg/kg/h infusion rates (Table 2).
Percentage Change in Plasma or Tissue Distribution of 11C Radioactivity Produced by CsA
For both CsA infusion rates, the profiles of injected dose (ID)–normalized plasma 11C time–activity curves did not differ significantly without and with CsA up to 40 min from verapamil injection (Fig. 4). In each animal, CsA administration increased the distribution of 11C-verapamil into the maternal brain and fetal liver (reporter of placental P-gp activity) (Fig. 4). In the maternal brain, CsA increased the ID-normalized peak concentration, time to peak concentration, and ID-normalized AUC0–9 by 209% ± 42% (n = 4, P < 0.05), 833% ± 410% (P < 0.05), and 238% ± 49% (P < 0.05), respectively (Supplemental Table 2). The ID-normalized AUC0–9 of the fetal liver increased by 108% (n = 4, P < 0.05) after CsA administration (Supplemental Table 2). CsA did not significantly affect the ID-normalized AUC0–9 of other organs, including the maternal liver and gallbladder. The impact of CsA on the uptake of radioactivity into the maternal brain and fetal liver is shown in Figure 5.

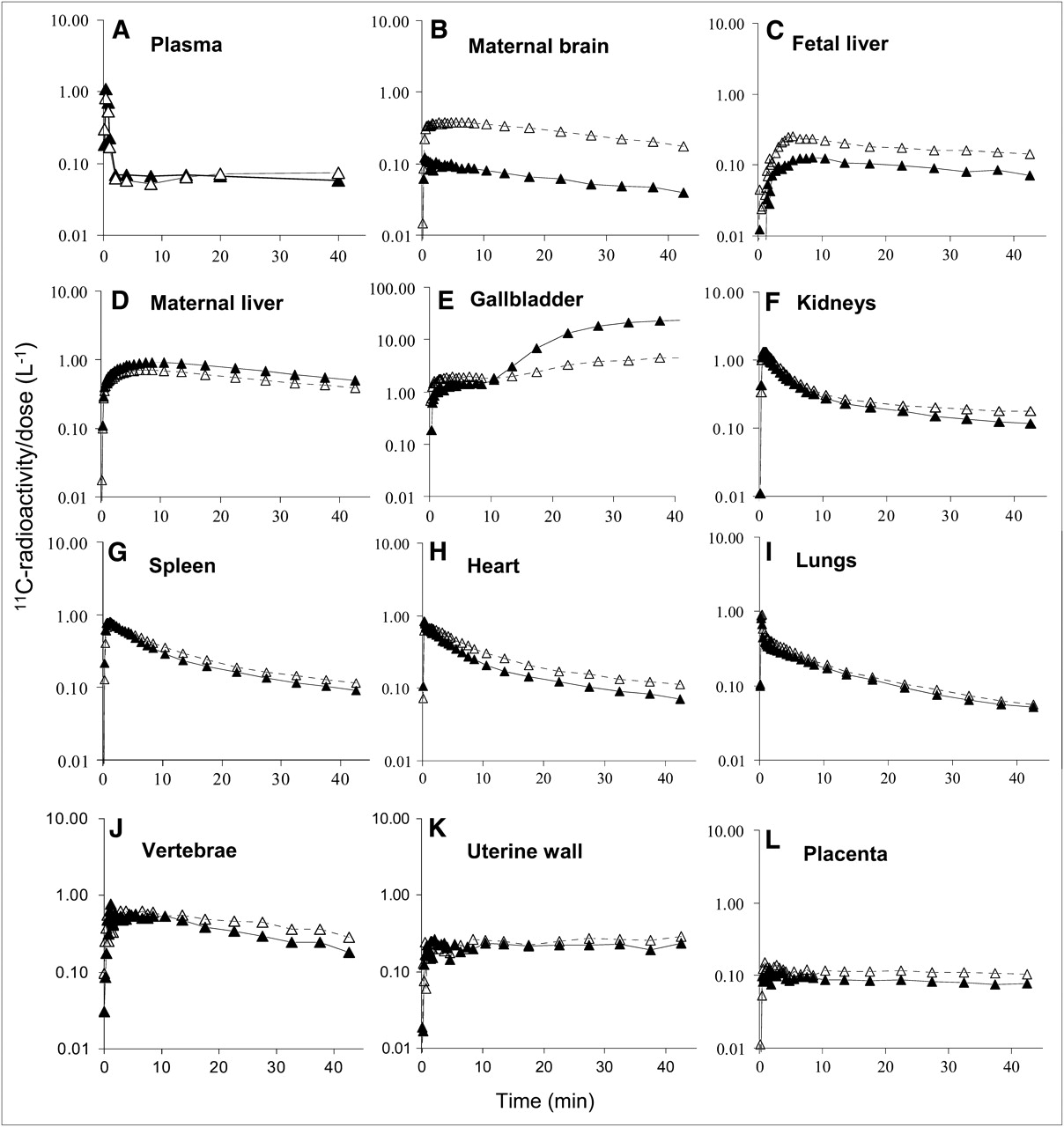
Plasma (A) and tissue (B–L) 11C radioactivity concentration time profiles in representative animal without (▴) and with (▵) CsA (12 mg/kg/h). With CsA, these profiles show greater distribution of radioactivity into maternal brain (B) and fetal liver (C) but lesser distribution into maternal liver (D). In maternal gallbladder (E), CsA initially increased accumulation of 11C radioactivity but reduced it at later time points. CsA did not change time–concentration profiles of radioactivity in plasma or other tissues.
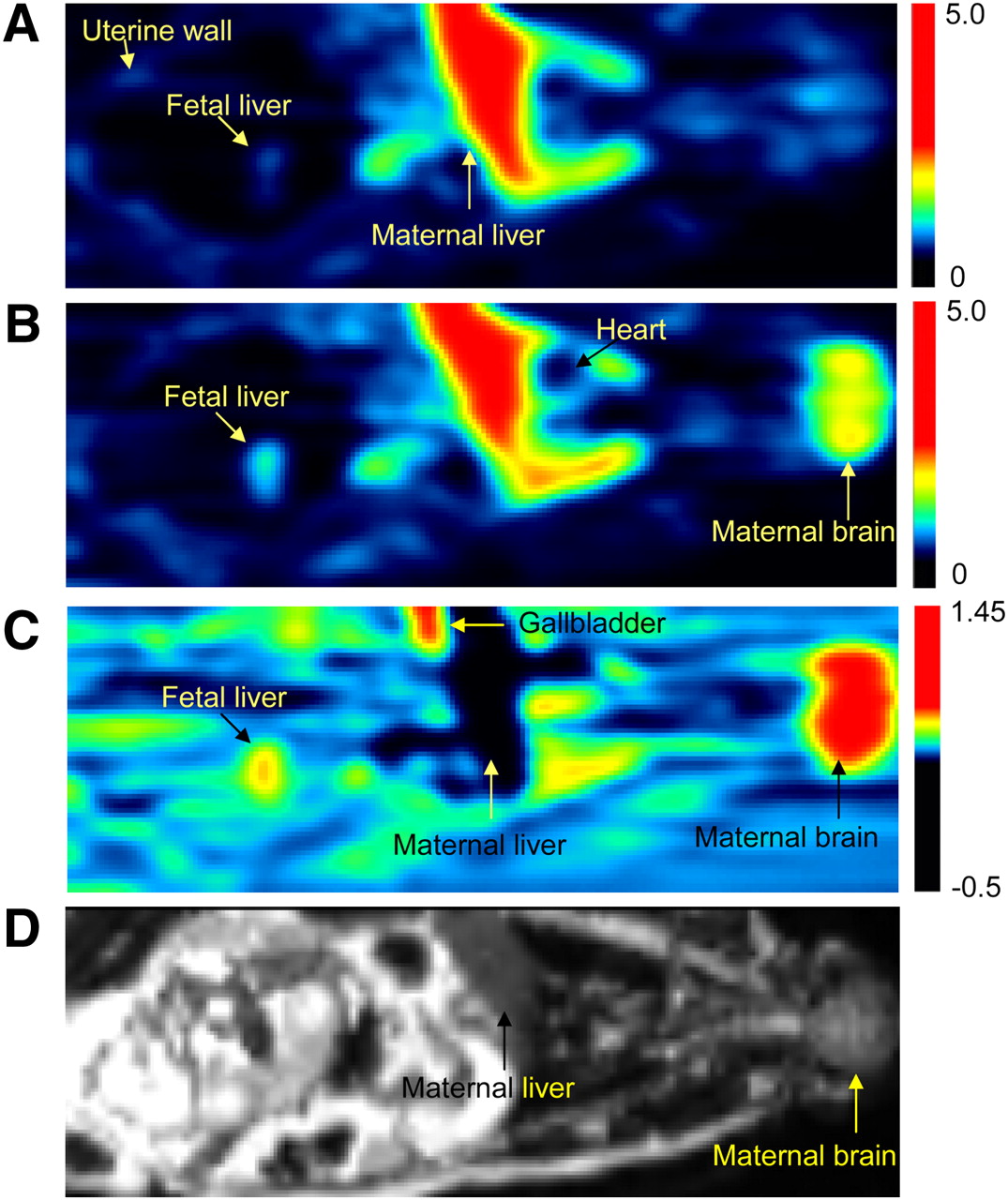
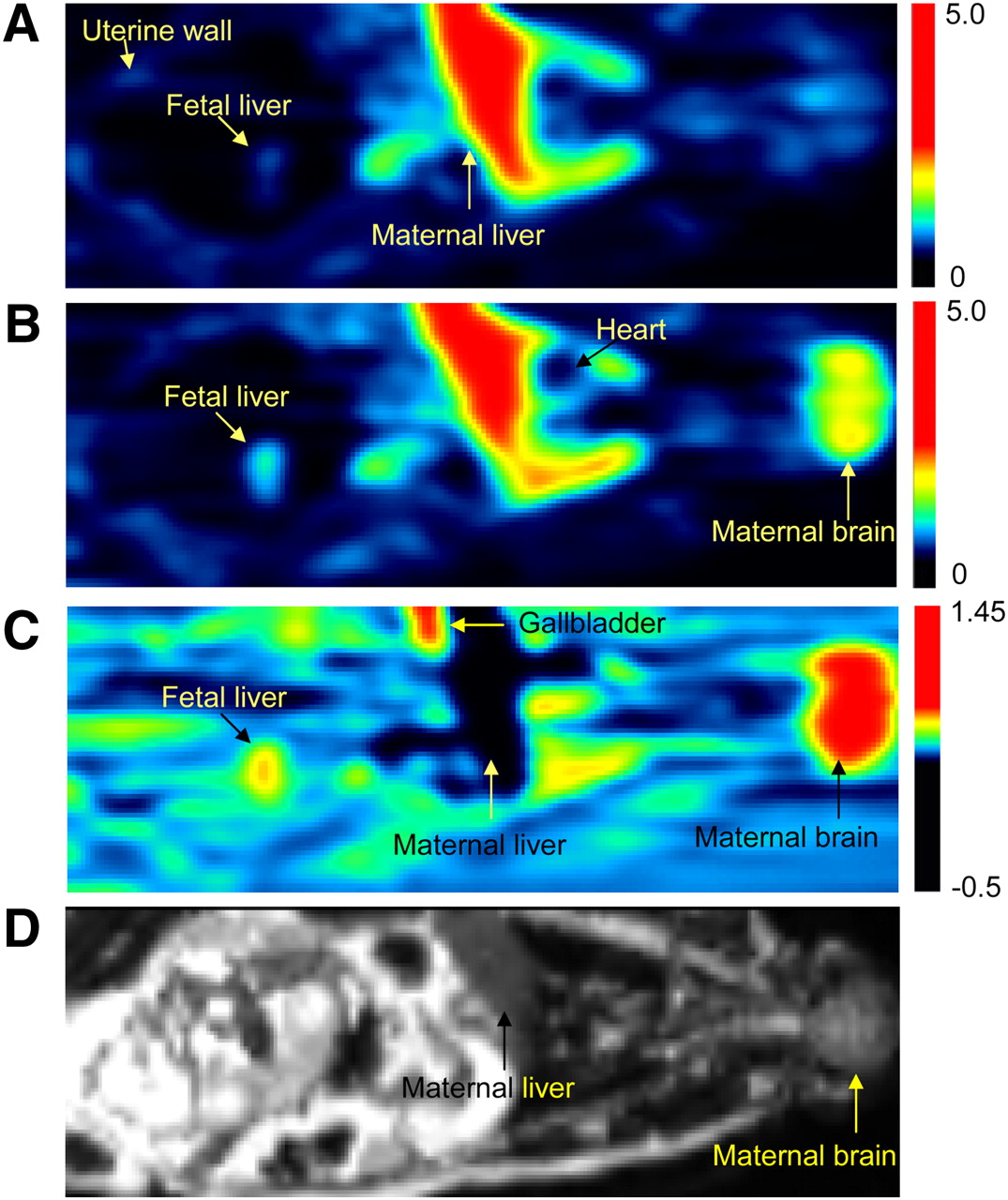
PET images of pregnant M. nemestrina before (A) and during (B) administration of CsA (12 mg/kg/h). PET scans (A and B) are SUV images summed over a period of 1–9 min after 11C-verapamil injection. Images A and B were scaled to the same pixel value. (C) Image produced by pixel-by-pixel subtraction of scan A from scan B. Fetal liver was reporter of 11C radioactivity that crossed placental barrier. Because of P-gp inhibition, CsA significantly increased distribution of 11C radioactivity into maternal brain and fetal liver (yellow to red areas in B and C). In maternal liver, CsA decreased amount of 11C radioactivity (darker area in C). Color scale reflects SUV as shown by thermometer. (D) T2-weighted MR image of the same animal. SUV = standardized uptake value.
A better measure of the effect of CsA on uptake of 11C radioactivity across blood–tissue barriers is obtained when the AUC values of individual organs are normalized by the AUC values of plasma (Fig. 6). CsA effect on AUC ratios did not increase proportionally with the mean blood concentrations of CsA (Table 2). As a result of P-gp inhibition at the BBB of the macaque, the maternal brain distribution of 11C radioactivity increased from 0.92 ± 0.15 to 3.40 ± 0.66 (276% ± 88%; n = 4, P < 0.05). The AUC ratios of the fetal liver across all animals increased from 0.90 ± 0.25 to 1.87 ± 0.34 (122% ± 75%; P < 0.05, n = 4). AUC ratios of other tissues were not significantly affected by CsA. Comparable results were obtained when tissue and plasma concentration–time curves were integrated over the first 0–20 min or 0–40 min from each verapamil administration (Fig. 6). Changes at 40 min were not statistically tested, because only 3 animals continued the imaging study beyond 20 min.
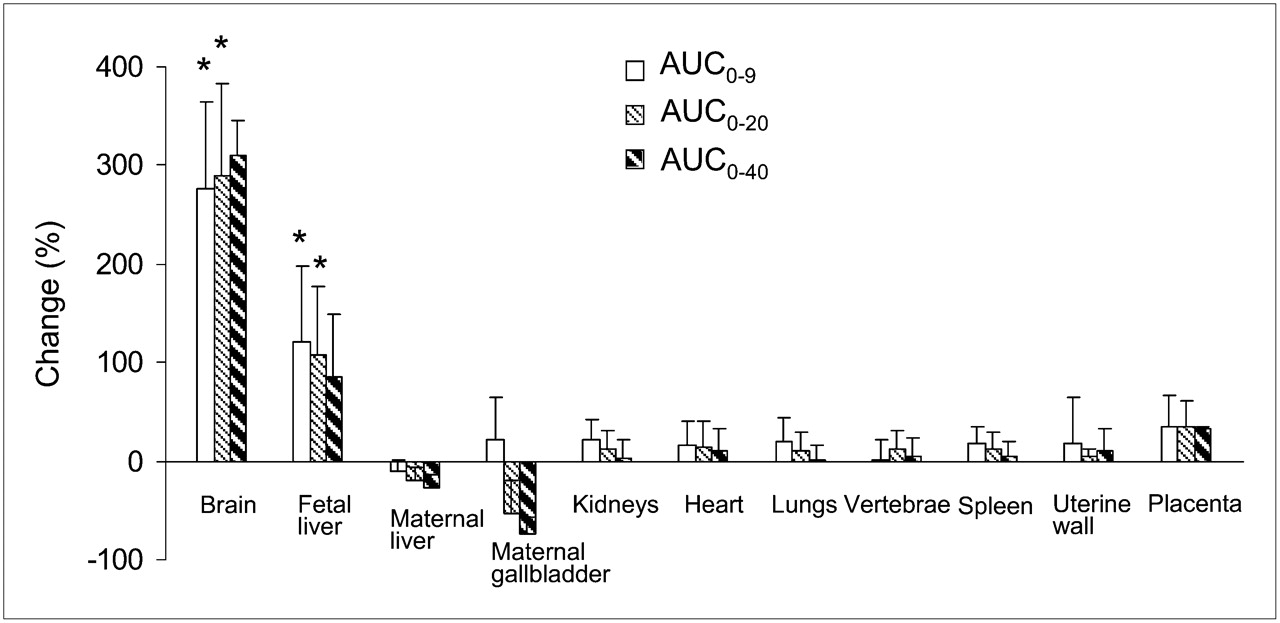
Effect of CsA on distribution (percentage change in AUCtissue/AUCplasma) of 11C radioactivity into maternal brain and fetal liver was large and significant. In contrast, its effect on distribution of 11C radioactivity into other tissues was insignificant. Data are expressed as mean ± SD (n = 4). *P < 0.05.
CsA increased mean maternal cerebral blood flow by 18% ± 38% (from 1.57 ± 0.57 to 1.65 ± 1.16 mL/mL/min, n = 3). On average, CsA decreased placental blood flow by 19%, but intersubject variability was high (SD, 56%).
DISCUSSION
Here we report the first, to our knowledge, simultaneous assessment of in vivo P-gp activity and inhibition in multiple tissues in the macaque. Our study was designed to test the feasibility of whole-body imaging to identify tissues in which inhibition of P-gp activity (by CsA) results in marked changes in tissue distribution of a P-gp substrate drug, 11C-verapamil. As was demonstrated by our studies and those of others, the percentage change induced by CsA in the tissue distribution of 11C-verapamil radioactivity was interpreted as the magnitude of P-gp inhibition (2,5–7).
Because this is a pilot study that compares P-gp inhibition among different organs, we targeted 2 different blood concentrations of CsA: one that approximates the half-maximal effective concentration (EC50) for P-gp inhibition in the rat (7.2 μM) (7) and the other that is higher than the rat EC50 and predicted to completely inhibit P-gp function (7). Although verapamil undergoes stereoselective metabolism (22,23), the R and S enantiomers of verapamil have approximately equal affinity for P-gp (24). Thus, P-gp–mediated distribution of 11C-verapamil radioactivity into tissues would be dependent only on the total 11C-racemic verapamil blood concentration and the tissue-specific P-gp activity, making it unnecessary to study the distribution of individual enantiomers.
The metabolism of verapamil was faster in pregnant macaques than it was in humans (Fig. 3) (5) or in male macaques (25) and was most likely enhanced by pregnancy, as a result of CYP3A induction (26,27). The major radioactive metabolites in macaques, similar to those in humans, were 11C-D617/D-717, formed by dealkylation, and polar metabolites, formed by N-demethylation. CsA did not change the fraction or the amount of radioactivity present in plasma as verapamil at 9 min after verapamil administration. By 40 min, verapamil accounted for less than 20% of the total radioactivity. For this reason, and because differential uptake of verapamil and its metabolites into individual tissues can exist, we limited our analysis to the first 9 min after each verapamil injection. Over this period, at least 70% of the observed radioactivity was related to verapamil and D-617/D-717, a P-gp substrate (28). Nevertheless, the contribution of metabolites to total tissue radioactivity did not confound our estimation of P-gp inhibition because AUC ratios and the effects of CsA on tissue radioactivity, with the exception of effects on the maternal gallbladder, were quantitatively comparable between measurements at 9, 20, and 40 min (Fig. 6).
The shape and location of the placenta make it difficult to reliably identify and measure tracer uptake. In addition, a substantial fraction (∼50%) of the placenta is maternal or fetal blood (29). The latter is not labeled by 11C-CO. However, the fetal liver is readily identifiable on PET. The substantial fetal hepatic accumulation of verapamil radioactivity served as a readily identifiable reporter of the net placental passage of verapamil into the fetal compartment (tracer concentrations in the extrahepatic fetal tissues were too low to determine their values with confidence). Although fetal movement between the PET and MRI scans complicated coregistration of PET and MR images, the exclusive concentration of radioactivity in the fetal liver made this organ readily identifiable.
In agreement with previous studies in mice and rats (2,6), the distribution of 11C-verapamil radioactivity (AUC0–9) into the maternal kidneys, heart, lungs, spleen, and uterus in our study was not significantly affected by CsA (Fig. 6). In contrast, the impact of CsA on P-gp activity was significant at the maternal BBB and placenta. These differences in activity likely reflect higher P-gp expression in these tissues. Our interpretation of tissue P-gp inhibition is based on the assumption that the combination of verapamil and CsA results in inhibition of P-gp only. This is not an unreasonable assumption because previous studies have shown that verapamil is not a substrate of the multidrug resistance-associated proteins or breast cancer resistance protein and, on the basis of its chemical structure, is unlikely to be a substrate of organic anion transporter families (5). However, we cannot exclude the possibility that in certain organs, verapamil and CsA could interact through organic cation transporters, the novel organic cation transporter 2, or other, yet unidentified efflux and influx transporters (30,31).
The largest effect at the BBB was likely due to the high level of P-gp expression and function at this barrier. At the higher mean CsA blood concentration (19.4 μM), the brain-to-plasma AUC ratio of 11C radioactivity changed 3.9-fold. These results are consistent with the 2.3-fold increase in the cerebrum-to-blood AUC of 11C-verapamil radioactivity in male Macaca mulatta after the administration of the CsA analog PSC833 (25). In contrast, our study in rats demonstrated an approximately 12-fold increase in the brain-to-plasma total 3H radioactivity ratio at comparable mean CsA blood concentrations (17.3 μM) (7). The lower maximal increase in the brain distribution of 11C radioactivity in the pregnant macaques might be explained by species differences in the contribution of BBB P-gp activity to the distribution of 3H-verapamil radioactivity into the brain, inhibition of an influx transporter by CsA, or pregnancy. However, because a similar degree of inhibition was observed in male, unanesthetized macaques with another P-gp inhibitor, the first explanation appears to be more likely. If this conclusion is extrapolated to humans, the rodent P-gp knockout models may overestimate the potential for P-gp–mediated drug interactions at the human BBB. This may seem at odds with our previous conclusion that the rodent is an excellent predictor of the verapamil–CsA interaction at the BBB (32). However, there is no discrepancy. Although there is an excellent agreement between the interaction observed at the rat and human BBBs at the lower CsA blood concentrations, our data suggest a divergence between the rat and the human as the inhibitor concentration is increased and as P-gp inhibition approaches a maximum. To test this hypothesis, additional drug interaction studies in humans with inhibitors of P-gp more potent than CsA (quinidine, itraconazole) are needed.
At the placental barrier, the significance of P-gp was demonstrated by the 2.2-fold (122%) increase in fetal liver radioactivity after CsA administration. Our results indicate that CsA has a greater inhibitory effect on the efflux of verapamil across the BBB than across the placenta, likely reflecting a tighter P-gp barrier at the brain, at least for the combination of verapamil and CsA. Mean placental blood flow was not significantly affected by CsA and thus could not explain the enhanced distribution of 11C radioactivity across the placenta. The contribution of P-gp to the placental barrier may change with gestational age. We have previously shown that P-gp expression in human placental syncytiotrophoblasts decreases approximately 40-fold with gestational age (10), but it is currently unknown whether this is paralleled by a change in P-gp function. This study has demonstrated the feasibility of PET in pregnant macaques to serially measure the effect of gestational age on placental P-gp functional activity. In addition, our data suggest that P-gp–mediated drug interactions at the placental P-gp barrier are possible; however, the magnitude of interaction, at therapeutic concentrations of CsA, is expected to be modest. Although CsA is transferred across the placenta (33) and P-gp is expressed in many tissues of the third-trimester human fetus (34), we could not assess the effect of CsA on fetal tissue P-gp activity because the tracer concentrations in the extrahepatic fetal tissues were too low to determine their values with confidence.
In contrast to the effects of CsA on tissue radioactivity in the brain and fetus, with the exception of effects on the gallbladder, CsA did not affect tissue distribution of 11C radioactivity into other tissues such as the liver and kidneys. These data are consistent with the rodent data, in which the contribution of P-gp at these blood–tissue barriers appears to be less than that at the BBB and blood–placental barrier. We speculate that the reduction in gallbladder radioactivity during CsA administration reflects the inhibition of P-gp in liver canaliculi.
CONCLUSION
The current study provides proof of concept that PET can be used in the pregnant macaque to noninvasively and simultaneously assess the impact of P-gp inhibition on the distribution of drugs into multiple tissues. The inhibition of P-gp is tissue-specific, and the greatest impact is at the BBB and the placental barrier, two important barriers affecting drug distribution into the brain and the fetus, respectively. In addition, the maximal inhibition of P-gp at supratherapeutic concentrations of CsA affects brain distribution of 11C-verapamil radioactivity in macaques less than it does in rodent models, in which P-gp function is genetically or chemically ablated. If the macaque is representative of humans, these data suggest that genetic or chemical knockout of P-gp in rodents will overestimate the maximum potential of drug interactions at the human BBB when potent inhibitors of P-gp are studied. To test this hypothesis, additional drug interaction studies in humans, with inhibitors of P-gp more potent than CsA (e.g., quinidine), are needed.
Acknowledgments
We are indebted to Steve Shoner, Tony Park, Kathryn Bray, Barbara Lewellen, Pam Pham, Thomas Lewellen, Lisa Flint, Jeff Stevenson, Paul Chu, and Jenne Hoard, from the Department of Radiology; Mike Gough, Ed Novak, Pat Delio, Bruce Brown, Keith Vogel, Cliff Astley, and Melinda Young, from WaNPRC; and Dale Whittington, from the Department of Pharmaceutics, for their expert technical assistance. We further thank Andrei Mikeev, Mary Blonski, Suresh Babu Naraharisetti, Brian Kirby, Christopher Enders, Andrew Bostrom, Huixia Zhang, and Xiaohui Wu, from the Department of Pharmaceutics, for their assistance in conducting this study and Duane Bloedow, from the Department of Pharmaceutics, for fruitful discussions. This study was supported by National Institute of Health grants U10HD047892 (Obstetric-Fetal Pharmacology Research Unit Network), P50HD044404, GM032165, and RR00166.
Footnotes
-
COPYRIGHT © 2009 by the Society of Nuclear Medicine, Inc.
References
- Received for publication October 23, 2008.
- Accepted for publication January 6, 2009.
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Transplacental Pharmacokinetic Model of Digoxin Based on Ex Vivo Human Placental Perfusion Study
- Impact of P-Glycoprotein-Mediated Active Efflux on Drug Distribution into Lumbar Cerebrospinal Fluid in Nonhuman Primates
- Drug Concentration Asymmetry in Tissues and Plasma for Small Molecule-Related Therapeutic Modalities
- Emerging Models of Drug Metabolism, Transporters, and Toxicity
- Total-Body PET: Maximizing Sensitivity to Create New Opportunities for Clinical Research and Patient Care
- Modulation of P-glycoprotein at the Human Blood-Brain Barrier by Quinidine or Rifampin Treatment: A Positron Emission Tomography Imaging Study
- Role of (Drug) Transporters in Imaging in Health and Disease
- Modeling Cyclosporine A Inhibition of the Distribution of a P-Glycoprotein PET Ligand, 11C-Verapamil, into the Maternal Brain and Fetal Liver of the Pregnant Nonhuman Primate: Impact of Tissue Blood Flow and Site of Inhibition
- Developmental Changes in P-Glycoprotein Function in the Blood-Brain Barrier of Nonhuman Primates: PET Study with R-11C-Verapamil and 11C-Oseltamivir
- Xenobiotic, Bile Acid, and Cholesterol Transporters: Function and Regulation