


Abstract
The 18F-labeled aromatic amino acid 6-fluoro-3,4-dihydroxy-l-phenylalanine (6-18F-fluoro-l-DOPA) is widely used as a radiopharmaceutical in neurologic and oncologic PET. In this study, a novel approach to the preparation of carrier-added (CA) 6-18F-fluoro-l-DOPA in 3 radiosynthesis steps was developed and evaluated; in this approach, direct nucleophilic 18F fluorination of a protected amino acid derivative was used. The method currently used for the routine preparation of 6-18F-fluoro-l-DOPA by electrophilic labeling is limited to the production of small amounts of activity at high costs. Alternative syntheses based on the advantage of large-scale production of nucleophilic 18F-fluoride, however, either have resulted in insufficient enantiomeric purity or are difficult to automate because of the complexity of the necessary multiple steps. Methods: An isotopic exchange reaction on the precursor (2S,5S)-tert-butyl-5-(4-benzyloxy-2-fluoro-5-formylbenzyl)-2-tert-butyl-3-methyl-4-oxoimidazolidine-1-carboxylate was used. The formyl group served as the activating group in the 18F-for-19F exchange with tetrabutylammonium bicarbonate for anion activation in N,N-dimethylformamide. The intermediate was converted to a hydroxy group by Baeyer–Villiger oxidation with meta-chloroperbenzoic acid. After final deprotection with hydrobromic acid, CA 6-18F-fluoro-l-DOPA was isolated by high-performance liquid chromatography. Results: The precursor was obtained by an 11-step organic synthesis. The optimized isotopic 18F exchange proceeded with a radiochemical yield of about 50%. The complete preparation and isolation of CA 6-18F-fluoro-l-DOPA thus far are possible with a radiochemical yield of about 22%, within a synthesis time of 105 min, and at a much higher specific activity than with the electrophilic method. The enantiomeric excess of the desired l-isomer was greater than 96%.Conclusion: The pathway to 6-18F-fluoro-l-DOPA by isotopic exchange not only is more efficient but also is suited to automation as a “one-pot” procedure.
An 18F-labeled analog of 3,4-dihydroxy-l-phenylalanine, 6-18F-fluoro-3,4-dihydroxy-l-phenylalanine (6-18F-fluoro-l-DOPA), is an established radiotracer for diagnosis of the integrity and function of the nigrostriatal dopaminergic system by PET (1,2). More recently, it was also found that 6-18F-fluoro-l-DOPA has potential for oncologic diagnosis (3,4), especially in PET studies of neuroendocrine tumors (5).
Because 6-18F-fluoro-l-DOPA can be applied in a carrier-added (CA) form (6), the commonly used method for its routine preparation is the electrophilic fluorodestannylation reaction (7,8), for which remote-controlled synthesis units are commercially available (9,10). Nevertheless, the electrophilic method is limited to the production of small amounts of activity at high costs, even when the 18O(p,n)18F nuclear reaction is used (11), and only half of the radioactivity of molecular 18F-F2 can be incorporated. Therefore, the use of this important tracer in clinical centers is limited mainly by the lack of a nucleophilic radiofluorination method based on the advantage of the large-scale production of 18F-fluoride.
The first nucleophilic approaches for the radiosynthesis of 6-18F-fluoro-l-DOPA were based on substitution with 18F-fluoride on small benzaldehyde derivatives (12). After the 18F-labeling step, the aldehyde functionality was converted to a benzyl halide group, subsequently coupled either to chiral auxiliaries (13) or to a prochiral auxiliary by chiral phase transfer alkylation (14). The latter strategy permits the large-scale production of 6-18F-fluoro-l-DOPA with excellent chemical, radiochemical, and enantiomeric purities (>96%). Furthermore, both methods yield 6-18F-fluoro-l-DOPA at the no-carrier-added (NCA) level. However, because of their complexity, these multistep syntheses are cumbersome to automate.
For improving the efficiency of the nucleophilic preparation of 6-18F-fluoro-l-DOPA, a direct nucleophilic isotopic exchange reaction on a formyl-activated masked aromatic amino acid derivative was studied. The required carbon skeleton and the chiral center with the desired configuration were constructed before introduction of the 18F-fluoride ion by use of a homochiral piperazine derivative as described by Schöllkopf' (15). After cryptand-mediated 18F introduction, the intermediate was transformed to 6-18F-fluoro-l-DOPA by Baeyer–Villiger oxidation and hydrolysis (16). This method, however, yielded 6-18F-fluoro-l-DOPA at an insufficient enantiomeric excess, only 70%.
Kuroda et al. (17) described the synthesis of a similar precursor with this strategy by choosing a homochiral imidazolidinone derivative (according to Seebach et al. (18)) as the masked amino acid functionality and iodide as the leaving group in the formyl-activated 18F exchange reaction. Unfortunately, no 18F-labeling results with this precursor have been reported so far. It can be presumed that the stability of the precursor is not sufficient for an 18F-labeling reaction.
In the present study, the advantages of the concepts of both Tierling et al. (16) and Kuroda et al. (17) were combined in a novel, efficient nucleophilic “one-pot” approach for the preparation of 6-18F-fluoro-l-DOPA; with this approach, direct nucleophilic 18F fluorination of a protected chiral amino acid precursor yielded the desired product at a high enantiomeric excess.
MATERIALS AND METHODS
All chemicals were purchased from Aldrich, Merck, or KMF in pro analysi quality and were used without further purification. 6-Fluoro-l-DOPA and 6-fluoro-d,l-DOPA standards were obtained from ABX.
High-performance liquid chromatography (HPLC) separations were achieved with a Sykam S1000 pump, a Knauer K2500 UV/VIS detector (254 nm), a manual Rheodyne injector (20- or 2,000-μL loop), and an NaI(Tl) well-type scintillation detector (EG&G Ortec; model 276 Photomultiplier Base) with an ACE Mate Amplifier and BIAS supply (Ortec) for radioactivity detection. Data acquisition and interpretation were performed with Nina software (Firma Nuklear Interface GmbH).
System A
Analytic HPLC of 18F-labeled compound 12 was performed with an analytic reverse-phase Kromasil 100-5 C18 column (250 × 4.6 mm; CS Chromatographieservice GmbH). Elution was performed at a constant flow rate of 1 mL/min with acetonitrile:water (80:20) (see supplemental materials [available online only at http://jnm.snmjournals.org]).
System B
Analytic HPLC of 6-18F-fluoro-l-DOPA was performed with an analytic reverse-phase Synergi 4μ Hydro-RP 80A column (250 × 4.6 mm; Phenomenex). The mobile phase was aqueous acetic acid (0.1%), and the flow rate was 1 mL/min.
System C
Preparative HPLC of 6-18F-fluoro-l-DOPA was conducted with a Synergi 4μ Hydro-RP 80A column (250 × 10 mm; Phenomenex). The mobile phase was aqueous acetic acid (0.1%), and the flow rate was 4 mL/min.
System D
The enantiomeric purity of the radiopharmaceutical was determined after HPLC separation. A Crownpak CR (+) 5μ column (150 × 4 mm; Daicel Chemical Industries) was eluted with aqueous HClO4 (0.02 M) at a flow rate of 1 mL/min, and ultraviolet light was monitored at a wavelength of 283 nm.
Thin-Layer Chromatography (TLC)
In optimization studies, TLC was performed for the determination of radiochemical yields. Aluminum roll Silica Gel 60 F254 sheets (Merck) were developed with diethyl ether:petroleum ether. The ratios of the mixtures and the corresponding Rf values are given later. Radioactivity on TLC plates was measured with an Instant Imager (Packard).
Precursor Synthesis
A description of precursor synthesis and analytic data, including a discussion, is available in the supplemental materials.
Labeling Procedure
Production of NCA 18F-Fluoride
NCA 18F-fluoride was produced by the 18O(p,n)18F nuclear reaction with the BC 1710 cyclotron (Japan Steel Works) at Forschungszentrum Jülich as described earlier (19). The 18F-fluoride produced was purified through electrostatic adsorption on a Sigradur-Anode (HTW Hochtemperatur-Werkstoffe GmbH) and desorption into 500 μL of pentadistilled water (20). This solution was added to a 130-μL (20-μmol) solution of 0.15 M tetrabutylammonium bicarbonate (TBAHCO3), and the mixture was diluted with 0.9 mL of dry acetonitrile (<0.03% water) and transferred via syringe to a 5-mL conical vial (Reactivial; Wheaton) closed with a silicon septum. The solvent was evaporated under a stream of nitrogen at 80°C and 750 mbar. This azeotropic evaporation was repeated twice with 800 μL of acetonitrile and was followed by evaporation of the dry Reactivial for 5 min at 20–30 mbar. Finally, dry tetrabutylammonium 18F-fluoride was obtained as the fluorinating agent.
CA 6-18F-Fluoro-l-DOPA (18F-14)
A solution of 5.7 mg (11.4 μmol) of (2S,5S)-tert-butyl-5-(4-(benzyloxy)-2-fluoro-5-formylbenzyl)-2-tert-butyl-3-methyl-4-oxoimidazolidine-1-carboxylate (compound 12) in 0.8 mL of N,N-dimethylformamide (DMF) was added to the freshly dried residue of tetrabutylammonium 18F-fluoride. The vial was then closed with a screw cap and heated at 110°C for 8 min in an oil bath. After labeling, the DMF solution was diluted with water (9 mL), and the entire solution was passed through a conditioned LiChrolut RP-18e (500-mg) cartridge (Merck); from this cartridge, the product (18F-12) was eluted back into the reaction vessel with 1.5 mL of acetonitrile. Analytic HPLC of the intermediate was performed with system A (k value, 3.15) or with TLC (ethyl acetate:petrol ether, 1:1; Rf, 0.78). The acetonitrile solution of (2S,5S)-tert-butyl-5-(4-(benzyloxy)-2-18F-fluoro-5-formylbenzyl)-2-tert-butyl-3-methyl-4-oxoimida-zolidine-1-carboxylate was azeotropically dried at 80°C and 800 mbar. A solution of 11 mg of meta-chloroperbenzoic acid (mCPBA; technical grade; 77%) in 1 mL of chloroform was added, and the mixture was stirred for 20 min at 60°C. The intermediate compound (18F-13) was not isolated.
After chloroform was evaporated at 60°C and 950 mbar, 1 mL of hydrobromic acid (HBr) (48%) was added to the residue. The vial was closed, and the solution was refluxed at 150°C for 30 min. After hydrolysis, the solution was cooled and diluted with 1.0 mL of HPLC eluent. Finally, 6-18F-fluoro-l-DOPA (18F-14) was purified by preparative HPLC (system C; k value for 6-18F-fluoro-l-DOPA [k value6-F-l-DOPA], 1.75). Analytic HPLC was performed with system B (k value6-F-l-DOPA, 1.62) for the determination of radiochemical yields. Enantiomeric purity was analyzed with system D (k value6-F-d-DOPA, 1.04; k value6-F-l-DOPA, 1.65).
RESULTS
The synthesis of 6-18F-fluoro-l-DOPA was performed in a manner similar to the 3-step procedure described by Tierling et al. (16): an 18F-for-19F displacement reaction and subsequent Baeyer–Villiger oxidation were followed by hydrolysis (Fig. 1).
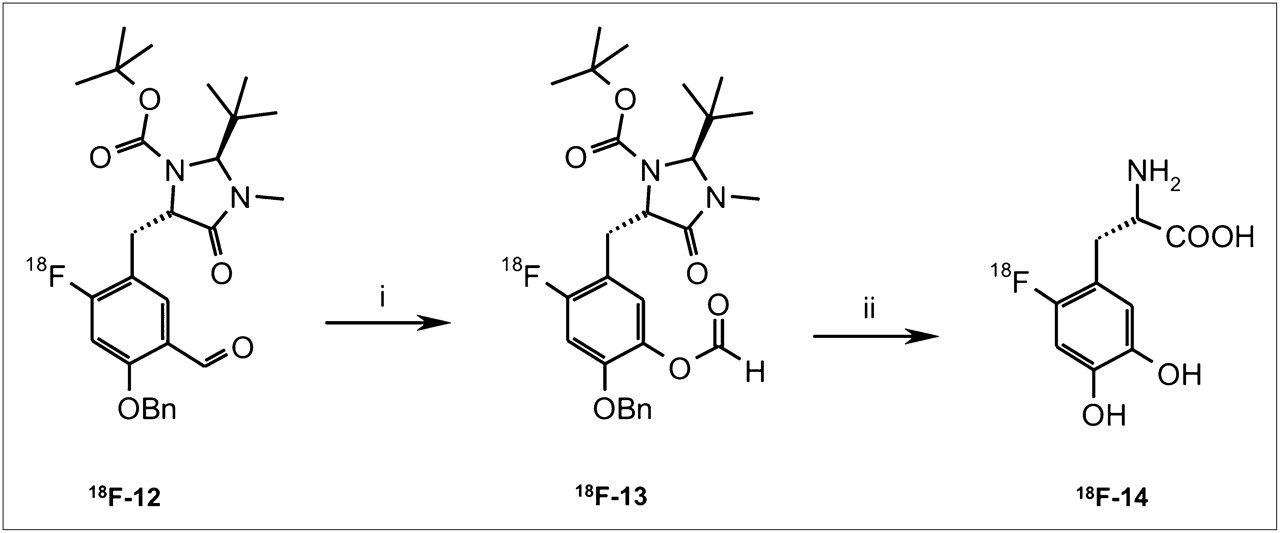
Radiosynthesis of 18F-fluoro-l-DOPA (18F-14) by Baeyer–Villiger oxidation of 18F-12 and subsequent acid hydrolysis. (i) mCPBA, CHCl3, 60°C, 20 min. (ii) HBr (47%), 150°C, 30 min.
In the first 18F exchange step, we observed the formation of an unwanted side product, which was identified as a decomposition product of the precursor. In order to avoid this product, we optimized the conditions of the radiofluorination reaction by systematically varying the anion activation phase transfer catalyst, solvent, temperature, and concentration of the precursor.
For optimization, initially different anion activators were studied. The standard Kryptofix 2.2.2 (Merck)/potassium carbonate system was examined with DMF as a solvent at 130°C to obtain information about the stability of the precursor (12) under the relatively strong basic labeling conditions. The isotopic exchange was very efficient, and after 15 min, the sum of radiochemical yields of all 18F-labeled products formed was about 90%. However, besides the desired product (18F-12), one major, as-yet-unidentified by-product (by-product A) was observed; the level of by-product A increased with the reaction time. The resulting radiochemical yield of the desired product (18F-12) was thus limited to 50%, as determined by TLC; by-product A exhibited an Rf value of 0.6 (ethyl acetate:petrol ether, 1:1), and the Rf value of 18F-12 was 0.78.
To avoid the decomposition of compound 18F-12, we also used cesium carbonate and the less basic mixed Kryptofix 2.2.2/potassium oxalate and potassium carbonate (ratio, 33:1) system (21) for 18F labeling. In both cases, no by-product was observed, but the radiochemical yield of 18F-12 was less than 10%. In the case of cesium carbonate for anion activation, the poor solubility of cesium carbonate in DMF was probably the reason for the relatively low radiochemical yield. With tetrabutylammonium hydroxide, the isotopic exchange was very efficient, but the unknown 18F-labeled by-product A became the main product. The radiochemical yield of 18F-12 amounted to only about 2%. Changing the strongly basic hydroxide anion to bicarbonate led to a better relationship between the desired product (18F-12) and by-product A, depending on the reaction time (Table 1). On the basis of these findings, all subsequent optimizations of the nucleophilic radiofluorination reactions were performed with TBAHCO3.
Radiochemical Yields of Product 18F-12 and By-Product A with Different Anion Activation Phase Transfer Catalysts
The precursor was soluble in acetonitrile, DMF, dimethylacetamide (DMAA), and dimethyl sulfoxide (DMSO). The 18F-labeling results obtained with acetonitrile were rather unsatisfactory, with a low yield, less than 1%. With DMAA and DMSO at 120°C, the radiochemical yield of the desired product (18F-12) was less than 30%, and a further by-product (by-product B) was observed (supplemental materials). As shown in Table 2, only DMF proved to be a suitable solvent for the aromatic 18F fluorination of 12, leading to good radiochemical yields and a satisfactory relationship between the product and by-product A only.
Radiochemical Yields of 18F-12 and By-Products in Different Solvents
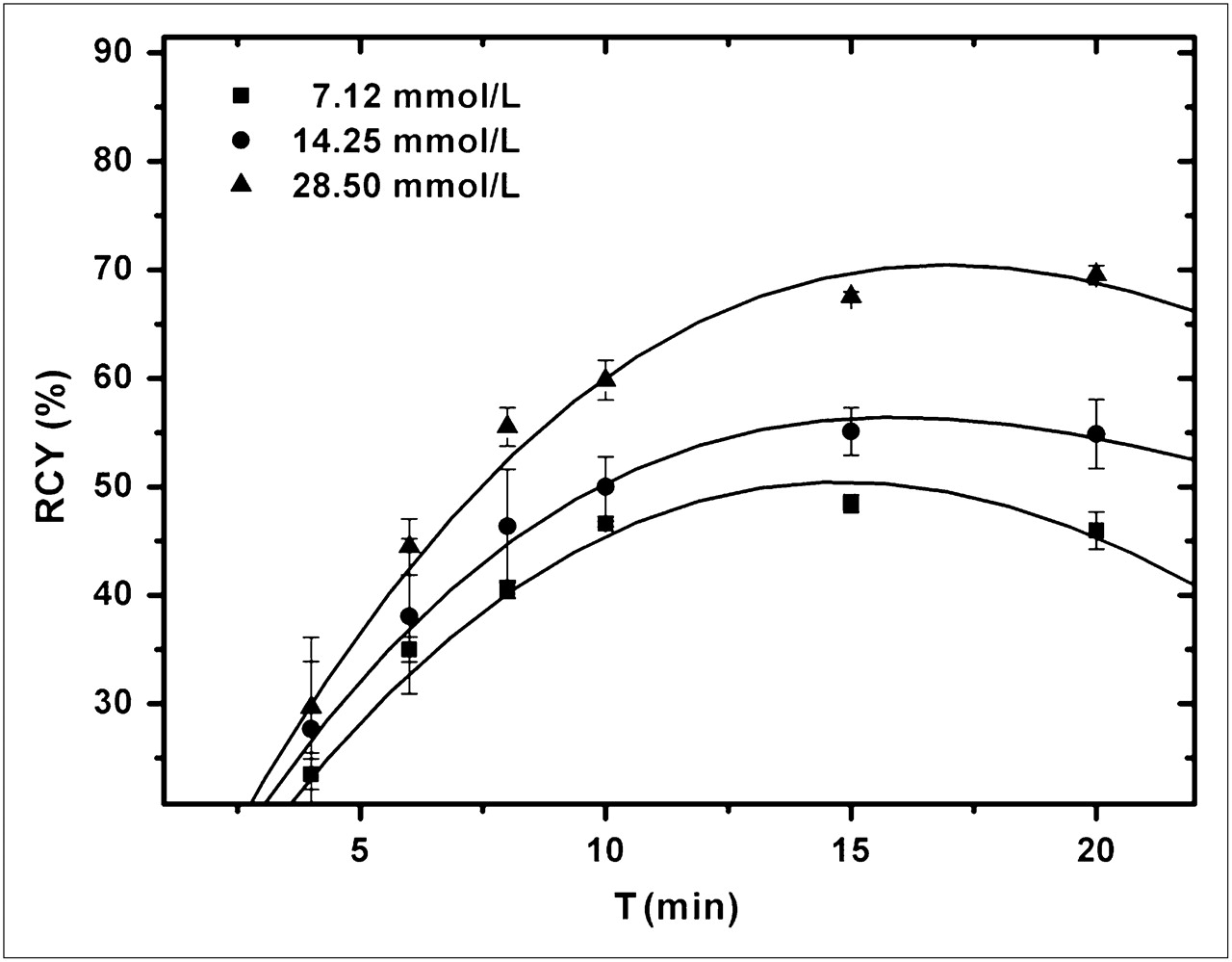
To find the limits of the useful concentration of 12, the concentration was varied between 7.12 and 28.5 mmol/L in DMF at a reaction temperature of 120°C. Figure 2 shows that radiochemical yields increased with higher concentrations, as expected, with a maximum radiochemical yield of about 65%.

Radiochemical yield (RCY) of 18F-12 as function of precursor concentration. [TBAHCO3] = 25 mmol/L, 800 μL of DMF, 120°C (n = 3), analyses by TLC. T = time.
The use of higher concentrations at 120°C did not prevent the formation of by-product A but improved the relative yield of product 18F-12; this result was caused by faster labeling than decomposition. The best results were achieved with a precursor concentration of 28.50 mmol/L at 120°C.
The use of various temperatures (100°C, 120°C, and 140°C) resulted in almost the same radiochemical yield of 18F-12, about 40%, after about 5 min of reaction time. After 15 min, however, the highest radiochemical yield of 18F-12, 65%, was achieved at 110°C (Fig. 2; Table 3).
Radiochemical Yields of 18F-12 and By-Product A with Variations in Temperature
As a result of these optimization studies, a precursor concentration of 14.25 mmol/L in 0.8 mL of DMF and a TBAHCO3 concentration of 25 mmol/L at 110°C resulted in a radiochemical yield of ∼55% of the desired product, (2S,5S)-tert-butyl-5-(4-benzyloxy-2-18F-fluoro-5-formylbenzyl)-2-tert-butyl-3-methyl-4-oxo-imidazolidine-1-carboxylate (18F-12), within 8 min of reaction time. Furthermore, under these conditions, the formation of the unwanted side product could be limited to a radiochemical yield of less than 13%. The resulting radiolabeled product was purified by solid-phase extraction to remove the phase transfer catalyst and the polar solvent.
The following Baeyer–Villiger oxidation of the formyl group and subsequent acid hydrolysis were performed in a one-pot reaction. The oxidation step was achieved with mCPBA in chloroform within 20 min at 60°C. A radiochemical yield of CA (2S,5S)-tert-butyl 5-(4-benzyloxy-2-18F-fluoro-5-(formyloxy)-benzyl)-2-tert-butyl-3-methyl-4-oxoimidazolidine-1-carboxylate (18F-13) of about 80% was achieved (16). Next, the solvent was evaporated, HBr was added, and hydrolysis was performed at 150°C for 30 min. Under these conditions, a total radiochemical yield of 6-18F-fluoro-l-DOPA of 22% was achieved within 105 min; in a standard experiment starting with 370 MBq of 18F-fluoride, approximately 37 MBq of 6-18F-fluoro-l-DOPA were obtained after 105 min. Product analysis with a chiral column (system D) revealed a high enantiomeric excess (>96%) of the final product but also a radiochemical impurity to an extent of less than 3.3% of the radiochemical yield, probably from by-product A, which was not separated by preparative HPLC (system C; supplemental materials). Thus, when this radiosynthesis procedure is scaled up on an automated system for routine production, the HPLC purification is subject to optimization.
DISCUSSION
The final radiosynthesis of 6-18F-fluoro-l-DOPA involved the incorporation of 18F-fluoride into the precursor (12), subsequent Baeyer–Villiger oxidation, and acid hydrolysis. The isotopic exchange reaction on compound 12 with NCA 18F-fluoride was the crucial step of this reaction sequence; therefore, different reaction conditions were examined. Most important was the choice of the anion activation system. The standard Kryptofix 2.2.2/potassium carbonate system, which is often used for nucleophilic aromatic 18F-substitution reactions (22), led to unsatisfactory results. A further 18F-labeled by-product (by-product A) was found in addition to the desired product (18F-12), limiting the radiochemical yield of the latter to 40%. These findings reveal the sensitivity of the precursor to the basic 18F-labeling conditions, which are necessary to perform a nucleophilic 18F exchange. For reducing the basicity of the 18F-labeling solution, other anion activation systems were studied. In particular, the use of TBAHCO3 fulfilled the requirement of a high yield in the exchange reaction and a small amount of by-product A.
The choice of the solvent for the 18F exchange reaction had an influence on the formation of further by-products. The use of DMSO or DMAA led to a second by-product that was not found when DMF or acetonitrile was used. With acetonitrile, however, the yield was very low (<1%), probably because of its low boiling point. Therefore, DMF proved to be an optimal solvent for the 18F displacement reaction.
Further, variations in temperature had a strong influence on decomposition and the formation of unwanted labeled by-product A. At temperatures of 100°C, 120°C, and 140°C, nearly the same radiochemical yield of 18F-12, about 40%, was reached after about 5 min of reaction time. Unexpectedly, at 110°C the highest radiochemical yield of 18F-12, 65%, was achieved after 15 min. When the temperature was increased to 140°C, faster isotopic exchange could be observed; however, the decomposition reaction dominated after just 4 min, leading to a radiochemical yield of about 40% for both 18F-12 and by-product A (data not shown in detail). The formation of by-product A was about 10% at 110°C and was not influenced in the temperature range up to 130°C (Table 3). In summary, a reaction time of 8 min at a temperature of 110°C appeared to be most suitable for obtaining high radiochemical yields of 18F-12 (∼65%) and relatively small amounts of radioactive by-product A (<10%).
Furthermore, at this temperature, the ratio of compound 18F-12 to the unknown side product was almost independent of the concentration of the precursor (12) in the range from 14.25 to 28.50 mmol/L. Thus, the lower concentration, 14.25 mmol/L, can be used for the 18F-labeling step, facilitating the isolation of the product. Of course, when isotopic exchange is used for labeling, the lower concentration of the precursor increases the specific activity of the final product.
After the isotopic exchange reaction, it was necessary to remove the polar solvent and the nonreacted 18F-fluoride from 18F-12 to make the following reaction sequence possible. This step was achieved by intermediate solid-phase extraction.
The Baeyer–Villiger reaction was performed under the conditions described by Tierling et al. (16) with mCPBA in chloroform and was nearly quantitative. To avoid further separation after the oxidation step, we performed the subsequent hydrolysis and deprotection steps after the evaporation of chloroform by directly adding hydroiodic acid or HBr. The reaction with hydroiodic acid was performed at 200°C for 30 min. Under these previously reported conditions (13), radiochemical yields of 6-18F-fluoro-l-DOPA (18F-14) of only 2%−5% were achieved. Changing to HBr under the same conditions led to almost the same results. However, decreasing the temperature to 170°C and 150°C increased the radiochemical yields to 5%−7% with hydroiodic acid and 22% with HBr, respectively. The reason was probably the excess of mCPBA, which oxidized hydroiodic acid and HBr to elemental iodine and bromine, respectively, under the harsh conditions used. In addition, many side reactions reduced the radiochemical yield of 6-18F-fluoro-l-DOPA (18F-14). The use of lower temperatures could prevent these side reactions to some extent.
In the preparation of 6-18F-fluoro-l-DOPA (18F-14), the maximum amount of carrier that can be expected from a starting quantity of precursor 12 of 11.5 μmol is less than 2 mg. This amount is in the lower range for electrophilic methods, in which up to 15 mg of 6-fluoro-l-DOPA are obtained and tolerated in tracer preparations for human injection (6). Although in production runs of electrophilic radiofluorine a specific activity of only about 0.3 GBq/μmol can be reached under optimal conditions (23), a value at least 5-fold higher can be attained with the isotopic exchange procedure, especially with the much larger amounts of 18F-fluoride activity that can be generated in one irradiation batch.
The parameters of various methods for the synthesis of 6-18F-fluoro-l-DOPA (18F-14) are summarized in Table 4. Compared with the electrophilic method, all nucleophilic procedures have more synthesis steps and thus are more time-consuming. The general advantage of nucleophilic 18F introduction is the possibility of a much higher starting activity. Until now, the nucleophilic approach of Lemaire et al. (14) has been the method of choice for obtaining 6-18F-fluoro-l-DOPA (18F-14) at the NCA level. Nevertheless, the number of steps involved prevents easy integration of the synthesis sequence into available standard commercial modules. The method of Tierling et al. (16) reduces the synthesis steps to only 3; however, the low enantiomeric excess of 6-18F-fluoro-l-DOPA (18F-14) would demand an additional chiral HPLC separation. Based on the same 18F-labeling concept, the new precursor described here (12) fulfils the requirements of efficient nucleophilic synthesis of 6-18F-fluoro-l-DOPA at a high enantiomeric excess.
Comparison of Methods for Synthesizing 6-18F-Fluoro-l-DOPA
CONCLUSION
The new precursor, (2S,5S)-tert-butyl-5-(4-benzyloxy-2-fluoro-5-formylbenzyl)-2-tert-butyl-3-methyl-4-oxoimidazolidine-1-carboxylate (12), enables the radiosynthesis of 6-18F-fluoro-l-DOPA by an isotopic exchange reaction with 18F-fluoride. After only 2 further radiosynthesis steps, namely, Baeyer–Villiger oxidation with mCPBA followed by hydrolysis with HBr, the desired l-isomer of CA 6-18F-fluoro-DOPA was isolated at an enantiomeric purity of more than 99%. The complete preparation and isolation of CA 6-18F-fluoro-l-DOPA under the conditions optimized thus far resulted in a radiochemical yield of about 22% after a synthesis time of 105 min. With other methods for the synthesis of 6-18F-fluoro-l-DOPA starting from 18F-fluoride, however, enantiomeric purity is insufficient or the necessary multistep syntheses are difficult to automate because of their complexity. Thus, the nucleophilic synthetic pathway to 6-18F-fluoro-l-DOPA described here not only is more efficient than known nucleophilic methods but also can be performed as a one-pot reaction. As a result, it is capable of being scaled up in existing automated synthesizer modules to offer reliable, large-scale production of 6-18F-fluoro-l-DOPA.
Acknowledgments
We thank Johnny Castillo Meléan for assistance, Kurt Hamacher and Marcus H. Holschbach (at INM-5, Forschungszentrum Jülich) for helpful discussions, Dr. Holschbach also for nuclear magnetic resonance measurements of organic compounds, and Sabine Willbold and Diana Hofmann (both at ZCH, Forschungszentrum Jülich) for assistance in the interpretation of the spectra and for recording high-resolution mass spectrometry.
Footnotes
-
COPYRIGHT © 2009 by the Society of Nuclear Medicine, Inc.
References
- Received for publication February 13, 2009.
- Accepted for publication July 10, 2009.





{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Iodonium Ylide-Mediated Radiofluorination of 18F-FPEB and Validation for Human Use
- In Vivo Biodistribution of No-Carrier-Added 6-18F-Fluoro-3,4-Dihydroxy-L-Phenylalanine (18F-DOPA), Produced by a New Nucleophilic Substitution Approach, Compared with Carrier-Added 18F-DOPA, Prepared by Conventional Electrophilic Substitution
- Production at the Curie Level of No-Carrier-Added 6-18F-Fluoro-L-Dopa