


Abstract
Translation of new methodologies for labeling nonactivated aromatic molecules with 18F remains a challenge. Here, we report a one-step, regioselective, metal-free 18F-labeling method that uses a hypervalent iodonium(III) ylide precursor, to prepare the radiopharmaceutical 18F-3-fluoro-5-[(pyridin-3-yl)ethynyl]benzonitrile (18F-FPEB). Methods: Automated radiosynthesis of 18F-FPEB was achieved by reaction of the ylide precursor (4 mg) with 18F-Et4NF in dimethylformamide at 80°C for 5 min and formulated for injection within 1 h. Results: 18F-FPEB was synthesized in 20% ± 5% (n = 3) uncorrected radiochemical yields relative to 18F-fluoride, with specific activities of 666 ± 51.8 GBq (18 ± 1.4 Ci)/μmol at the end of synthesis and was validated for human use. Conclusion: Radiofluorination of iodonium (III) ylides proved to be an efficient radiosynthetic strategy for synthesis of 18F-labeled radiopharmaceuticals.
Historically, the formation of aromatic C–F bonds has been challenging in the field of synthetic organic chemistry, and even more so in radiochemistry, with the short-lived radionuclide 18F (half-life, 109.7 min) for molecular imaging by PET. Electrophilic fluorination reactions with carrier-added 18F-F2 gas and its derivatives (e.g., 18F-CH3CO2F) have enabled the development of 18F-labeled aromatics by direct electrophilic substitution or demetalation reactions with organometallic reagents such as aryl stannanes (1,2). Electrophilic radiosynthesis with 18F-F2 and its derivatives involves the use of carrier-added fluorine gas and consequently results in products with relatively low specific activities. Such reactions also require specialized equipment as well as technical expertise for the safe handling of F2(g). Commercial availability of high-specific-activity, no-carrier-added 18F-fluoride has led to this reagent becoming the most widely used radiofluorinating species. Aromatic molecules with 18F-fluoride are typically synthesized by nucleophilic aromatic substitution reactions with electron-deficient (activated) aromatics, and these reactions have been used extensively to prepare high-specific-activity radiopharmaceuticals (3). However, labeling of electron-rich (nonactivated or deactivated) aromatics with 18F-fluoride remains a long-standing and unmet challenge in routine PET radiopharmaceutical production.
Early efforts to expand the scope of reactions of electron-rich aromatics with 18F-fluoride include inefficient and low-yielding thermal decomposition processes such as the Balz–Schiemann and Wallach reactions (3). Isotopic exchange via 18F-for-19F displacement reactions can be used to label electron-rich aromatics and have been applied to prepare low-specific-activity radiotracers (3,4). Strategies that convert electron-withdrawing aryl substituents to electron-donating groups after radiofluorination (e.g., reduction of fluoronitrobenzene or fluorobenzaldehydes and decarbonylation reactions) are useful to access electron-rich 18F-fluoroaromatics in high specific activity but are known to have limited chemical scope, involve multistep labeling approaches, and are challenging to adapt to radiopharmaceutical production. Recent strategies for reactions of 18F-fluoride with electron-rich aromatics include the development of transition metal-mediated reactions with isolable aryl palladium or nickel complexes, copper-mediated fluorinations with aryl borate esters, oxidative fluorination with phenolic substrates, and fluorination of diaryliodonium or sulfonium salts as well as diarylsulfoxides, and most of these methods have been recently reviewed (5). These newer methods appear promising for preparing radiolabeled aromatics. However, drawbacks include the use of air-sensitive or toxic transition metals, limited substrate scope, or poor regioselectivity. Furthermore, radiotracer syntheses by the above-mentioned methods generally result in low isolated radiochemical yields. Such limitations have hampered the use of these alternative labeling reactions for clinical translation. To our knowledge, none of these methodologies have been applied to radiolabel a nonactivated aromatic ring with 18F-fluoride for synthesis of a radiopharmaceutical and validate it for human use.
Nonactivated aromatics can be radiofluorinated with aryliodonium ylide–based precursors bearing various β-dicarbonyl auxiliaries (6). This technology was introduced by Satyamurthy and Barrio using barbituric or Meldrum acid auxiliaries (7). A recent study by Cardinale et al. demonstrated the satisfactory application of this method using Meldrum acid auxiliaries but led to nonregiospecific labeling and formation of several byproducts (8). Concurrently, we explored the application of iodonium ylide precursors with barbituric acid and Meldrum acid auxiliaries for 18F-labeling but also found yields to be suboptimal (9). We discovered that spirocyclic iodonium(III) ylide precursors, in conjunction with systematic optimization of the reaction conditions, led to efficient, regiospecific, 1-step radiosyntheses of nonactivated aromatics with 18F-fluoride (9). These bench-stable precursors involved facile synthesis and demonstrated a broad substrate scope for nonactivated 18F-aromatics including hindered alkyl substituents, benzyl azides, anisoles, amides, heterocycles, and halogenated aromatics. The radiofluorination is operationally simple and was shown to be suitable for routine and automated production of 18F-labeled aromatics. The conceptual advantages of excellent regioselectivity and viability of incorporating 18F into a wide array of nonactivated (hetero)arenes makes this methodology attractive for routine radiopharmaceutical production.
18F-3-fluoro-5-[(pyridin-3-yl)ethynyl]benzonitrile (18F-FPEB) is a metabotropic glutamate receptor subtype 5 (mGlu5) antagonist used in preclinical (10,11) and clinical PET neuroimaging research (12,13). Radiosynthesis of 18F-FPEB and several structurally related radiotracers for mGlu5 has been challenging as nucleophilic aromatic substitution by 18F-fluoride is not a favored reaction because of the absence of electron-withdrawing groups located ortho or para to the labeling site. High temperatures are generally required, and several chemical impurities are generated during the labeling reactions. Isolated 18F-FPEB is typically obtained in low radiochemical yields (1%−5% uncorrected yield relative to 18F-fluoride) (10,14–16).

Manual optimization of 18F-FPEB radiosynthesis based on crude reaction mixtures. Conditions: precursor 1 (2 mg), Et4NHCO3, DMF (400 μL). †Incorporation: percentage radiochemical conversion as determined by radio–thin-layer chromatography (n = 3). Product identity was confirmed by coinjections of authentic 1 and 2 via radio–high-performance liquid chromatography.
The goals of the present work were to exploit our new spirocyclic iodonium(III) ylide precursor technology to develop a high-yield radiosynthesis of 18F-FPEB and to demonstrate that this methodology is suitable for routine radiopharmaceutical production.
MATERIALS AND METHODS
Full details of precursor synthesis and characterization, radioisotope production, analytic methods, spectra, and radiosynthesis with a TRACERlab FXFN radiosynthesis module (GE Healthcare), as well as human validation data, are provided in the supplemental materials (available at http://jnm.snmjournals.org).
Manual and Automated Radiosynthesis of 18F-FPEB
Precursor (1, 2 or 4 mg) was dissolved in N,N-dimethylformamide (DMF, 400 μL) and added to a glass V-vial containing azeotropically dried 18F-Et4NF (typically 37–111 MBq [1–3 mCi]). The reaction was heated at 80°C for 5 min. The reaction mixture was cooled for 3 min and then quenched with a 60:40 CH3CN:H2O + 0.1N ammonium formate solution (mobile phase; 2 mL). The reaction was further diluted with water (16 mL) and passed through a preactivated (ethanol [1 mL] and water [5 mL]) solid-phase extraction (C18 Sep-Pak; Waters) cartridge. The solid-phase extraction cartridge was flushed with water (2 mL), and the product was eluted with ethanol (1 mL). Product identity and purity were confirmed by radio–high-performance liquid chromatography and radio–thin-layer chromatography (100% EtOAc). The product was more than 99% radiochemically pure. Radiochemical yield was determined as the percentage of radioactivity that was isolated as the final product from the amount of activity present in the V-vial before addition of iodonium precursor to dried 18F-Et4NF and was not decay-corrected.
Automated synthesis of 18F-FPEB was performed on a TRACERlab FXFN radiosynthesis module. The final product was formulated and found suitable for injection in compliance with the quality control protocols and guidelines of the International Conference of Harmonization of Technical Requirement of Pharmaceuticals for Human Use (supplemental materials).
RESULTS
The iodonium ylide precursor, 1, was synthesized in 6 steps starting with 4-amino-3,5-diiodobenzoic acid. A Sandmeyer reaction furnished 3,5-diiodobenzoic acid, which was converted to the nitrile via acid chloride formation, amidation, and dehydration. Sonogashira coupling with 3-ethynylpyridine provided the aryl iodide (IPEB). We anticipated that oxidation of the iodine could be problematic in the presence of a pyridine functional group, which is known to undergo efficient transformation to the pyridine N-oxide under conditions analogous to those used in 3-iodo-5-(pyridin-2-ylethynyl)benzonitrile. Oxidants such as m-chloroperoxybenzoic acid and H2O2/urea in acetic acid resulted in complex mixtures of products. To our delight, Oxone (DuPont) in trifluoroacetic acid was successfully used to oxidize IPEB, partially attributed to a protonation event on nitrogen of pyridine, which prevented the formation of N-oxide. Removal of trifluoroacetic acid, resolubilization in ethanol, treatment with the auxiliary in aqueous sodium carbonate, and subsequent purification with silica gel chromatography provided 1 in 40% yield (Fig. 1).
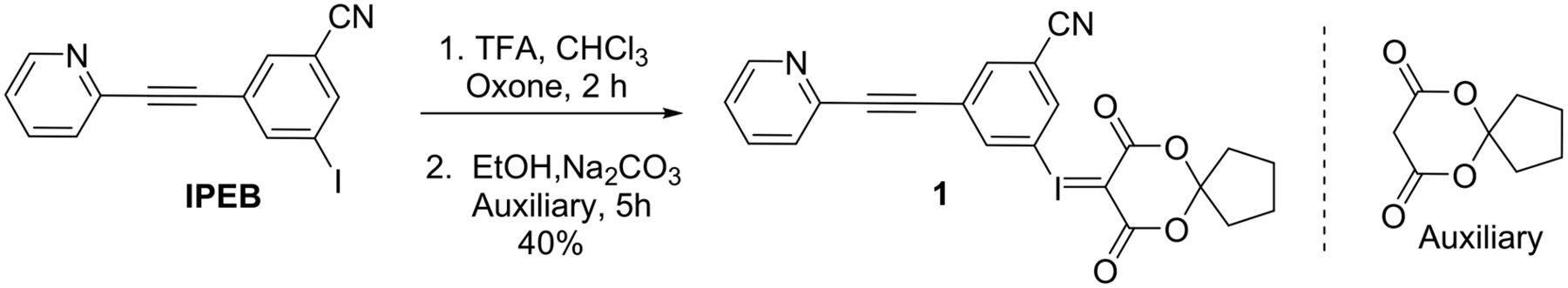
Synthesis of iodonium ylide precursor (1) for radiofluorination. IPEB = 3-iodo-5-(pyridin-2-ylethynyl)benzonitrile; TFA = trifluoroacetic acid.
Initial efforts toward the radiosynthesis of 18F-FPEB using precursor 1 (2 mg) were performed manually with 37–111 MBq (1–3 mCi) of starting 18F-fluoride. Using our previously optimized radiolabeling conditions for these reactions (9), with Et4NHCO3 (7 mg) in DMF (400 μL) at 120°C, only 6% radiochemical conversion (via radio–thin-layer chromatography) of 18F-fluoride to 18F-FPEB was attained, along with formation of a radioactive byproduct (2; see below) in 18% radiochemical conversion (Scheme 1, entry I). Monitoring the reaction over time indicated that 18F-FPEB formed with high conversions during the initial 3 min of the reaction and subsequently decomposed. Control reactions with nonradioactive FPEB demonstrated that the compound was stable in DMF at 120°C. However, in the presence of an excess (2 equivalents) of Et4NHCO3 at the same temperature, FPEB underwent a rapid, quantitative base-mediated hydrolysis of the nitrile group to form 3-fluoro-5-(pyridin-2-ylethynyl)benzamide, 2. To suppress amide formation, the concentration of Et4NHCO3 was reduced from 90 to 40 mM. Reduced base concentration resulted in increased 18F-fluoride incorporation into products and favored distribution between 18F-FPEB and byproduct 2. Increased radiochemical conversion from 6% to 27% for 18F-FPEB was observed, and formation of byproduct 2 decreased to 12% (Scheme 1, entry II). Furthermore, the temperature was lowered to 80°C and the reaction time was reduced to 5 min, affording 18F-FPEB in excellent radiochemical conversion of 49% ± 6% (n = 3; Scheme 1, entry III).
In light of this promising result—automated radiosynthesis of 18F-FPEB—validation was subsequently performed to demonstrate the utility of the iodonium(III) ylide precursor for clinical translation. Three consecutive productions of 18F-FPEB were isolated with more than 7,400 MBq (200 mCi) at the end of synthesis and formulated for injection within 1 h. Analysis of the formulated product (10% ethanol in 0.9% sodium chloride) by high-performance liquid chromatography showed high specific activity (666 ± 51.8 GBq/μmol [18 ± 1.4 Ci/μmol]) as well high radiochemical purity (≥99%) and chemical purity (≥98%). Validation via an established quality control protocol (16) demonstrated that 18F-FPEB synthesized from iodonium ylide precursor 1 is suitable for human injection (the supplemental materials present full validation data).
DISCUSSION
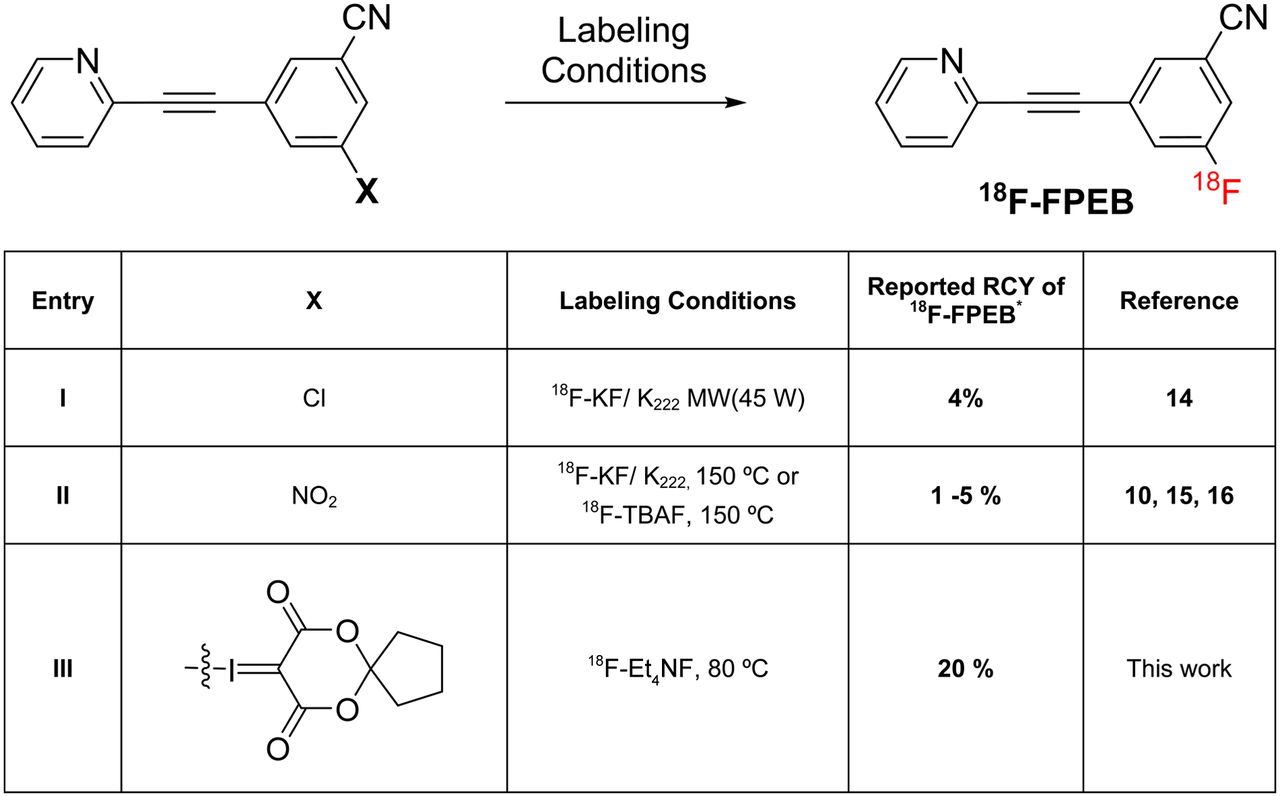
Radiosynthesis of 18F-FPEB is low yielding by most traditional nucleophilic aromatic substitution reactions (<5% radiochemical yields), because nucleophilic displacement of common leaving groups (e.g., Cl, Br, or NO2) by 18F-fluoride is not favored when the electron-withdrawing group, that is, nitrile, is at the meta position. Harsh conditions, including high temperatures and prolonged reaction times, are generally required and several chemical and radiochemical impurities are usually generated during these reactions, thereby complicating purification. The original radiosynthesis of 18F-FPEB used a chlorinated precursor (Scheme 2, entry I) (14). We and other laboratories (10,15,16) have validated a reproducible radiosynthesis of 18F-FPEB via 3-nitro-5-(pyridin-2-ylethynyl)benzonitrile (Scheme 2, entry II), which resulted in 1%–5% radiochemical yield for clinical research studies. Notably, our efforts to further optimize the radiochemical yield of 18F-FPEB by use of the nitro-precursor in the presence of reduced base concentrations still required high temperatures to proceed (∼150°C) and continued to yield a problematic 18F-labeled hydrolysis product as well as chemical byproducts that are difficult to separate (comparison of semipreparative high-performance liquid chromatograms is provided in the supplemental materials). Use of the bromo-precursor or use of microfluidic technologies demonstrated that the radiotracer could be prepared suitably for human use by conventional radiofluorination or flow chemistry, albeit without an increase in isolated radiochemical yield or simplified purification (16). In the present work, attempts to prepare a trimethyl ammonium triflate precursor (17) proved to be a chemical challenge and consistently led to the formation of an undesired methyl pyridinium salt, as predicted to be the thermodynamically favored product (supplemental materials). The spirocyclic iodonium ylide (1) was explored as a novel precursor for 18F-FPEB on the basis of our recent demonstration of the viability of this strategy for radiolabeling a wide range of compounds (Scheme 1, entry III) (9).

Comparison of 18F-FPEB production yields for clinical research. *Non–decay-corrected radiochemical yield at end of synthesis relative to starting 18F-fluoride.
Radiofluorination of nonsymmetric diaryliodonium compounds is believed to involve a distinct mechanistic pathway compared with traditional nucleophilic aromatic substitution–type reactions, that is, 18F-fluoride capture followed by reductive elimination to produce 18F-labeled aromatics with C−18F bond formation occurring at the more electron-deficient substituent (18). Diaryliodonium precursors for structurally related mGlu5 radiotracers have been radiolabeled (19) but were not pursued herein because of the lack of regioselectivity. Nonetheless, diaryliodonium salts have been widely used for preclinical studies and were recently shown to be suitable for human use with an activated (electron-deficient) aromatic precursor of 18F-flumazenil (20). Unlike diaryliodonium salts, which rely on an aryl auxiliary, diaryliodonium ylides use an electron-rich β-dicarbonyl auxiliary, resulting in a selective C−18F bond formation and increased chemical stability (9). Iodonium ylides were easily purified by silica flash chromatography, which is a challenge with diaryliodonium salts, and are bench-stable compounds at room temperature.
A high-yield radiosynthesis of 18F-FPEB, via the iodonium ylide–based precursor 1, resulted in 20% ± 5% (n = 3) radiochemical yield ready for injection (Scheme 2, entry III) and represents a 10-fold increase over our previous methodology based on the NO2 precursor (16). The present method is achieved (ready for injection) in 60 min, compared with 90 min by our previous method, with more than a 2-fold increase in specific activity (18 Ci/μmol). The production was easily automated and validated for routine radiopharmaceuticals, passing our quality control protocol (16). A reduced base concentration and temperature was able to suppress formation of an amide impurity (2), identified as a major byproduct formed by base-promoted hydrolysis of 18F-FPEB (Scheme 1). Compound 1 has been stored at room temperature for 2 mo and has not shown signs of decomposition by structural characterization and chromatography and has not lost labeling efficiency when reacted with 18F-fluoride. The radiochemical methodology demonstrated herein should prove to be widely applicable to several diagnostic PET imaging agents for mGlu5 that share a similar structural scaffold to 18F-FPEB (13).
CONCLUSION
The use of a spirocyclic hypervalent iodine(III)-mediated radiofluorination was shown to provide a high-yielding synthesis of the nonactivated aromatic ring of 18F-FPEB and was validated for human imaging studies. A 10-fold increase in radiochemical yield (20%, non–decay-corrected) and more than a 2-fold increase in specific activity (666 GBq [18 Ci]/μmol) compared with our established clinical production procedure was achieved. The methodology described herein not only should facilitate widespread preclinical and clinical use of 18F-FPEB but also represents the utility of iodonium ylides as a viable strategy for the practical radiofluorination of nonactivated aromatics with 18F-fluoride and is suitable for human use.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18-USC section 1734. We thank the Alzheimer’s Drug Discovery Foundation as well as Dr. Ivan Greguric and the Australian National Science and Technology Organisation for financial support. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Drs. T. Lee Collier and Benjamin Rotstein for helpful discussions, as well as Din Avdic, Peter Rice, Raul Jackson, and Erin Gomes for technical assistance. We thank Dr. Jack A. Correia and David F. Lee, Jr., and the Massachusetts General Hospital PET Core facility for 18F-fluoride production.
Footnotes
Published online Feb. 5, 2015.
- © 2015 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication November 10, 2014.
- Accepted for publication January 5, 2015.





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Understanding Exposure-Receptor Occupancy Relationships for Metabotropic Glutamate Receptor 5 Negative Allosteric Modulators across a Range of Preclinical and Clinical Studies
- Recent Advances in 18F Radiochemistry: A Focus on B-18F, Si-18F, Al-18F, and C-18F Radiofluorination via Spirocyclic Iodonium Ylides