


Abstract
Recent studies have shown that indirect effects of ionizing radiation may contribute significantly to the effectiveness of radiotherapy by sterilizing malignant cells that are not directly hit by the radiation. However, there have been few investigations of the importance of indirect effects in targeted radionuclide treatment. Our purpose was to compare the induction of bystander effects by external beam γ-radiation with those resultant from exposure to 3 radiohaloanalogs of metaiodobenzylguanidine (MIBG): 131I-MIBG (low-linear-energy-transfer [LET] β-emitter), 123I-MIBG (potentially high-LET Auger electron emitter), and meta-211At-astatobenzylguanidine (211At-MABG) (high-LET α-emitter). Methods: Two human tumor cell lines—UVW (glioma) and EJ138 (transitional cell carcinoma of bladder)—were transfected with the noradrenaline transporter (NAT) gene to enable active uptake of MIBG. Medium from cells that accumulated the radiopharmaceuticals or were treated with external beam radiation was transferred to cells that had not been exposed to radioactivity, and clonogenic survival was determined in donor and recipient cultures. Results: Over the dose range 0–9 Gy of external beam radiation of donor cells, 2 Gy caused 30%–40% clonogenic cell kill in recipient cultures. This potency was maintained but not increased by higher dosage. In contrast, no corresponding saturation of bystander cell kill was observed after treatment with a range of activity concentrations of 131I-MIBG, which resulted in up to 97% death of donor cells. Cellular uptake of 123I-MIBG and 211At-MABG induced increasing recipient cell kill up to levels that resulted in direct kill of 35%–70% of clonogens. Thereafter, the administration of higher activity concentrations of these high-LET emitters was inversely related to the kill of recipient cells. Over the range of activity concentrations examined, neither direct nor indirect kill was observed in cultures of cells not expressing the NAT and, thus, incapable of active uptake of MIBG. Conclusion: Potent toxins are generated specifically by cells that concentrate radiohalogenated MIBG. These may be LET dependent and distinct from those elicited by conventional radiotherapy.
Radiation-induced biologic bystander effects (RIBBEs) derive from the cellular processing of the physical radiation insult, which need not interact directly with DNA, into factors that cause damage to neighboring unirradiated cells (1–6). The outcome may be cell death, mutation, chromosomal aberrations, or long-term genomic instability. Only recently has the potential importance of bystander phenomena after irradiation for cancer treatment been widely appreciated.
As with drug therapy, the main limitation to external beam radiation treatment is damage to normal tissues. For the management of some tumor types, this problem may be overcome by targeted radiotherapy: the administration of radionuclides conjugated to molecules that are concentrated specifically in malignant cells. One such tumor-targeting molecule is metaiodobenzylguanidine (MIBG), an analog of adrenergic neuron blockers. The radiolabeled drug 131I-MIBG is selectively concentrated in neuroendocrine tumors via the noradrenaline transporter (NAT), resulting in selective irradiation of the target tumor cells with relative sparing of normal tissues not expressing NAT. Radioiodinated MIBG is used for the imaging and treatment of tumors arising from the neural crest (7,8). It has provided good remissions and palliation, though long-term remedy remains elusive (9–12).
Induction of RIBBEs is prevalent at low radiation dose and low dose rate (13,14), features that are characteristic of targeted radionuclide treatment of cancer. Therefore, bystander effects induced by targeted radionuclides could have a strong impact on therapeutic efficacy, as well as radiation dosimetry and protection, and should be considered in the design of radiotherapy protocols. To date, the majority of studies of bystander effects have involved external radiation sources that produce microbeams or γ-rays. In contrast, there is little knowledge of the RIBBE derived from targeted radionuclides of different radiation qualities. The issue of bystander effects is particularly relevant to the consequences of nonuniform distribution of radioactivity that impacts on risk estimation and clinical outcome of targeted radionuclide therapy.
Previous studies with labeled compounds as the radiation source have demonstrated bystander effects for 3H-thymidine–labeled cells in 3-dimensional tissue culture models (15) and for 5-125I-iodo-2′-deoxyuridine (125I-IUdR) in an in vivo tumor model (16). Further, we have previously demonstrated sterilization of multicellular tumor spheroids containing only 5% NAT-transfected cells exposed to meta-211At-astatobenzylguanidine (211At-MABG) or 131I-MIBG (17). These studies indicate the potential for bystander-mediated cell kill and improved clinical efficacy of tumor targeting when only a small proportion of the tumor mass expresses the radiotherapeutic molecular target, in this case, introduced via gene modification. RIBBEs could compensate for the low levels of gene delivery currently achievable in vivo in cancer gene therapy strategies when married with targeted radionuclide therapy. However, the magnitude of bystander kill and the underlying mechanisms remain unidentified. Likewise, the dependence on radiation quality is unknown.
In the current study, we have applied a media transfer protocol, similar to that described for the investigation of indirect effects of external beam irradiation (18) but amended to enable the evaluation of RIBBE resulting from the cellular accumulation of radionuclides. We determined direct and indirect clonogenic cell kill after exposure of a human glioma cell line (UVW) and a cell line derived from a human transitional cell carcinoma of bladder (EJ138), both transfected with the NAT gene, to β-particle–emitting 131I-MIBG, α-particle–emitting 211At-MABG, and Auger electron–emitting 123I-MIBG. Results were compared with those obtained after exposure to 60Co γ-rays.
MATERIALS AND METHODS
Plasmids
The bovine NAT (bNAT) complementary DNA (cDNA) was kindly provided by Dr. Michael Bruss and Prof. Heinz Bonisch (University of Bonn, Bonn, Germany). Recombinant plasmids, containing the bNAT gene under control of the strong viral promoter cytomegalovirus (CMV) for transfection into UVW glioma cells or the human RNA component telomerase promoter hTR for transfection into EJ138 cells, were constructed as previously described (19). Plasmid purification was performed using a Plasmid Maxi Kit (Qiagen Ltd.) according to the manufacturer's instructions.
Cell Lines
The transgene hosts were the UVW human glioma cell line (17) and the EJ138 human bladder carcinoma cell line (19). All tissue culture reagents were obtained from Gibco/Invitrogen Ltd. UVW cells were maintained in 75-cm2 flasks containing Eagle's minimum essential medium (MEM) with 10% (v/v) fetal bovine serum, penicillin/streptomycin (100 U/mL), fungizone (2 μg/mL), and l-glutamine (2 mmol/L). EJ138 cells were maintained in 75-cm2 flasks containing Eagle's MEM with 25 mmol/L N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) buffer and Earle's Salt, supplemented with 10% (v/v) fetal bovine serum, penicillin/streptomycin (100 U/mL), fungizone (2 μg/mL), l-glutamine (2 mmol/L), and nonessential amino acids (0.1 mmol/L). Cells were cultured at 37°C in a 5% CO2 atmosphere.
UVW and EJ138 cells were stably transfected with plasmids containing bNAT cDNA under control of the CMV (UVW/NAT) and hTR (EJ138/NAT) promoters using the Effectine transfection reagent (Qiagen Ltd.). These promoters were chosen for the respective cell lines because previous studies have shown that they enabled greater MIBG uptake than alternative promoter elements (19). Stable transfectants were not further selected into clones derived from single transfected cells. This provided a model representative of the heterogeneity of transgene expression expected after in vivo gene delivery. The transfectants were maintained by geneticin (G418) treatment at every passage. Stably transfected cell populations were obtained approximately 4 wk after gene transfer.
Radiopharmaceuticals
No-carrier-added 131I-MIBG and 123I-MIBG were synthesized by iododestannylation of tributylstannlybenzylguanidine (20) using a solid-phase system where the tin precursor was attached to an insoluble polymer via the tin-aryl bond (21). No-carrier-added 211At-MABG was synthesized as previously described (22).
Radiopharmaceutical Uptake
Transfectants were assessed for 131I-MIBG uptake as previously described (23). Briefly, monolayers were prepared by seeding the appropriate numbers of cells in 6-well plates at an initial density of 0.5 × 105 cells per well and culturing for 48 h. MIBG incorporation was measured by incubating the cells for 2 h at 37°C in complete medium with a range of activity concentrations of 131I-MIBG. Nonspecific uptake was measured in the presence of 1.5 mmol/L desmethylimipramine (DMI) (Sigma-Aldrich), a specific inhibitor of NAT. After incubation, medium was removed, the cells were washed with phosphate-buffered saline (PBS), and radioactivity was extracted using 2 aliquots of 10% (w/v) trichloroacetic acid. The activities of the extracts were then measured in a γ-counter (Cobra II Auto-γ-Counting System; Packard Instrument Co.).
Media Transfer Assay: 60Co γ-Irradiated Cells
This technique has been described in detail (18). UVW/NAT and EJ138/NAT cells were seeded in 10 mL complete medium in 25-cm2 flasks (Nunc Plastics), at 2 × 105 cells per flask. The cells were irradiated, at room temperature, 24 h later when the cultures were 60%–70% confluent. Three sets of test cultures were prepared. These were designated donor cells (donor), which were directly irradiated but had the medium transferred to the recipient cells (recipient), which were not directly irradiated but received medium from irradiated cells, and cells (direct), which were directly irradiated and from which the medium was not removed. Consequently the direct cells experienced the physical and biologic effects of the radiation treatment. Controls for the transfer of medium only and for the transfer of irradiated medium only were included in each experiment.
On the day of the irradiation, the medium was removed from all cells and replaced with 5 mL fresh medium. Irradiation at a dose rate of 0.25 Gy/min (the output from a 60Co irradiator) was performed at room temperature. Donor and direct cultures were irradiated at a range of doses from 0 to 9 Gy. Immediately after irradiation, all cells were returned to the incubator, maintained at 37°C and 5% CO2. One hour later, regardless of dose, the medium was removed from the recipient cells and replaced with the medium from the donor cells after having been passed through a 0.22-μm filter to ensure that no cell was present in the transferred medium (3,18). One hour after irradiation was the period chosen for the transfer of medium because this corresponds to a time point analyzed by others (18).
Thereafter 5 mL of fresh medium were added to the donor cells. The direct cells were not subjected to such manipulation. All cultures were equilibrated with 5% CO2 and incubated for 24 h at 37°C. Thereafter the cells from each culture were removed by treatment with a 0.05% (w/v) solution of trysin-ethylenediaminetetraacetic acid in PBS (Gibco/Invitrogen Ltd.), counted using a hemocytometer, and seeded for clonogenic assay in triplicate at 2.5 × 103, 5 × 103, and 7.5 × 103 cells per 35-mm Petri dish (Nunc Plastics). The cells were incubated at 37°C and, after 10–14 d, colonies were fixed in methanol/PBS (50:50) and stained with 10% Gram's crystal violet solution (BDH Laboratories). Clusters containing 50 or more cells were scored as colonies. Surviving fraction (SF) was determined by dividing the plating efficiencies (PE, defined as the number of colonies counted divided by the number of cells seeded) of the treated cultures by the PE for the untreated cultures. Each experiment was performed 6 times in triplicate for each cell line.
To control for the irradiation procedure, a culture of control donor cells was sham-irradiated and its medium was transferred to corresponding recipient cells. Controls for the transfer of medium only and for the transfer of irradiated medium only were also included in each experiment.
Media Transfer Experiments: Radiopharmaceutical-Treated Cells
Cells were seeded into 25-cm2 flasks as described for the γ-irradiation experiments. Five sets of cultures were prepared for each cell line. Those designated donor cells were incubated with radiopharmaceutical before removal of the medium and transfer to recipient cultures. Recipient cells were not directly irradiated but received medium from radiopharmaceutical-treated cells. Direct cells were incubated with radiopharmaceutical and no medium was removed from these cells. On the day of exposure to the labeled compounds, medium was removed from cultures and replaced with 1 mL fresh medium. Radiopharmaceutical was added to the donor and direct cultures. The activity concentrations, based on our previous experiments with these radiopharmaceuticals (17,19,22–24), ranged from 0 to 11 MBq/mL (131I-MIBG and 123I-MIBG) and from 0 to 45 kBq/mL (211At-MABG). All cells, whether treated with radiopharmaceutical or not treated, were incubated at 37°C. After 2 h, medium was removed from the donor and direct cells and the cultures were washed twice with PBS to remove unincorporated radiopharmaceutical. Five milliliters of fresh medium were added to all cells, which were then incubated for a further 1 h to allow bystander factors to accrue. The medium from the donor cells was then removed, passed through a 0.22-μm filter to ensure that no cell was present in the transferred medium, and added to the recipient cells from which the medium had been discarded. The medium was not removed from the direct cells, which were incubated for a further 24 h. Therefore, the direct cells suffered toxicity due to irradiation plus bystander effects. Five milliliters of fresh medium were added to the donor cells, which were incubated for a further 24 h at 37°C.
The intracellular concentration of 131I-MIBG, 123I-MIBG, and 211At-MABG by NAT gene-transfected cells occurs by the active uptake of radiopharmaceutical as well as reuptake of egressed radiopharmaceutical. During the 1-h incubation period after the initial 2-h uptake phase of the experiment, a small amount of accumulated activity leaked from the cells into the nutrient medium. To control for killing of recipient cells due to the transfer of effluxed radiopharmaceutical, an aliquot of medium from the donor flasks was removed after the 1-h incubation and the activity was measured in a γ-counter. The egressed activity was ≤1% of the initial activity added to the medium in all cases. To determine the cytotoxicity of the effluxed radioactivity, a fourth set of cells, designated activity controls, was prepared. Their medium received radiopharmaceutical activity equivalent to that which had leaked from donor cultures (treated with a range of activity concentrations) and would have been transferred to recipient cells. The activity control cultures were then incubated for 24 h after administration of these small amounts of radiopharmaceutical. The fifth set of cultures was used to determine whether bystander effects were induced by radiopharmaceutical treatment of untransfected UVW and EJ138 cells. The medium from these cultures of parental cells (i.e., not transfected with the NAT gene) was transferred to NAT gene-transfected UVW or EJ138 recipients and to untransfected UVW and EJ138 parental recipients. These were designated as parental.
RESULTS
MIBG Uptake
NAT-specific cellular uptake of 131I-MIBG was determined by comparison with the uptake that occurred in control cells treated with DMI, a specific inhibitor of NAT-mediated transport. Figure 1 shows the uptake capacity of untransfected UVW and EJ138 cells and NAT gene transfectants derived from these cell lines, designated as UVW/NAT and EJ138/NAT, respectively. Untransfected UVW and EJ138 cells exhibited no active uptake of 131I-MIBG, consistent with an absence of NAT expression. In contrast, UVW and EJ138 cells transfected with the NAT transgene under control of the CMV and hTR promoters, respectively, showed 50- and 36-fold enhancement of 131I-MIBG active uptake compared with DMI-treated cells. The cell accumulated activity as a percentage of the activity in the incubation medium was 21.4% ± 1.9% for EJ138/NAT cells and 31.4% ± 3.7% for UVW/NAT cells. At the activity concentrations applied in cytotoxicity experiments, the molar amounts of radiopharmaceutical were below the levels at which saturation of the transporter is observed (24). Throughout the range of radioactivity concentrations examined, there was a linear relationship between accumulated and administered activity for both EJ138/NAT (r = 0.962) and UVW/NAT cells (r = 0.986) (Fig. 2).

131I-MIBG uptake by UVW cells and EJ138 cells, untransfected and transfected with the NAT gene. Uptake evaluation was performed after administration of 7 kBq 131I-MIBG in the presence or absence of 1.5 mmol/L DMI, a specific inhibitor of NAT-mediated uptake. Data are mean ± SD of 3 experiments performed in triplicate.
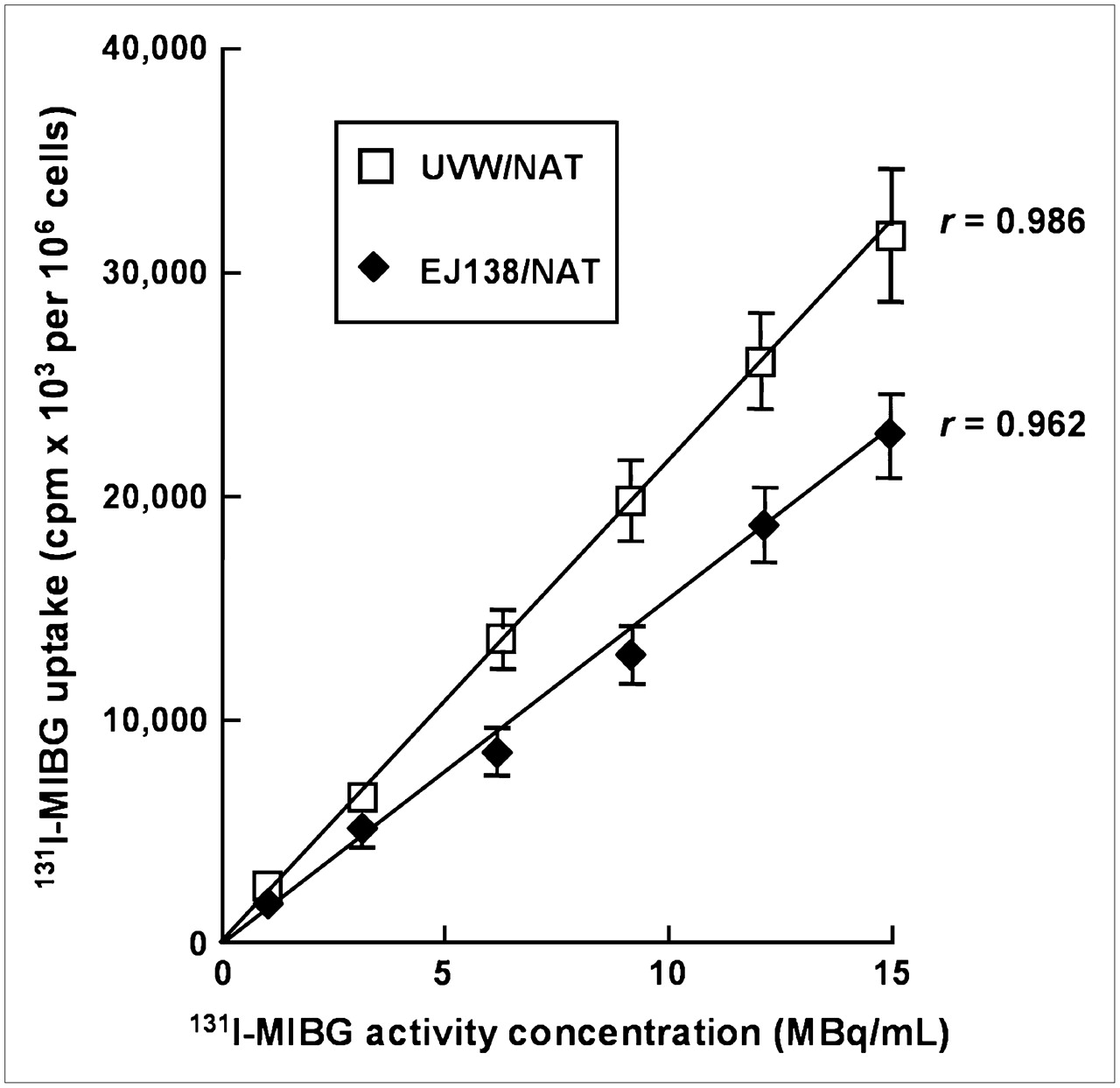
Uptake of 131I-MIBG over a range of activity concentrations by UVW cells and EJ138 cells transfected with NAT gene. Data are mean ± SD of 3 experiments performed in triplicate.
Bystander Activity After External Beam Irradiation
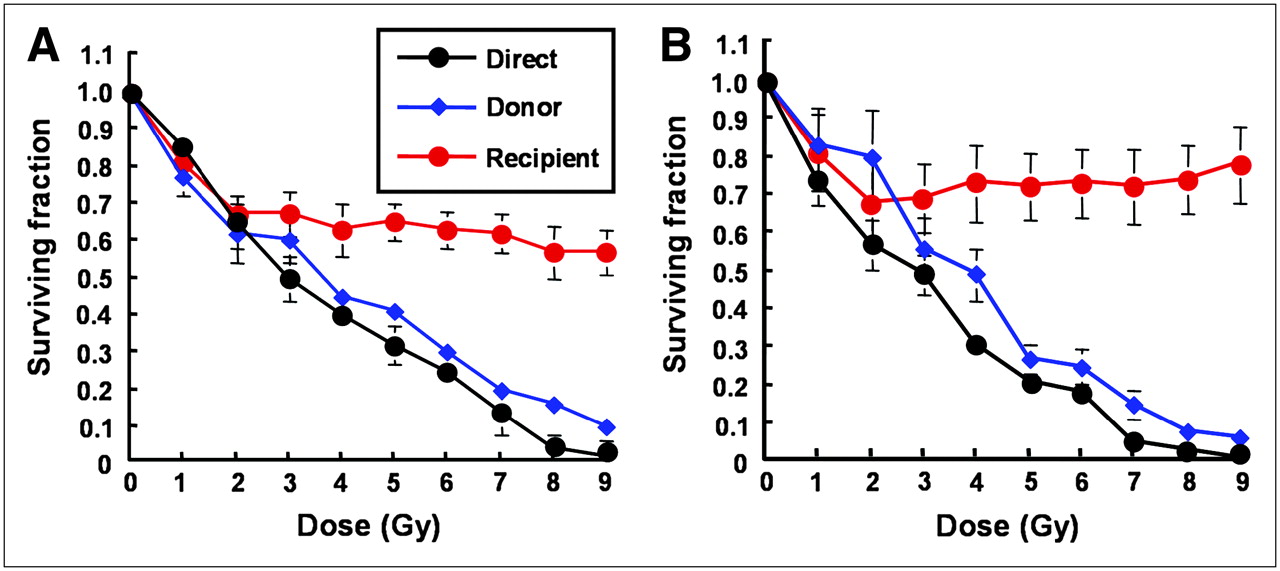
The survival of UVW/NAT and EJ138/NAT clonogens after direct γ-irradiation and after the media transfer procedure are presented in Figure 3. Throughout the dose range, the survival of cells that were directly irradiated and from which no medium was removed (direct, depicted in black) was lower than that of donor cells (depicted in blue) from which medium was removed. For both UVW/NAT and EJ138/NAT cells, exposure to the harvested medium from external beam-irradiated cells reduced the clonogenic survival of nonirradiated recipients (depicted in red). These results indicated that, in response to γ-irradiation, both cell lines produced and responded to bystander signal effects. In contrast, sham-irradiation or exposure of cultures to either irradiated or unirradiated medium in the absence of cells resulted in no reduction in SF, demonstrating that neither irradiation of the medium alone nor the medium transfer protocol contributed to the reduction in survival fraction (results not shown).

Survival of UVW/NAT cells (A) and EJ138/NAT cells (B) after treatment with γ-radiation or medium from irradiated cells. Data are mean ± SD of 6 experiments performed in triplicate.
Although the SF of donor (blue) and direct (black) cells decreased in a dose-dependent manner, recipient (red) cells that were not irradiated and, instead, received medium from irradiated cells showed an initial dose-responsive reduction in SF followed by a plateau in cell kill at higher radiation doses. This was in agreement with previous reports of cellular response to irradiated cell–conditioned medium (13) and implied that the magnitude of the effect saturated in these cell lines at doses of approximately 2 Gy. The maximum kill of recipient cells that received medium from donor cells was 40% and 35% for UVW/NAT and EJ138/NAT cell lines, respectively. The response of these cell lines to γ-irradiation was consistent with earlier reports of RIBBEs in vitro (13,25,26), indicating the suitability of UVW and EJ138 cells for a comparison of bystander effects elicited by γ-rays and targeted radionuclides.
Bystander Activity After Targeted Radiopharmaceutical Treatment
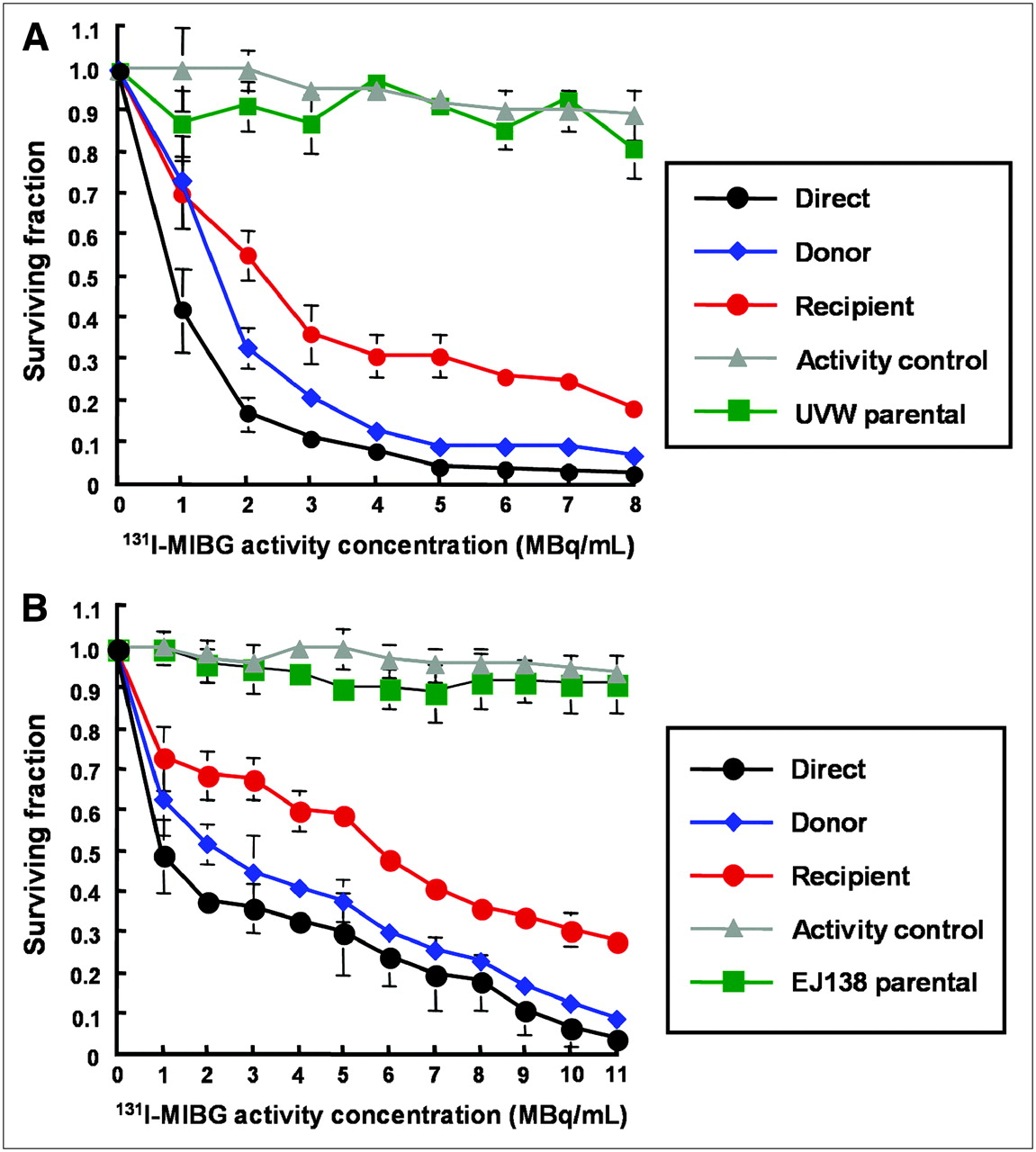
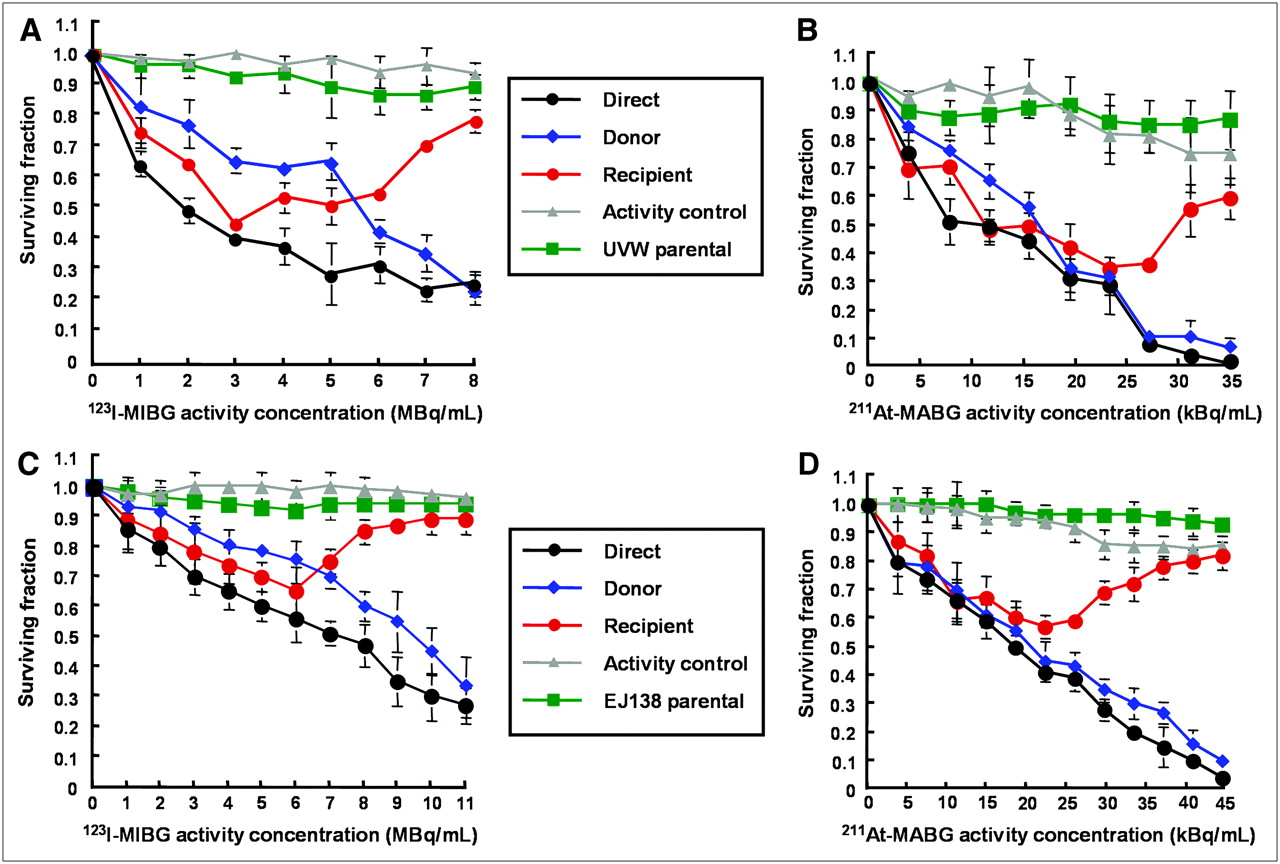
Figure 4 shows the response of UVW/NAT and EJ138/NAT cells to targeted 131I-MIBG treatment and to the bystander toxins created by cells accumulating the radiopharmaceutical. Whereas γ-irradiation produced no significant increase in RIBBE-mediated cell kill at doses greater than 2 Gy, a level that killed 35%−45% of clonogens by the direct mechanism (depicted in black, Fig. 3), no corresponding plateau in toxicity was observed after the exposure of cells to the medium from β-irradiated cells. In contrast, at 8 MBq/mL 131I-MIBG, which sterilized 98% of targeted UVW/NAT cells (depicted in black, Fig. 4A), bystander effects were sufficient to kill 80% of recipients (depicted in red, Fig. 4A). Similarly, 11 MBq/mL of 131I-MIBG killed 97% of EJ138/NAT cells directly (depicted in black, Fig. 4B) and 72% of cells via bystander-mediated processes. At all activity concentrations of 131I-MIBG investigated, we observed a bystander effect that was related to the amount of radioactivity taken up by donor cells. The magnitude of the RIBBE-induced cell killing from this β-particle emitter (depicted in red, Figs. 4A and 4B) was almost as high as that arising directly from the cellular concentration of 131I-MIBG (depicted in black, Figs. 4A and 4B).
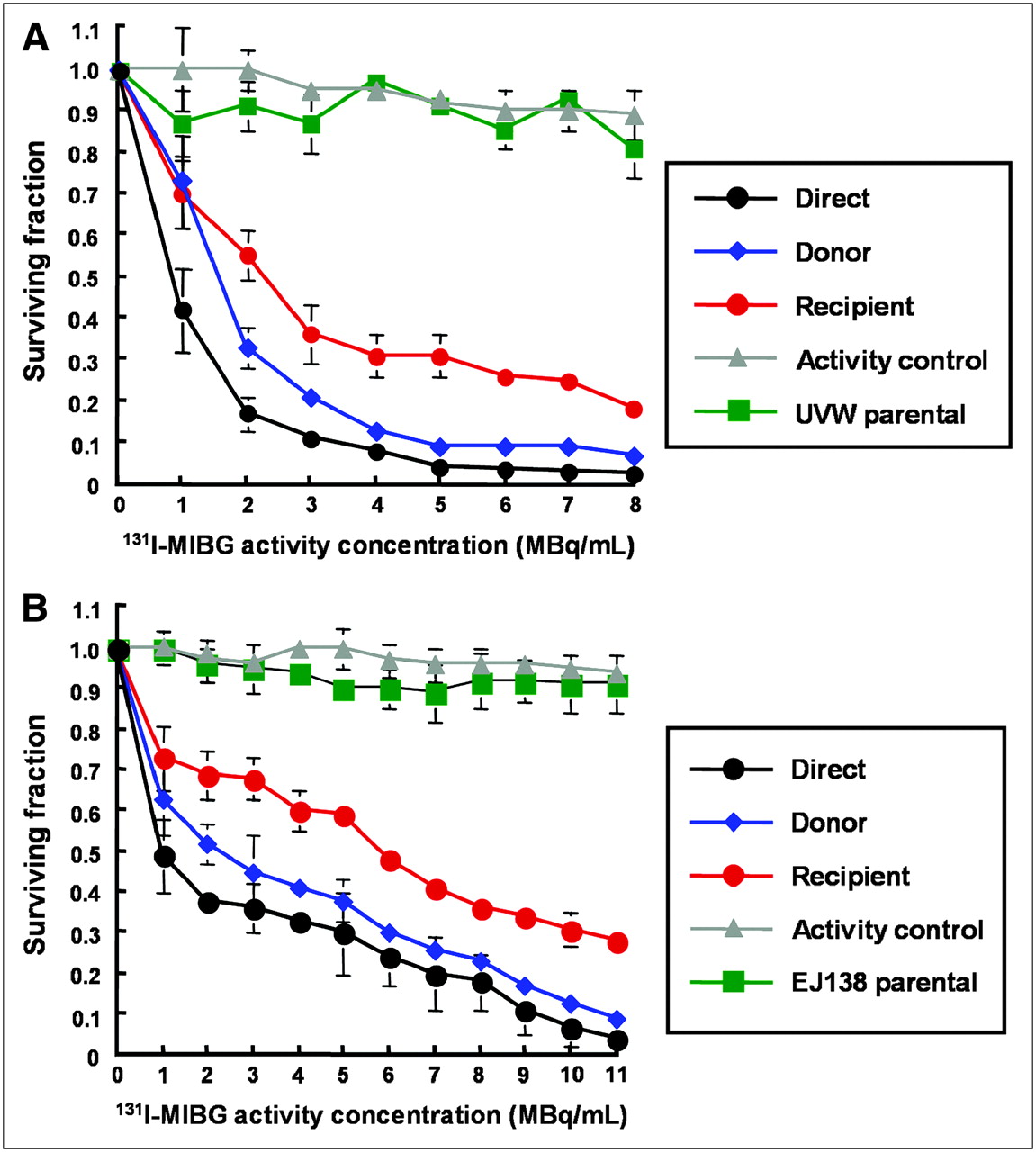
Survival of UVW/NAT cells (A) and EJ138/NAT cells (B) after treatment with 131I-MIBG or medium from 131I-MIBG-treated cells. Data are mean ± SD of 6 experiments performed in triplicate.
In contrast to the extended dose–response relationship observed for the RIBBE produced by cellular exposure and incorporation of MIBG labeled with the low-LET, β-particle emitter 131I, treatment of UVW/NAT and EJ138/NAT cells with 123I-MIBG and 211At-MABG, which emit high-LET Auger electrons and α-particles, respectively, yielded U-shaped survival curves for RIBBE-induced cell killing (depicted in red, Fig. 5) in both cell lines. For these radiopharmaceuticals, dose-related cytotoxicity was apparent at low activity concentrations but, with increasing activity, a nadir with respect to RIBBE was attained, and at higher activity concentrations the potency of bystander kill diminished. The RIBBE elicited by the prenadir activity range of these high-LET targeted radionuclides resulted in a magnitude of cell kill similar to that caused by direct irradiation (depicted in black, Figs. 5A–5D). This suggests that bystander effects may be important contributors to the cytotoxicity of 123I-MIBG and 211At-MABG at low activity concentrations.

Survival of UVW/NAT cells (A and B) and EJ138/NAT cells (C and D) after treatment with 123I-MIBG and 211At-MABG, respectively, or medium from radiopharmaceutical-treated cells. Data are mean ± SD of 6 experiments performed in triplicate.
We have demonstrated previously that radiohalogenated benzylguanidines are not cytotoxic to cells not expressing NAT and, hence, incapable of actively concentrating these radiopharmaceuticals (17,19). In the present study, no significant toxicity was observed in cells that received medium from cultures of wild-type (NAT negative) UVW cells (UVW parental, depicted in green in Figs. 4A, 5A, and 5B) or wild-type (NAT negative) EJ138 cells (EJ138 parental, depicted in green in Figs. 4B, 5C, and 5D) that had been exposed to radiohalobenzylguanidines. In Figures 4 and 5, the inconsequential effects on survival, after transfer of medium from NAT-negative cells exposed to radiopharmaceutical, are shown for NAT-transfected recipients. Similarly, a negligible effect on survival of untransfected recipients cells was observed. For the sake of clarity, the latter results have been omitted from the figures. This indicated that intracellular concentration of radiopharmaceutical was required not only for direct cell kill but also for the production of cytotoxic bystander effectors.
We also observed insignificant cell kill resulting from the transfer of the very low levels of radiopharmaceutical that had leaked from targeted cells during the 1-h incubation phase of the experimental protocol and were inadvertently transferred to recipient cultures (activity control, depicted in gray in Figs. 4 and 5). The latter finding demonstrated that the observed reduction in cell survival in recipient cultures was not due to the transfer of radioactivity but to the transfer of bystander factors.
DISCUSSION
The successful application of targeted radionuclide cancer therapy is critically dependent on the delivery of cytotoxic doses of radiation to the vast majority of the malignant cell population. Achieving this goal can be confounded by multiple factors, including variations in target molecule expression and tumor hemodynamic parameters, which can lead to heterogeneity in radiopharmaceutical delivery. However, it has been widely appreciated that cells not accumulating the labeled molecule can be killed as a result of being hit by radiation emitted from neighboring cells. Consideration of this process, known as the (physical) bystander effect, has played an important role in the design of radiotherapeutic strategies—for example, the selection of a long-range β-particle emitter in situations in which heterogeneous radiopharmaceutical delivery is anticipated.
A second type of bystander effect that could have important implications for targeted radionuclide therapy is the RIBBE, which results in the killing of cells not directly exposed to radiation. The mechanisms involved are as yet undefined. However, studies using γ-ray and α-particle beams have provided some insight into possible factors. These include oxidative stress leading to increased radical formation (27,28), nitric oxide release (29,30), cytokine release (31), and gap junctional intracellular communication (32). RIBBE resultant from targeted radionuclides has largely been unexplored, and the effects of radiation quality remain unknown (33).
The goal of the current study was to use standard techniques (18) to evaluate the potential role of RIBBEs in the treatment of cancer with targeted radionuclides. Radiohalobenzylguanidines were selected for these experiments for several reasons. First, chemically and biologically similar analogs are available labeled with radionuclides emitting β-particles, α-particles, or Auger electrons. Second, no-carrier-added syntheses are available (20–22), with the result that radiopharmaceutical uptake is not saturated over the activity concentrations investigated. Third, these radiopharmaceuticals either are being used currently or are being considered for use in the treatment of tumors that express NAT either naturally (neuroendocrine) or after gene transfection (17,19). We did not attempt to determine the mechanism underlying the bystander effect elicited by targeted radionuclide therapy nor did we examine bystander effects other than cell death. Our purpose was to compare the cytotoxic RIBBE elicited by different types of radionuclide decay after intracellular accumulation.
Our results demonstrated that exposure of 2 human tumor cell lines to media derived from external beam–irradiated cells produced a dose-dependent reduction in SF, in the dose range 0–2 Gy, followed by a plateau in clonogenic cell kill at levels greater than 2 Gy. Similarly, other reports of medium transfer experiments, after treatment with γ-rays or soft x-rays, have indicated that the dose–response in bystander cells reached a plateau at low doses (13,18,34).
In contrast, no such plateau with respect to clonogenic cell kill was evident in recipients of medium from NAT-expressing cells incubated with 131I-MIBG. The potency of RIBBE produced by NAT-expressing cells after treatment with Auger electron–emitting 123I-MIBG or α-particle–emitting 211At-MABG increased with activity up to levels that resulted in a direct kill of 35%–45% (EJ138 cells) or 60%–70% (UVW cells) of clonogens. At higher activity concentrations of 123I-MIBG or 211At-MABG, RIBBE became progressively weaker. This suggests that after intracellular bombardment by high-LET radionuclides, the RIBBE-generating apparatus is inhibited above a threshold radiation dose.
Elucidation of the pathways involved in this process could indicate ways of manipulating RIBBE production to reduce toxicity to normal tissues that are inadvertently irradiated during the course of a targeted radiotherapy regime. Careful choice of radionuclides and dose administered in clinical scenarios for targeted radionuclide therapy of tumors, which naturally accumulate targeted radionuclides or have been genetically manipulated to do so, will allow factors such as inefficient gene transfer and heterogeneous uptake to be compensated for, thus optimizing the cell kill potential of this therapeutic scheme.
Importantly, neither direct nor indirect kill was observed at any activity concentration in cultures of cells that did not express the NAT and were incapable of active uptake of MIBG. Thus, these RIBBEs are not related to decays of unbound radiopharmaceutical present in the media. These findings suggest that potent toxins are generated specifically by cells that concentrate targeted radionuclides. Furthermore, a comparison of the dose dependence of these effects with those observed after exposure to 60Co γ-rays suggests that RIBBEs from targeted radiotherapeutics may be distinct from those elicited by conventional external beam radiotherapy.
Whatever the mechanism, RIBBE could be important not only in relation to radiation protection and safety but also with respect to the therapeutic use of ionizing radiation. Exploitation of RIBBE could be especially relevant to the efficacy of targeted radiotherapy because this treatment is limited by heterogeneous uptake of radionuclides by tumors. Freely diffusible toxic bystander signals could overcome the inefficiency of tumor control due to nonuniform distribution of radiation dose. However, this presumes that the RIBBE observed under in vitro conditions also occurs in vivo.
For this reason, it is important to note that there is evidence that suggests that RIBBEs are not exclusive to cells in culture. Brooks (35) demonstrated that after exposure of a small proportion of hamster liver cells to α-irradiation, all of the cells in the liver, whether directly exposed or not, were at the same risk for induction of chromosomal damage. Khan et al. (36) showed that γ-irradiation of the base of the rat lung induced DNA damage in the lung apex. Moreover, cytotoxic effects observed in solid tumors located at sites distant from those targeted by radiation have been reported in patients (37).
Watson et al. (38) subjected murine bone marrow cells to neutron bombardment before transplantation into sex-mismatch recipients and provided the first demonstration of radiation-induced genomic instability in hemopoietic bystander cells in vivo. More recently, Xue et al. (16) showed that cells preloaded with Auger electron–emitting 125I-IUdR and mixed with unlabeled cells exerted a damaging effect on neighboring unlabeled tumor cells growing subcutaneously in nude mice. This study demonstrated the potential of internalized Auger electron emitters to generate bystander effects in vivo. In the current investigation, we observed that the Auger electron–emitter 123I induced the release of bystander cytotoxins in vitro, highlighting the possible application of 123I-MIBG for the treatment of patients with neural crest-derived tumors.
Because α-particles are densely ionizing radiation, their effectiveness for killing cancer cells is high and is not diminished by hypoxia, making α-emitters such as 211At most promising radionuclides for targeted cancer radiotherapy (39,40). For example, 211At is an attractive alternative to 131I as a radiolabel for halobenzylguanidine (17). However, because the path length of 211At α-particles is only 55–70 μm, cross-fire irradiation from targeted to untargeted cells would be considerably less extensive than that from a β-emitter such as 131I. Therefore, it is encouraging that our results suggest that this potential limitation to the efficacy 211At-MABG may be overcome by the substantial RIBBE generated by this radiopharmaceutical.
CONCLUSION
Our results suggest that RIBBE resultant from the accumulation of radiolabeled MIBG analogs can effectively kill cancer cells not directly exposed to these agents. Furthermore, the mechanisms controlling the production of toxic bystander effectors from targeted radiotherapeutics may be distinct from those elicited by conventional radiotherapy. We seek now to investigate these RIBBE signals using radiopharmaceuticals localized to different subcellular regions. The efficiency of this mode of kill in tumor and normal cells and the dependence of genetic background and tumor microenvironment will also be assessed. The identification of RIBBE factors will stimulate the design of strategies to maximize damage to tumor cells while minimizing damage to normal cells.
Acknowledgments
This work was supported in part by grants from the Department of Health, The Clerk Maxwell Cancer Research Trust, Cancer Research United Kingdom, the Neuroblastoma Society, the British Urological Foundation, the Royal College of Physicians and Surgeons of Glasgow (Ian Sunter Charitable Trust Research Fellowship), the National Institutes of Health (grant CA42324), and the Pediatric Brain Tumor Foundation. The authors thank Drs. Ganesan Vaidyanathan (Department of Radiology, Duke University Medical Center, Durham, NC) and Sally L. Pimlott (Radionuclide Dispensary, Western Infirmary, Glasgow, U.K.) for synthesis of the radiopharmaceuticals used in these experiments.
References
- Received for publication November 18, 2005.
- Accepted for publication January 9, 2006.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Marshalling the Potential of Auger Electron Radiopharmaceutical Therapy
- Radium-223-Induced Bystander Effects Cause DNA Damage and Apoptosis in Disseminated Tumor Cells in Bone Marrow
- Radiation-Mediated Up-Regulation of Gene Expression from Replication-Defective Adenoviral Vectors: Implications for Sodium Iodide Symporter Gene Therapy
- MIRD Continuing Education: Bystander and Low Dose-Rate Effects: Are These Relevant to Radionuclide Therapy?
- A Transfectant Mosaic Xenograft Model for Evaluation of Targeted Radiotherapy in Combination with Gene Therapy In Vivo
- Targeted Radiotherapy: Is the "Holy Grail" in Sight?