


Abstract
Peptides are useful tools for directing radioisotopes into tumors. We evaluated the ability of a bacterial peptide display system to isolate new prostate tumor-specific peptides. Methods: We used the bacterial FliTrx system to identify a new cyclic peptide that binds to prostate carcinoma. Serum stability and binding affinities of the 125I-labeled peptide were tested. Furthermore, the 131I-labeled peptide was used to evaluate its biodistribution. Results: Several peptides showing a potential consensus motif were identified. The new peptide MM-2 is stable in serum for up to 24 h. It binds to PC-3 cells, and this binding can be inhibited more than 70% with the unlabeled peptide. Binding to human umbilical vein endothelial cells (HUVECs) and PNT-2 cells is weaker, and competition (27%) in HUVECs is less efficient. The biodistribution showed moderate accumulation in tumor. Conclusion: Bacterial peptide display, an alternative to phage peptide display, can allow the identification of specific binding and stable peptides.
Compared with antibodies, peptides show advantages for radionuclide imaging that are mainly due to their small size: Localization of the peptide is not limited by diffusion, and clearance from the circulation is rapid, resulting in low background activity. Furthermore, peptides have binding affinities similar to those observed with antibodies. High affine receptors for peptides, such as somatostatin (1,2), gastrin-releasing peptide (3), and vasoactive intestinal peptide (4), have been observed in a variety of tumors and can be used as molecular targets for isotope-based diagnosis and treatment of tumors. Phage display techniques have successfully been applied to identify target-specific molecules such as antibodies or peptides (5–7). In phage display, large numbers (up to 1010) of different peptides are expressed on the surface of bacteriophages. In M13 phages, the peptides can be expressed as fusion proteins, with the coat proteins p3 or p8. This leads to the presentation of 5 copies of the peptide if fused to p3 or of more than 100 copies if fused to p8. This coupling of genetic and proteomic information allows binding of the phages to target structures via the expressed peptide and enrichment of the peptide via phage amplification during several selection rounds. In the bacterial FliTrx system (Invitrogen) (8), a phagemid vector (pFliTrx) allows the display of peptides directly on the surface of E. coli by using 2 proteins: the major bacterial flagellar protein (FliC) and thioredoxin (TrxA). Peptide libraries are cloned in frame within the active site loop of thioredoxin, which is inserted into the dispensable region of the FliC gene. The resulting fusion protein assembles into the flagella on the bacterial cell surface protruding from the cells. The dodecamer peptide library is constrained by a disulfide bridge, which should result in a stable conformation for the synthetic peptide as well as resistance to degradation (9–11). In the present study, we used this system to identify a peptide that binds to human prostate carcinoma cells PC-3. The peptide was less affine to primary human umbilical vein endothelial cells (HUVECs) and benign prostate cells (PNT-2). The unlabeled peptide can compete for its binding. The synthetic peptide was stable in serum for more than 24 h and accumulated into PC-3 tumors in a nude mouse model.
MATERIALS AND METHODS
Cell Lines
The human prostate carcinoma cell line PC-3 and the human prostate cell line PNT-2 were cultivated at 37°C in a 5% CO2 incubator in RPMI 1640 (Invitrogen) with Glutamax (Invitrogen) containing 10% fetal calf serum and a 25 mmol/L concentration of N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid). HUVECs were isolated as described in the literature (12). Cultivation was on gelatin (1%)-coated cell culture flasks using medium 199 (Invitrogen) containing 20% fetal calf serum, 2 mmol of glutamine per liter, 100 IU of penicillin per milliliter, 100 IU of streptomycin per milliliter, and 2 ng of basic fibroblast growth factor per milliliter (Roche Diagnostics).
Selection of Tumor Cell-Binding Peptides
The bacterial peptide display system FliTrx was used. The bacteria were treated according to the manufacturer’s instructions. The panning was performed after induction of the bacterial library with tryptophan, which activates the transcription of the flagellin/thioredoxin peptide fusion proteins. A confluent dish with PC-3 cells was blocked with IMC medium containing 1% bovine serum albumin and 1% α-methyl mannoside (Sigma-Aldrich). Then, the cells were incubated with 10 mL of induced bacterial suspension in IMC containing 1% bovine serum albumin and 1% α-methyl mannoside for 1 h. Unbound bacteria were washed off in 4 washes for 5 min with IMC containing 1% α-methyl mannoside. Bound bacteria were recovered by stirring in a vortex mixer for 30 s and subsequently amplified in IMC medium containing ampicillin. After 4 selection rounds, single clones were isolated and sequenced.
Peptide
The MM-2 peptide (CPGDRGQRRLFSKIEGPC) was obtained by solid-phase peptide synthesis using Fmoc chemistry. At the C terminus, a tyrosine was added to allow iodination (CPGDRGQRRLFSKIEGPCY). After cleavage from the resin with trifluoroacetic acid and purification by high-pressure liquid chromatography (HPLC), the linear peptide was dissolved in 5 mmol of ammonium acetate per liter. The pH was adjusted to 7–7.5 with saturated NaHCO3 solution. Hydrogen peroxide was added, and cyclization was complete within 30 min. The crude product was purified by HPLC. The radiolabeling was achieved by iodination using the chloramine-T method (13). The specific activities obtained were 90 GBq/μmol for the 125I-labeled peptide and 110 GBq/μmol for the 131I-labeled peptide.
In Vitro Binding Experiments
A quantity of 200,000 cells was seeded into 6-well plates and cultivated for 24 h. The medium was replaced by 1 mL of fresh medium (without fetal calf serum). When the competitor was used, unlabeled peptide (10−4 mol/L) was preincubated for 30 min, and 125I-labeled peptide was then added to the medium (1–2 × 106 cpm/well) and incubated for 1 h. The cells were washed 3 times with 1 mL of phosphate-buffered saline and subsequently lysed with 0.5 mL of a 0.3 mol/L concentration of NaOH. Radioactivity was determined with a γ-counter and calculated as percentage applied dose per 106 cells.
Peptide Serum Stability Assay
The MM-2 peptide, 5 × 10−4mol/L, was dissolved in 1 mL of heparinized human serum and incubated at 37°C. After the indicated times, 25 μL of serum were removed, 25 μL of acetonitrile were added, 50 μL of distilled water were added, and finally 50 μL of acetonitrile were added. The sample was centrifuged for 5 min at 13,000 rpm, the supernatant was transferred to a new tube, and the centrifugation step was repeated twice. Stability was analyzed by HPLC.
Animals and Tumor Growth
Male 6-wk-old BALB/c nu/nu mice were obtained from Charles River WIGA and placed in VentiRack housing (BioZone). For inoculation of the tumors in nude mice, a MatriGel (Becton, Dickinson and Co.) matrix cell suspension (5 × 106 cells) was injected subcutaneously into the anterior region of the mouse trunk. Tumors were grown to approximately 1.0 cm3. All animals were cared for according to the national animal guidelines.
Organ Distribution with Radioiodinated MM-2
131I-MM-2 was intravenously injected into male nu/nu mice (2.8 × 107 cpm/mouse) carrying the subcutaneously transplanted human prostate tumor PC-3. At 15 min, 45 min, 135 min, and 24 h after injection, the mice were sacrificed. The organs were removed and weighed, and the radioactivity was determined using an automated NaI(Tl) well counter (Cobra II; Canberra Packard). The percentage of injected dose per gram of tissue was calculated.
RESULTS
Selection of a Peptide Binding to the Prostate Carcinoma Cell Line PC-3
For the selection of tumor-specific peptides, in vitro selection rounds were performed on PC-3 cells. For each round, 10 mL of bacterial library were added to a cell culture dish for positive selection for 60 min. The unbound bacteria were washed off, and bound bacteria were recovered. After 4 rounds, single clones were selected and amplified, and DNA was isolated for sequencing. Among 14 clones sequenced, 4 peptides showed the same sequence (CPGDRGQRRLFSKIEGPC) (Fig. 1). Four other peptides shared several amino acids, suggesting a possible motif. No identity to this sequence was found in available databases.
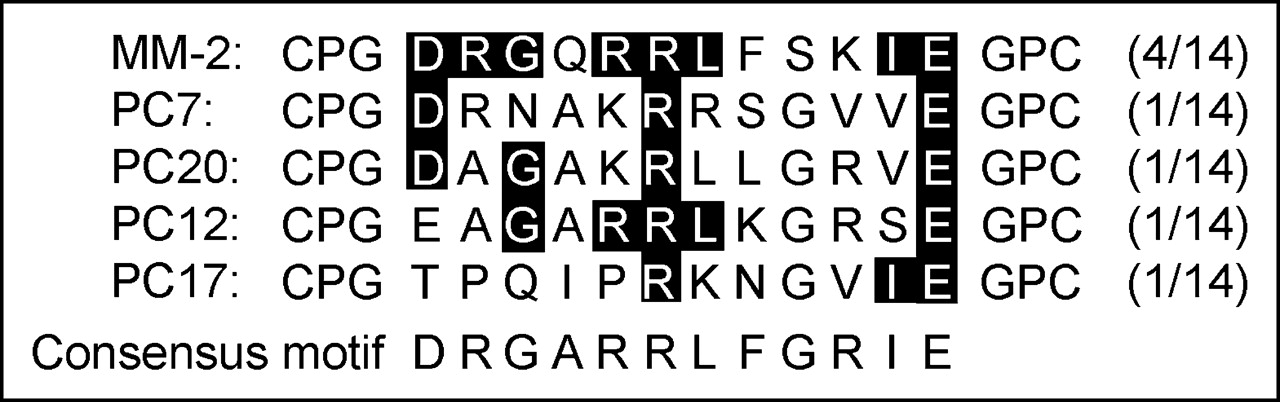
Amino acid sequence of bacterial clones enriched through selection rounds and identified by sequencing. The part of the thioredoxin loop sequence CPG…GPC forming the loop via disulfide bridge is shown.
Binding and Competition of MM-2
The peptide MM-2 was prepared by Fmoc solid-phase synthesis, and a tyrosine residue was added at the C terminus to allow labeling with 125I. PC-3 cells, HUVECs and PNT-2 cells were incubated for 1 h with the 125I-labeled peptide with or without unlabeled peptide as competitor (10−4 mol/L). Radioactivity of the lysed cells was calculated as percentage applied dose per 106 cells. The prostate tumor cells PC-3 showed uptake of 1.16% (Fig. 2). Preincubation with unlabeled peptide MM-2 allowed competition for more than 70% of this binding. In contrast, only 0.78% cell uptake could be observed by primary cultures of HUVECs and only 27% of this binding was inhibited by the peptide itself. The immortalized benign prostate cells PNT-2 showed 0.86% uptake, and about 60% of this binding was displaceable.

In vitro binding assay with MM-2Y. 125I-MM-2Y was added to wells with PC-3, HUVEC, or PNT-2 cells and incubated for 1 h. Experiments were performed in triplicate; mean value of 5 independent experiments (n = 3 for PNT-2 cells) and SE are shown. − = no competitor was added; + = 10−4 mol of unlabeled MM-2 per liter was added as competitor 30 min before incubation with labeled MM-2.
Peptide Stability
To evaluate susceptibility to degradation, we incubated the peptide in human serum and analyzed for degradation products by HPLC (Fig. 3). A slight decrease in the amount of peptide was observed after 10 min, but no degradation products appeared even after 4 h. After 24 h of incubation in serum, the peptide was still intact.
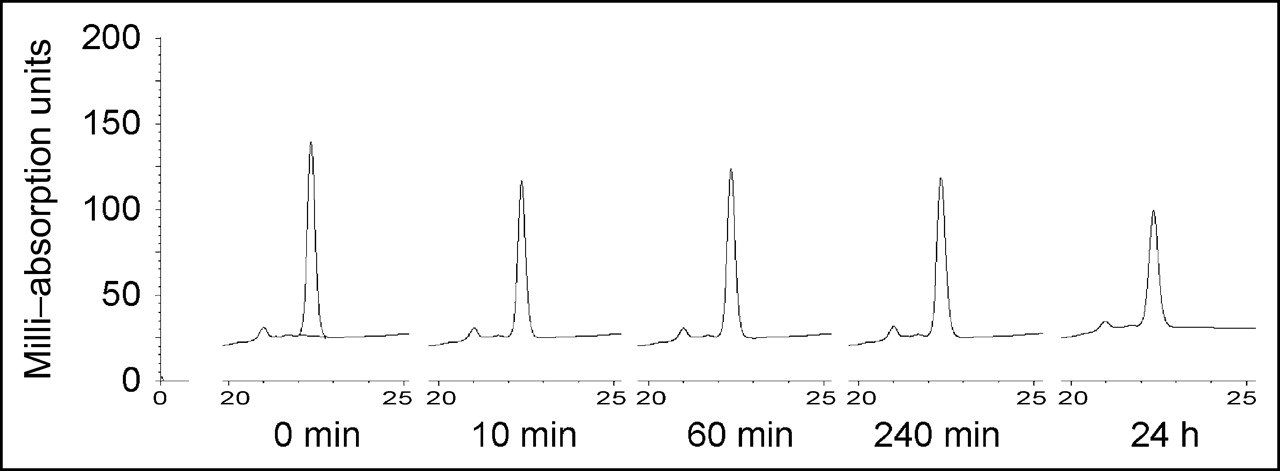
Serum stability of MM-2. MM-2 peptide was incubated with human serum. After the indicated incubation times, samples were analyzed with HPLC to show possible degradation of peptide.
Biodistribution of Radiolabeled MM-2
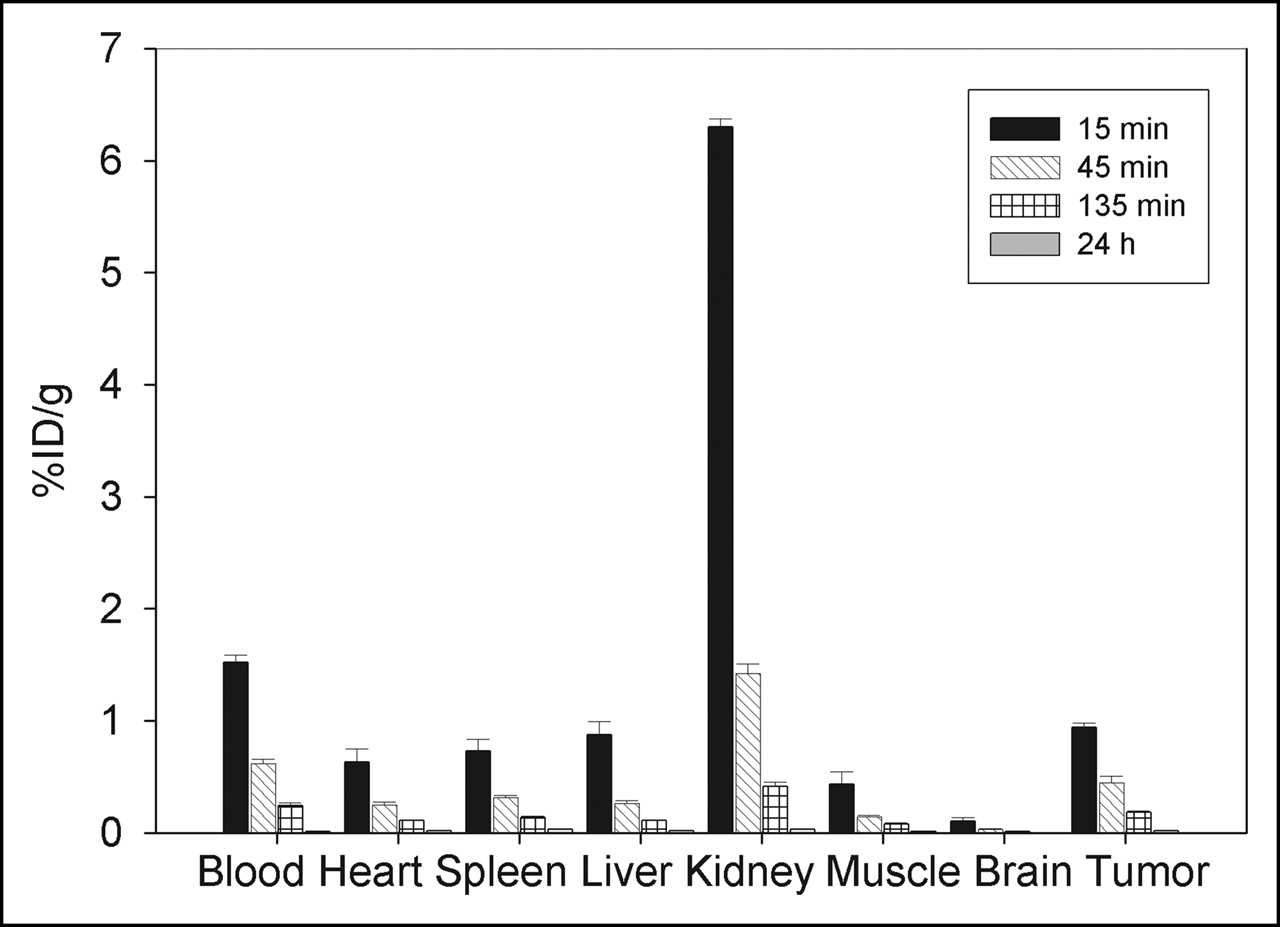
To investigate the distribution of MM-2 in vivo, we labeled the peptide with 131I and intravenously injected into male nu/nu mice carrying human prostate tumors PC-3. The biodistribution in mice showed that MM-2 accumulated in the tumor to a level of approximately 1% injected dose per gram of tissue—a higher level than in the other organs, with the exception of kidney and blood (Fig. 4). The tumor-to-muscle ratio was 2.16 at 15 min after injection and increased to 3.07 at 45 min and 2.43 at 135 min (Table 1). The tumor-to-blood ratio increased slightly with time. Over time, a decrease in radioactivity was observed in all organs. The strongest decrease was in the kidney, indicating elimination via the renal route. The tumors showed the lowest decrease over time (53% from 15 to 45 min), in comparison to that in the kidneys (a 77% decrease) or the blood (a 60% decrease). After 24 h, no significant radioactivity was found in any organ or blood.
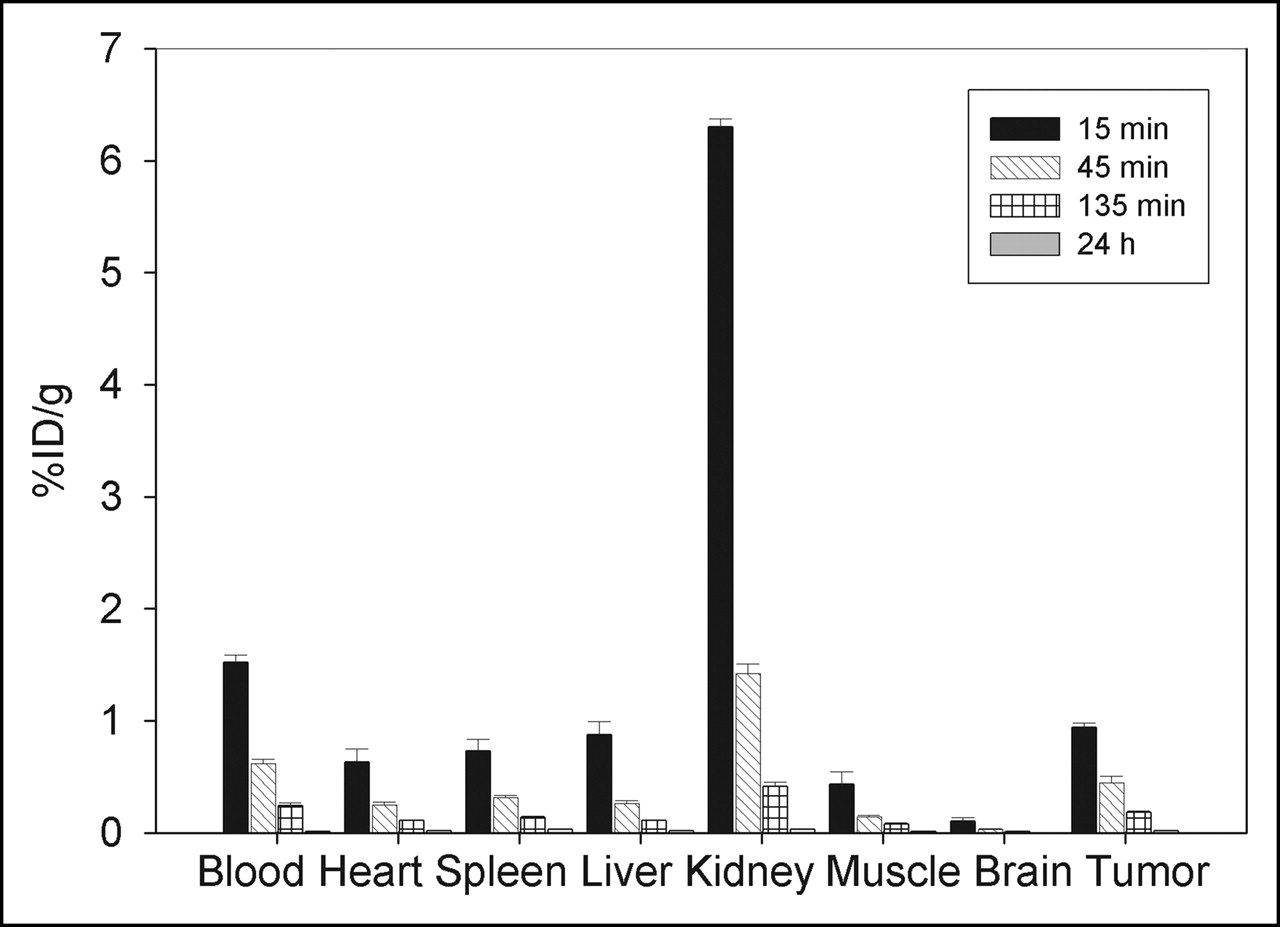
Biodistribution of MM-2Y in male BALB/c nu/nu mice carrying PC-3 tumors (n = 3 animals per time point). Animals were injected intravenously with 131I-MM-2Y, and percentage injected dose per gram of tissue (%ID/g) was measured in tumor and control organs after 15 min, 45 min, 135 min, and 24 h.
Biodistribution of MM-2 in Mice
DISCUSSION
This study used a bacterial peptide display system to identify a new prostate carcinoma-binding peptide that is stable in serum and binds to prostate carcinoma cells in vivo. Bacterial surface display systems are alternative tools in bioscience that have a wide scope of research application (14). Besides having some disadvantages, this system also offers specific advantages. One advantage is the insertion of the peptide library into the thioredoxin loop, extruding the peptide to allow easy access. This loop gives a constricted conformation through the disulfide bond that most likely leads to stable peptides. Consequently, we found that the MM-2 peptide is stable in serum for more than 24 h. Another advantage of the localization of the peptide in a loop is the localization within the protein, whereas in phage display systems the peptide is predominantly attached to a terminus, potentially leading to problems using the peptide within a protein structure. The thioredoxin loop peptide is easily integrated, such as in a viral capsid or other surface structures.
MM-2 binds to prostate carcinoma cells PC-3, and this binding can be inhibited by the unlabeled peptide, indicating a specific binding to the cells. A similar competition was observed in benign prostate cells, although in these cells binding was lower (by 26%) than in tumor cells. Binding to HUVECs was weaker and not significantly inhibited by unlabeled MM-2, indicating specificity of the peptide toward prostate tissue.
MM-2 has no sequence similarity to bombesin, luteinizing hormone-releasing hormone, prostate-specific antigen, or any other peptide or protein sequence available, as confirmed by a search of various protein databases such as the European Molecular Biology Laboratory and SwissProt databases. The fact that MM-2 shares several amino acids with other peptides that accumulated in the selection suggests a common motif. Also, MM-2 might not contain the optimal binding motif since the other peptides share amino acids with each other but not with MM-2, pointing toward possible improvements of the binding motif. Therefore, we expect that modifications of the leader sequence in MM-2 should result in peptides with improved imaging properties.
The serum stability assay showed not degradation of the peptide but only a slight decrease in the amount of MM-2. This slight decrease could be due to an unspecific interaction or adhesion of MM-2 with serum proteins or, since no degradation products were observed, variations in the injected amount of MM-2. In the biodistribution studies, the peptide showed decreasing accumulation in the tumor as well as in the other organs with time, although the decrease in tumor was slower than in organs. This leads to an increase of tumor-to-organ ratios for all organs. That decrease could indicate degradation of the peptide in vivo despite the observed serum stability. Alternatively, deiodination may occur while the peptide is still intact. Because the stability test used in our experiments showed resistance to proteases and peptidases occurring in serum, the possibility of degradation through other mechanisms in vivo is not excluded. This possibility is supported by the observation that no activity is left after 24 h.
CONCLUSION
Bacterial peptide display can identify stable peptides that can be used in tumor targeting. This technology is less time consuming since phage precipitation and titering can be avoided and the handling of only bacteria is easier than is phage display. The thioredoxin part leads to a stable conformation, and cyclization enhances the serum stability of the resulting peptides. Furthermore, the thioredoxin loop confers a greater versatility for peptide integration into surface structures without changing peptide conformation as it would be the case for N-terminal expressed phage display peptides. However, as shown in these experiments, stability in serum does not have to result in stability in vivo, and further modifications such as changes in the peptide sequence are needed. Thus, the peptide MM-2 can be the origin of the development of tracers that may be used for coupling with radioactive isotopes, anticancer agents or viral particles.
Acknowledgments
The authors acknowledge the contribution of Helmut Eskerski and Uschi Schierbaum to the animal experiments and specially thank Kerstin Schmidt for sharing her HUVEC cultures. This work was supported by grants HA2901/2–1 and 2–2 from Deutsche Forschungsgemeinschaft.
Footnotes
Received Aug. 18, 2004; revision accepted Oct. 21, 2004.
For correspondence or reprints contact: Sabine Zitzmann, PhD, German Cancer Research Center, Clinical Cooperation Unit Nuclear Medicine, Im Neuenheimer Feld 280, D-69120 Heidelberg, Germany.
E-mail: s.zitzmann{at}dkfz.de
REFERENCES
In this issue





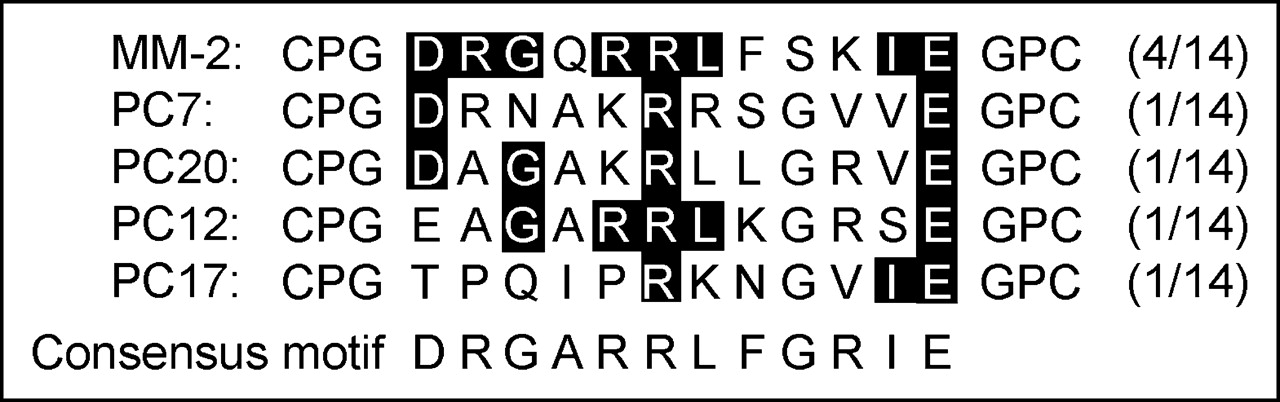{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Humanized ADEPT comprised of an engineered human purine nucleoside phosphorylase and a tumor targeting peptide for treatment of cancer
- TMTP1, a Novel Tumor-Homing Peptide Specifically Targeting Metastasis
- 111In-Benzyl-DTPA-ZHER2:342, an Affibody-Based Conjugate for In Vivo Imaging of HER2 Expression in Malignant Tumors