


Abstract
We have developed an efficient pretargeting strategy for renal cell carcinoma (RCC) based on a biologically produced bispecific monoclonal antibody (bs-mAb). Tumor targeting with this 2-step pretargeting strategy in the NU-12 mouse RCC model was very efficient compared with other pretargeting strategies, possibly due to unique characteristics of the NU-12 tumor used in our studies. Here we describe the bs-mAb G250xDTIn-1 pretargeting strategy in 3 different RCC nude mouse models. Methods: Three different human RCC xenografts in nude mice (NU-12, SK-RC-1, and SK-RC-52 tumors) were pretargeted with 100 pmol bs-mAb G250xDTIn-1. Three days after administration of the bs-mAb, mice were injected intravenously with 4 pmol 111In-labeled bivalent peptide, diDTPA-FKYK (DTPA is diethylenetriaminepentaacetic acid). At 1, 4, 24, 48, and 72 h after injection of the radiolabeled peptide, the biodistribution of the radiolabel was determined. The 3 RCC tumors were characterized in vivo and in vitro for G250 antigen expression, vessel density, vascular volume, and vascular permeability and by targeting with 111In-/125I-labeled cG250 mAb. Results: Using the pretargeting strategy, relatively high uptake of the radiolabel was observed in all 3 tumor models (maximum uptake: SK-RC-1 [278 ± 130 %ID/g (percentage injected dose per gram), 1 h after injection] > NU-12 [93 ± 41 %ID/g, 72 h after injection] > SK-RC-52 [54 ± 9 %ID/g, 4 h after injection]). Remarkably, uptake of the radiolabel in the tumor did not correlate with G250 antigen expression. The kinetics of the radiolabel in the tumor varied largely in the 3 RCC tumors: SK-RC-1 and SK-RC-52 tumors showed a washout of the 111In label from the tumor with time: only 30% of the radiolabel was retained in the tumor 3 d after injection, whereas the 111In label was fully retained in NU-12 tumors. Physiologic characteristics (vessel density, vascular volume, and vascular permeability) of the tumors may explain these differences. Conclusion: G250 antigen–expressing tumors can be pretargeted very efficiently with the bs-mAb G250xDTIn-1. There was no correlation between G250 antigen expression and uptake of the radiolabel in the tumor using the pretargeting strategy or with directly labeled mAbs. Therefore, these studies show that physiologic characteristics of the tumor, such as vascular permeability, play a significant role in pretargeting.
Radioimmunotherapy (RIT) as well as radioimmunoscintigrapy are based on selective localization of radiolabeled monoclonal antibodies (mAbs) in tumors expressing tumor-associated antigens (TAAs) on the tumor cell surface (1). However, accumulation of mAbs in solid tumors is a relatively slow and inefficient process and, for diagnostic applications with directly labeled mAbs, optimal tumor-to-background ratios are obtained only after several days. This unfavorable pharmacokinetic behavior of mAbs leads to relatively high radiation doses to normal tissues in RIT, while a relatively small fraction of the injected dose eventually localizes in the tumor (1,2). So far, RIT is only effective in hematologic tumors, probably due to the relatively good accessibility of the TAA in these tumors and their relatively high radiosensitivity (3).
In 1986, Goodwin et al. (4) were the first to propose the separation of the administration of the long-circulating antitumor mAb from the radiolabel. In this so-called pretargeting strategy, tumor cells are pretargeted with a nonlabeled bispecific antibody (bs-mAb) followed by a fast-clearing radiolabeled hapten. We have developed a pretargeting strategy using a biologically produced bs-mAb with a high affinity for the renal cell carcinoma (RCC) TAA (G250 antigen) and for indium-labeled diethylenetriaminepentaacetic acid (DTPA) (bs-mAb: G250xDTIn-1) for targeting of RCC (5). In a nude mouse RCC model with this system, high uptake (80 %ID/g [percentage injected dose per gram]) and good retention of the radiolabel in the tumor were observed when an 111In-labeled bivalent peptide was used (6).
Using the pretargeting strategy in the NU-12 mouse tumor model, the uptake of the radiolabel was exceptionally high compared with other pretargeting strategies (7–10), possibly due to unique characteristics of the NU-12 tumor.
Here we studied bs-mAb G250xDTIn-1–based pretargeting in 3 different RCC nude mouse models to determine whether this strategy would allow efficient targeting of other G250 antigen–expressing tumors. In addition, several tumor characteristics—such as G250 antigen density, vascular characteristics, internalization, and processing of the antibody—were determined to gain more insight into the features that may affect the targeting of RCC using a pretargeting system.
MATERIALS AND METHODS
RCC Cell Lines
The SK-RC-1 cell line was derived from a primary clear-cell RCC specimen. SK-RC-52 was derived from a clear-cell RCC metastatic lesion in the mediastinum (11). Both cell lines were obtained from the Memorial Sloan-Kettering Cancer Center, New York. The NU-12 cell line was derived from a primary clear-cell RCC and was characterized as described previously (12).
bs-mAb
The characteristics of mAb anti-G250 (IgG1), directed against the RCC-associated antigen G250, identified as carbonic anhydrase isoform IX (MN/CA IX), have been described elsewhere (13). The CA IX antigen is expressed on the cell surface of virtually all clear-cell RCCs (14).
The characteristics of mAb anti-DTPA-In (DTIn-1, IgG2a) have been described elsewhere (5,14). The isolation of bs-mAb–producing quadroma cells and the characterization and purification of bs-mAb anti-RCC x anti-DTPA(In) (bs-mAb: G250xDTIn-1) have been described in detail previously (5).
Bivalent Peptide
IMP156.
The ε-amino groups of both lysine residues of this tetrapeptide (Phe-Lys-Tyr-Lys) were conjugated with mono-activated DTPA to obtain a bivalent peptide as described previously (6,15). The N-terminus of the peptide was acetylated and the C-terminus was amidated to reduce degradation by exopeptidases. The peptide, Ac-Phe-Lys(DTPA)-Tyr-Lys(DTPA)-NH2 (diDTPA-FKYK; molecular weight 1,377 Da), was prepared by solid-phase synthesis and formulated in a lyophilized labeling kit, containing 11 μg diDTPA-FKYK, 50 mg 2-hydroxypropyl-β-cyclodextrin, and 4.4 mg citrate (pH 4.2).
Radiolabeling
111In-diDTPA-FKYK.
Eleven micrograms of lyophilized diDTPA-FKYK (IMP156) were reconstituted in 1 mL saline. To 15 μL of the peptide solution, 15 μL 40 mmol/L HCl, 375 μL H2O, and 55.5 MBq (1.5 mCi) 111InCl3 (Tyco Health Care) were added. The reaction mixture was incubated for 60 min at room temperature. The radiochemical purity (RCP) was determined by instant thin-layer chromatography (ITLC) on silica gel strips with methanol:water (55:45) and 0.15 mol/L citrate buffer (pH 6.0) as the mobile phase. When the RCP exceeded 95%, a 3-fold molar excess of InCl3 was added to saturate the remaining DTPA moieties with stable In3+.
cG250-DTPA-111In.
Conjugation of mAb with p-isothiocyanatobenzyl-DTPA (ITC-DTPA) was performed essentially as described by Ruegg et al. (16). Briefly, 1 mL mAb cG250 (10 mg/mL) was mixed with 110 μL of 1.0 mol/L NaHCO3 (pH 9.5) and 1.76 mg ITC-DTPA (50-fold molar excess). After incubation at room temperature for 1 h, the reaction mixture was dialyzed in a Slide-A-Lyzer (10-kDa cutoff; Pierce) overnight against phosphate-buffered saline (PBS). Subsequently, the cG250-ITC-DTPA conjugate was diluted in PBS to 1 mg/mL and aliquots were stored at −20°C.
Radiolabeling of cG250-ITC-DTPA.
Eight microliters 50 mmol/L NH4Ac, pH 5.4, and 7.4 MBq (200 μCi) 111InCl3 were added to 30 μL (1 mg/mL) cG250-ITC-DTPA and incubated at room temperature for 1 h. The RCP of the mAb was determined by ITLC on silica gel strips with 0.15 mol/L citrate buffer (pH 6.0) as the mobile phase.
125I-cG250.
Iodination of the mAb was performed according to the IODO-GEN method (17). To a tube coated with 25 μg of IODO-GEN, 5 μL of 0.5 mol/L phosphate buffer (pH 7.4), 15 μL of 50 mmol/L phosphate buffer (pH 7.4), 26 μL cG250 (0.5 mg/mL), and 7.4 MBq (200 μCi) 125I (Amersham Cygne) were added. The mixture was incubated for 10 min at room temperature and immediately followed by a purification step on a PD-10 column eluted with PBS and 0.5% bovine serum albumin (BSA). The RCP of the mAb was determined by ITLC on silica gel strips with 0.15 mol/L citrate buffer (pH 6.0) as the mobile phase.
Quantitative Determination of Antigen Expression
The G250 antigen density on the tumor cell surface was determined in an in vitro saturation assay, essentially as described by van Oosterhout et al. (18) with minor modifications. Briefly, tumor cells were incubated with various concentrations of 125I-cG250 (1.7–5,000 ng/100 μL mAb). After 4-h incubation at 4°C (to minimize internalization), cells were washed twice and the cell-bound fraction was determined in each vial using a γ-counter (Wallac Wizard 3″ 1480 automatic γ-counter).
Vascular Characteristics of Tumors
The relative vascular density of the tumors was determined immunohistochemically by staining the endothelial cells using a rat mAb specifically directed against murine endothelial cells: mAb 9F1 (19). Frozen sections (4 μm) of the tumors were air dried and fixed in cold acetone (−20°C) for 10 min. Subsequently, slides were rinsed with PBS and incubated for 1 h at room temperature with mAb 9F1 (1:50) in PBS and 0.1% BSA. Slides were rinsed with PBS, incubated with peroxidase-labeled goat anti-rat IgG (1:100) (GαRa-PO; Sigma-Aldrich), washed (PBS), and developed with 3,3′-diaminobenzidine (DAB) and 0.01% H2O2 (Sigma). Finally, the sections were counterstained with hematoxylin and mounted.
The vascular volume and vascular permeability of xenografted NU-12, SK-RC-1, and SK-RC-52 tumors were determined as described (20,21). Briefly, to determine the vascular volume of the tumors, the erythrocytes of the mice were labeled in vivo with 99mTc: Tumor-bearing mice were injected intravenously with 0.8 μg stannous pyrophosphate 30 min before the administration of 555 kBq (15 μCi) 99mTcO4−. To determine the vascular permeability of the tumors, the tumor-bearing mice were injected intravenously with 3 μg 125I-labeled MOP-C21 (an irrelevant mAb; 1 μCi/μg) simultaneously with the administration of 99mTcO4−. One hour after administration of the radioactivity, mice were killed by CO2 asphyxiation, and the radioactive concentrations of 99mTc and 125I in the blood and in the tumors were determined (vascular volume expressed as μL blood/g tumor; vascular permeability expressed as μL plasma/h/g tumor).
Murine RCC Tumor Models
Athymic female BALB/c nu/nu mice (6- to 8-wk old) were subcutaneously xenografted with 1 of the 3 RCC tumors (SK-RC-52: 1.5 × 106 cells per 200 μL; SK-RC-1: 2 × 106 cells per 200 μL; and NU-12: by serial subcutaneous transplantation). Two to 4 wk later, when the tumors were palpable (tumor size, 50–300 mg), mice were used in the biodistribution studies and determination of the vascular volume and permeability or tumors were dissected for the determination of G250 antigen expression.
Determination of G250 Antigen Expression by Fluorescence-Activated Cell Sorting (FACS) Analysis
Tumors were cut into small pieces, incubated overnight with collagenase, DNAse, and hyaluronidase RPMI + penicillin/streptomycin solution (Gibco). Subsequently, a cell suspension was produced using a cell strainer (70 μm; Falcon). Erythrocytes were lysed by a hypotonic shock in lysis buffer (0.8% NH4Cl, 0.1% KHCO3, and 3.7% ethylenediaminetetraacetic acid) for 10 min at 37°C.
Relative G250 antigen expression on human cells was determined by FACS analysis: human tumor cell suspensions were stained with α-CAR mAb (mouse anti-Coxsackie–Adenovirus Receptor; Upstate Biotechnology) (SK-RC-52) or with α-HLA-1 mAb (mouse anti-Human-Leukocyte-Associated 1; DAKO) (SK-RC-1). Cells were also stained with cG250 mAb (Centocor). GαM-PE (goat anti-mouse IgG/phycoerythrin; Southern Biotechnology Associates, Inc.) and GαHu-FITC (goat anti-human IgG/fluorescence isothiocyanate; DAKO) were used as conjugates. Cells were selected for CAR or HLA-1 expression and G250 antigen expression, and the fraction of the G250 antigen–positive cells was determined. The FACS analysis of cultured cells was performed similarly.
Biodistribution Studies
For the pretargeting experiments, animals were injected intravenously with 0.1 nmol (15 μg) bs-mAb G250xDTIn-1. Three days later, 4 pmol (6 ng) of the 111In-labeled peptide were administered. All reagents were injected intravenously via the tail vein (200 μL). At various time points after the injection of the radiolabeled peptide, mice were killed by CO2 asphyxiation. Blood was obtained by heart puncture. Tissues (tumor, muscle, lung, spleen, kidney, liver, and small intestine) were dissected and weighed, and their radioactivity content was determined in a γ-counter. To permit calculation of the radioactive uptake in each organ as a fraction of the injected dose, an aliquot of the injection dose was counted simultaneously. Results are expressed as % ID/g. All groups consisted of 4 or 5 mice. The biodistribution of 125I- and 111In-cG250 was determined in the 3 nude mouse tumor models. Mice were injected intravenously with 25 pmol (3.75 μg) mAb cG250 and, 72 h later, mice were killed and the biodistribution of the radiolabel was determined.
The studies were approved by the local Animal Welfare Committee and performed in accordance with their guidelines.
Statistical Analysis
All mean values are given ± SD. Statistical analysis was performed using the unpaired t test when 2 groups were analyzed, and the 1-way ANOVA was used when >2 groups were analyzed. The level of significance was set at P < 0.05.
RESULTS
G250 Antigen Expression of RCC cells
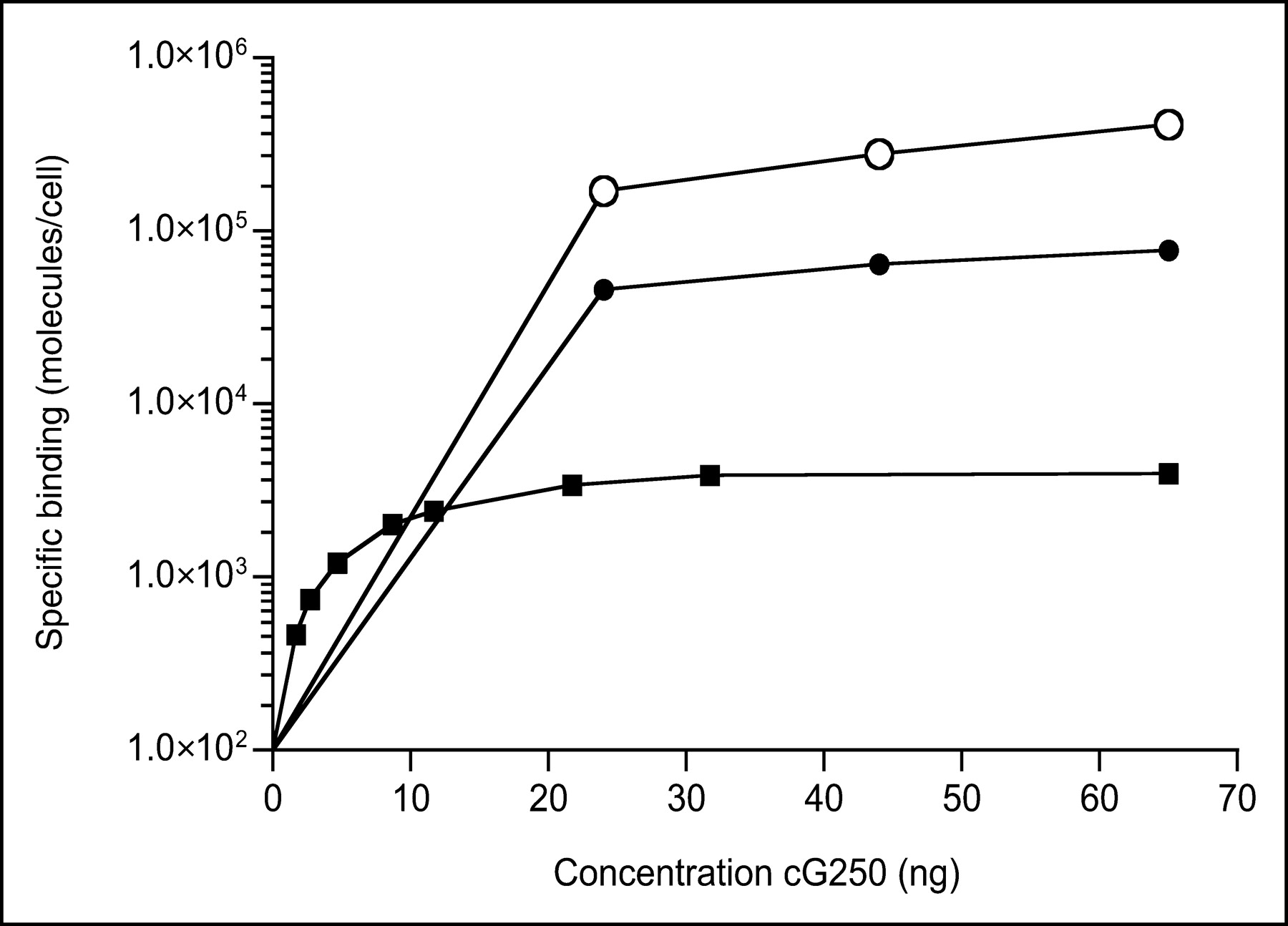
Determination of the G250 antigen density on the tumor cell surface by in vitro saturation assays indicated that the G250 antigen expression levels of the 3 RCC cell lines varied considerably (Fig. 1): SK-RC-52, 6 × 105 epitopes per cell; NU-12, 8 × 104 epitopes per cell; and SK-RC-1, 4 × 103 epitopes per cell.
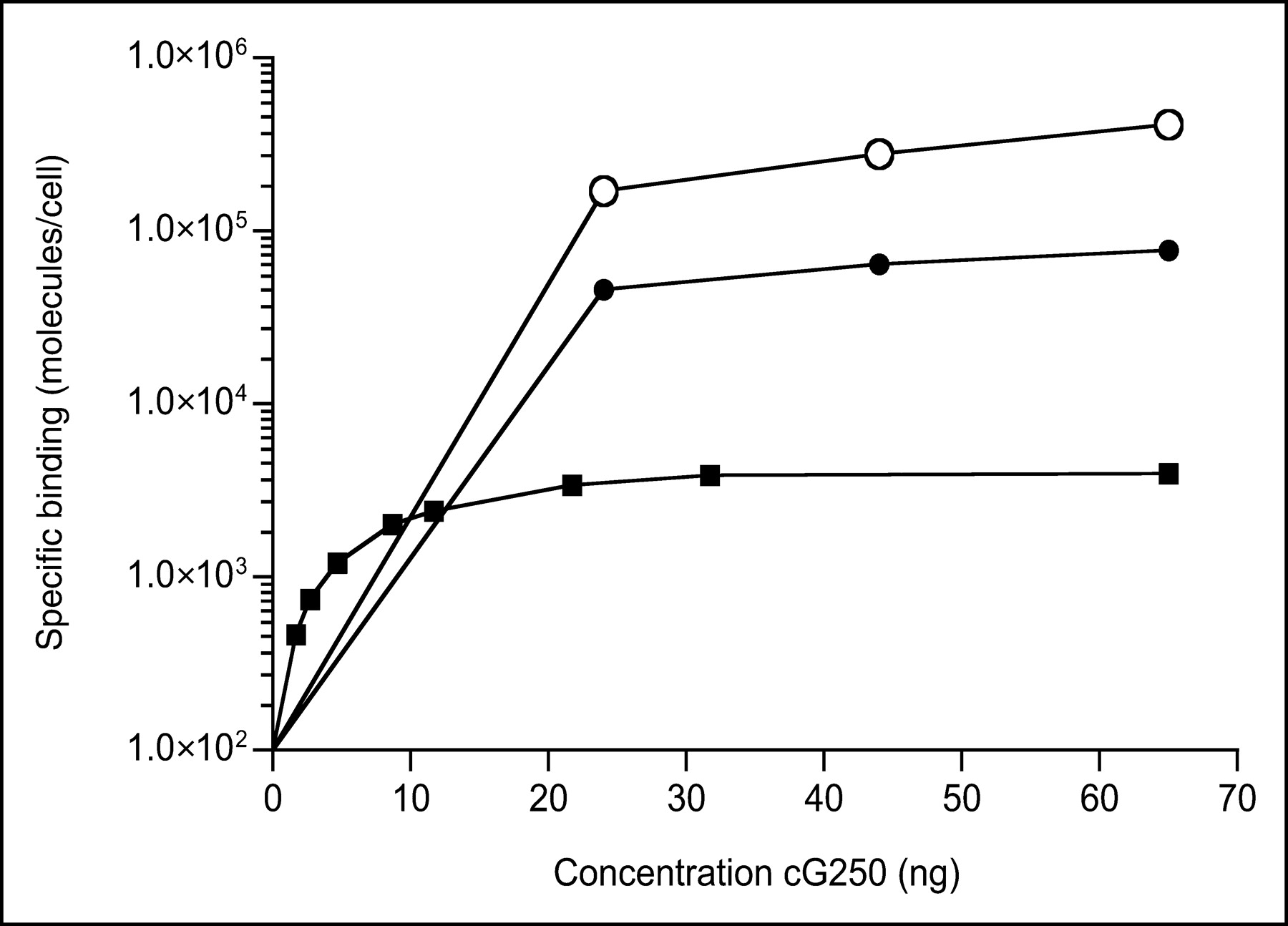
Antigen binding saturation test was performed to determine G250 antigen expression at cell surface of SK-RC-52 (○), NU-12 (•), and SK-RC-1 (▪) tumor cells. An increasing amount of cG250 mAb was added to a fixed number of tumor cells. After 4-h incubation at 4°C, samples were examined and antigen expression was calculated and expressed as number of epitopes per cell.
The mean fluorescence intensity (MFI), a relative measure of antigen expression, measured in the FACS analysis, indicated that the different expression levels measured on the in vitro cultured cells were also found on the cells from the tumor xenografts (Table 1). NU-12 cells showed moderate G250 antigen expression in vitro (MFI = 77). In vitro, approximately 75% of the SK-RC-1 cells expressed the G250 antigen, with an MFI of 43. To assess the G250 antigen expression levels correlated with the expression levels in vivo, FACS analysis was also performed on cells isolated from tumor xenografts. In vivo, approximately half of the SK-RC-1 cells (75% of the cells in the tumor) were G250 antigen positive, with an MFI index of 27. All SK-RC-52 cells cultured in vitro were G250 antigen positive, with an MFI index of 446. Approximately 90% of the SK-RC-52 cells (58% of the cells in the tumor), obtained from a xenografted tumor, were G250 antigen positive, with an MFI index of 100.
Antigen Expression of RCC Cells
The relative G250 antigen expression levels on cells isolated from the xenografts were confirmed by immunohistochemical staining using DAB-conjugated cG250 mAb. Slides were scored on the intensity of DAB staining: SK-RC-52 > NU-12 > SK-RC-1 (results not shown).
The vascular volume of the tested RCC models was in the same order of magnitude and at least twice as high as compared with the vascular volume of the 2 tested colon carcinoma models (Table 2). The vascular permeability of the RCC models was significantly different: SK-RC-1 tumor had a 3-fold higher vascular permeability compared with the SK-RC-52 tumor (159 ± 24 vs. 53.0 ± 5.0 μL plasma/h/g tumor, respectively). The vascular permeability of NU-12 and SK-RC-52 was in the same range (vascular permeability: NU-12, 63.1 ± 15.7 μL plasma/h/g tumor).
Vascular Characteristics of 3 RCC Tumors and 2 Colon Cancer Tumors
Immunohistochemical Analyses
Results of the endothelial cell staining of the RCC xenografts are shown in Figure 2. SK-RC-52 tumors showed a moderately high vessel density in the peripheral area of the tumors, whereas the center of the tumor was moderately necrotic. The vessel density in NU-12 tumors was similar to that of SK-RC-52 tumors, but the vessels were more homogeneously distributed throughout the whole tumor. SK-RC-1 tumors showed a very high vessel density at the tumor periphery, with central necrosis.
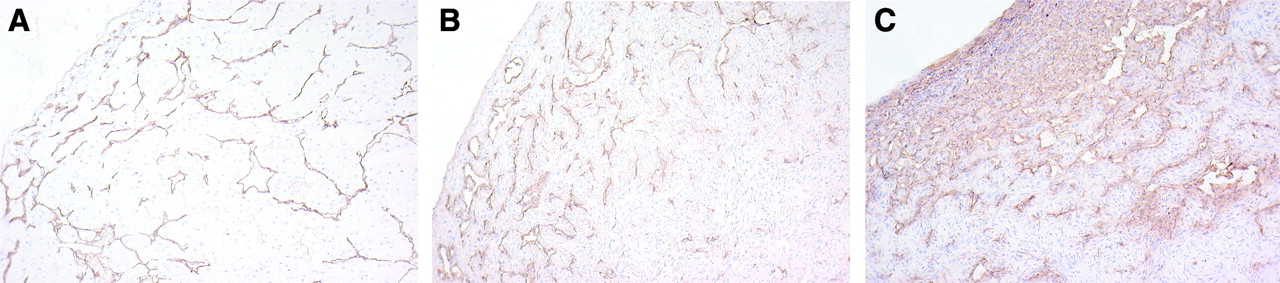
Endothelial staining of tested RCCs: NU-12 (A), SK-RC-52 (B), and SK-RC-1 (C). Sections of tumors were treated with GαRa-PO after treatment with 9F1 mAb. Nuclei of cells are stained blue by hematoxylin treatment. Detailed photographs (original magnification, 100× focused) of peripheral area of tumors are shown.
Pretargeting of RCC in 3 Tumor Models
The results of the pretargeting studies are summarized in Figure 3. In each model the tumor uptake of the 111In-labeled peptide was high. However, there was a marked difference in the maximum uptake in the tumor found in each model: SK-RC-1 (278 ± 130 %ID/g at 1 h after injection) ≫ NU-12 (93 ± 41 %ID/g at 72 h after injection) > SK-RC-52 (54 ± 9 %ID/g at 4 h after injection). Major differences were also found in the retention of the 111In label in the 3 tumors. The 111In label was retained in NU-12 tumors from 4 to 72 h after injection (78 ± 41 %ID/g and 93 ± 41 %ID/g, respectively). In contrast, in the other 2 RCC models, the uptake of the radiolabel in the tumor decreased with time: In SK-RC-52 tumors a decrease from 54 ± 9 %ID/g at 4 h after injection to 17 ± 2 %ID/g at 72 h after injection was observed, and the SK-RC-1 tumors showed a decrease from 287 ± 130 %ID/g at 1 h after injection to 75 ± 38 %ID/g at 72 h after injection. The radioactivity in the normal tissues was similar and low in all 3 RCC models, resulting in very high tumor-to-blood ratios (T/B) at 72 h after injection (NU-12, T/B = 3,490 ± 1,930; SK-RC-52, T/B = 1,337 ± 236; SK-RC-1, T/B = 4,829 ± 1,636). To demonstrate the specificity of the tumor targeting, in a separate group of mice the tumor was pretargeted with 15 μg irrelevant bs-mAb (MN14xDTIn-1) and 72 h later mice were injected intravenously with 6 ng 111In-labeled peptide. Mice were killed 24 h after injection of the radiolabel. The tumor uptake was 0.51 ± 0.16 %ID/g and the blood level was 0.52 ± 0.14 %ID/g. The radioactivity levels of the normal organs were very low (<0.75 %ID/g).

Biodistribution of subcutaneous NU-12 (A), SK-RC-52 (B), or SK-RC-1 (C) tumor-bearing mice, pretargeted with 0.1 nmol (15 μg) bs-mAb G250xDTIn-1, and 72 h later 4 pmol (6 ng) 111In-diDTPA were administered. Mice were killed at various time points after injection of radiolabel, and biodistribution of radiolabel was determined. At all time points, groups consisted of 5 mice. Uptake is expressed as %ID/g.
Biodistribution of Directly Labeled cG250 mAb in 3 RCC Models
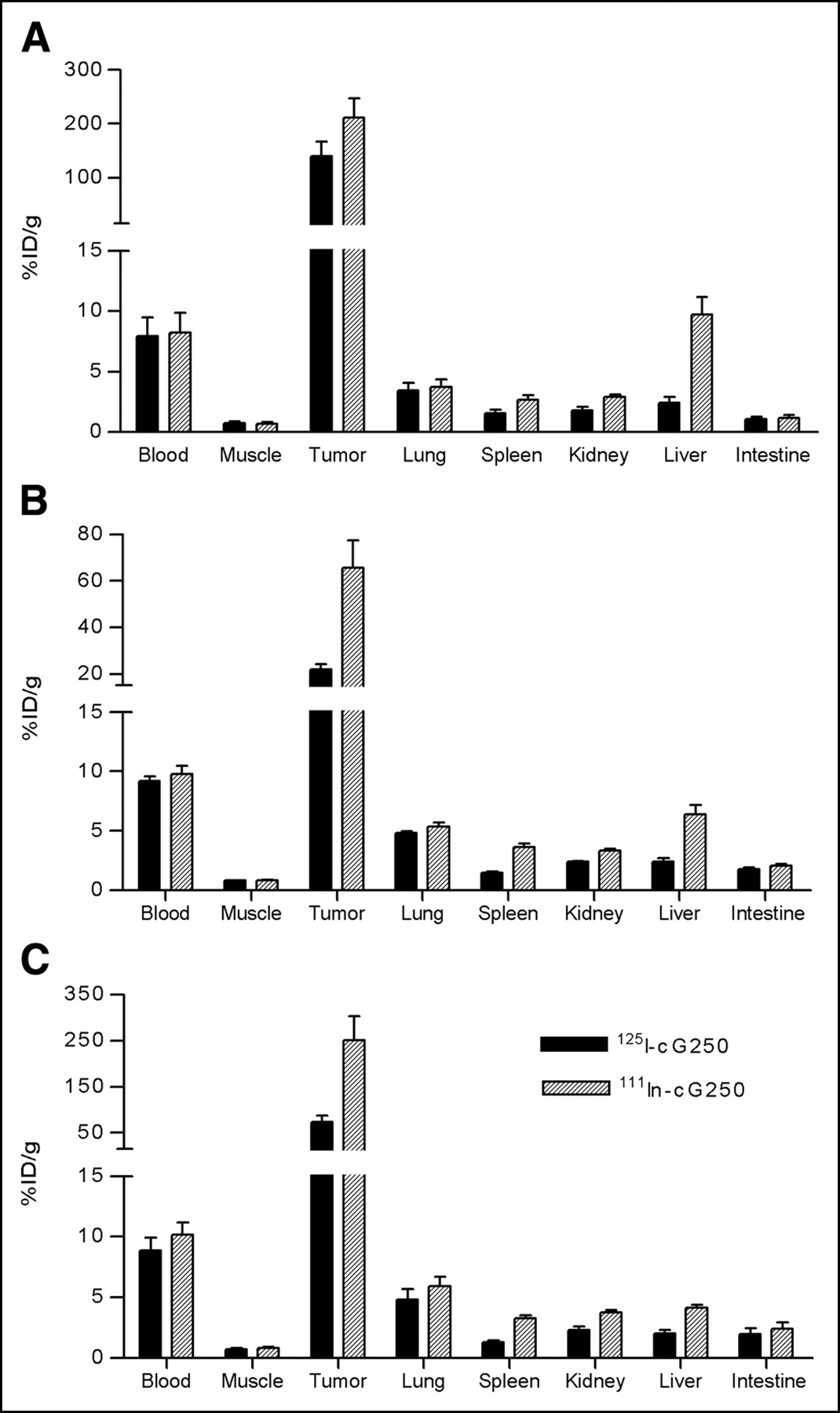
The biodistribution results of the 111In- and 125I-labeled cG250 mAb (specific activity, 11.1 GBq/μmol [0.3 Ci/μmol] and 14.8 GBq/μmol [0.4 Ci/μmol], respectively) are summarized in Figure 4. At 3 d after injection of the 111In-labeled mAb, uptake of the 111In label in the tumor was relatively high in each RCC model. The highest tumor uptake was observed in SK-RC-1 tumors (251 ± 90 %ID/g) and NU-12 tumors (211 ± 79 %ID/g). The uptake in SK-RC-52 tumors (66 ± 23 %ID/g) was significantly lower. To investigate the effect of internalization and subsequently metabolization of the targeted antibody, the biodistribution of the radioiodinated cG250 mAb was also studied in the 3 tumor models. For SK-RC-52 and SK-RC-1 tumors, the uptake of 125I-cG250 was 3-fold lower than that of 111In-cG250, whereas in NU-12 tumors, uptake of 125I-cG250 was only slightly lower than that of 111In-cG250 (137 ± 36 %ID/g vs. 218 ± 56 %ID/g).
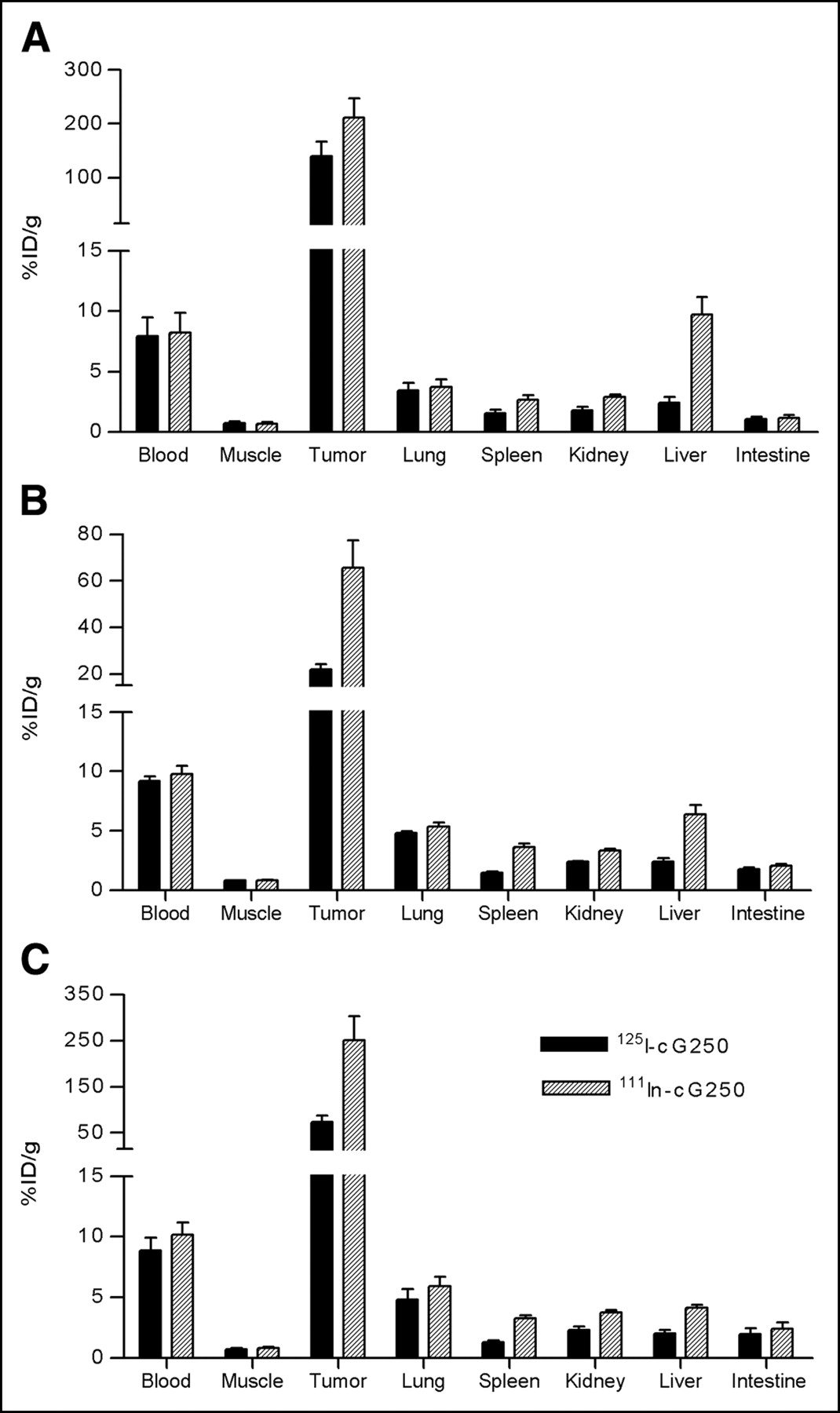
Biodistribution of subcutaneous NU-12 (A), SK-RC-52 (B), or SK-RC-1 (C) tumor-bearing mice, targeted with 25 pmol (3.75 μg) dual-label 111In-/125I-labeled cG250 mAb. Mice were killed at 72 h after injection of labeled mAb, and biodistribution of radiolabel was determined. Groups consisted of 4 or 5 mice. Uptake is expressed as %ID/g.
DISCUSSION
In these studies we tested the efficiency of tumor targeting using the pretargeting system with bs-mAb G250xDTIn-1 in different RCC tumor models. The studies were performed to further examine the excellent targeting obtained in our earlier studies. The biodistribution of an 111In-labeled peptide after pretargeting with bs-mAb G250xDTIn-1 was studied in 3 RCC models. Uptake of the radiolabeled peptide in the G250 antigen–expressing tumor models using the pretargeting strategy was considerably higher than that reported in other pretargeting studies using other mouse tumor models (8–10). Unexpectedly, in the pretargeting studies, the 3 RCC models showed no relation between G250 antigen expression and uptake of the radiolabel in the tumor. Paradoxically, the tumor with the lowest G250 antigen expression (SK-RC-1) showed the highest uptake of the radiolabel in the tumor, whereas the SK-RC-52 tumor model with the highest G250 antigen expression level showed the lowest uptake.
To exclude that this paradox was caused by a difference of antigen expression levels between in vitro cultured cells and in vivo growing cells (22), the in vitro and in vivo grown cells were analyzed with a FACS. To discriminate between human and murine cells in the tumors, FACS analysis was performed by double-fluorescence FACS staining. Less than 60% of the xenografted SK-RC-52 tumors were of human origin, whereas 75% of the SK-RC-1 tumors were tumor cells. These results contributed only partially to the paradoxical findings between tumor targeting and G250 antigen expression. FACS analyses indicated that for each cell line the MFI index (a measure of relative G250 antigen expression) of cells isolated from the subcutaneous growing tumor was lower than that of the in vitro cultured cells. However, the relative expression of the G250 antigen in the subcutaneous growing SK-RC-52 tumor cells was higher than that of the subcutaneous growing SK-RC-1 tumor cells.
Targeting of the tumor with the radiolabeled peptide was specific when performed with bs-mAb G250xDTIn-1. No specific tumor targeting of the radiolabeled bivalent peptide was observed using the bs-mAb MN14xDTIn-1.
It has been recognized that many physiologic characteristics of the tumor can affect mAb targeting (for example, vascular volume and permeability, blood flow, stromal components and the presence or absence of necrosis). As shown in Figure 2, immunohistochemical analysis indicated that the vessel density of SK-RC-52 and NU-12 tumors was in the same range, whereas SK-RC-1 tumors with the lowest G250 antigen expression had a higher vessel density, suggesting that the differences in tumor vascularization might explain the tumor-targeting differences. Further analysis of the vascular characteristics indicated that the vascular volumes of NU-12, SK-RC-1, and SK-RC-52 were in the same range (33.7 ± 5.9, 20.8 ± 4.1, and 21.2 ± 3.8 μL blood/g tumor, respectively), indicating that the variation in tumor uptake could not be attributed to the differences in vascular volume. The vascular permeability of the SK-RC-1 tumors was 3-fold higher than that of the SK-RC-52 and NU-12 tumors (159 ± 24 vs. 53 ± 5 and 63 ± 16 μL plasma/h/g tumor, respectively). This finding suggests that relatively efficient targeting of SK-RC-1 with the pretargeting system might be due to the enhanced vascular permeability. Other physiologic characteristics of the tumors, such as the interstitial fluid pressure, could also contribute to mAb targeting by limiting the penetration of mAbs into the tumor tissue (23,24). The lack of correlation between the G250 antigen expression and the tumor-targeting efficiency in the tested tumor models was also found with directly 125I-/111In-labeled cG250 mAb. Targeting of SK-RC-1 RCC with directly labeled cG250 exceeded targeting of NU-12 RCC, whereas SK-RC-52 RCC showed a relatively low uptake. In general, 125I-cG250 uptake was lower than 111In-cG250 uptake, suggesting internalization and, subsequently, degradation of the labeled mAb. It appeared that internalization and subsequently metabolization did play a minor role in the NU-12 tumor model.
The kinetics of the targeted radiolabel in the examined tumor models varied considerably: SK-RC-1 and SK-RC-52 showed a distinct washout of the 111In-labeled peptide from the tumor with time, whereas the 111In label was retained in NU-12 tumors with time. The washout of the radiolabel (Fig. 3) suggests that the binding of the 111In-labeled peptide to the tumor cell surface via a bs-mAb in SK-RC-1 and SK-RC-52 renal cell tumors might be reversible. This could be due to a monovalent binding of the bivalent peptide to the cell surface, instead of the more-avid bivalent binding. This delicate balance, to obtain bivalent binding, most likely depends on the G250 antigen density, the amount of bs-mAb G250xDTIn-1, the amount of 111In-diDTPA, and the internalization rate of the bs-mAb at the tumor cell surface.
These studies show that excellent tumor targeting via bs-mAb G250xDTIn-1 can be achieved in G250 antigen–expressing tumor models. Clinical studies with this pretargeting approach in RCC patients are warranted. Pretargeting would allow rapid imaging, early after injection of the radiolabel, and, for therapeutic applications, a higher radiation dose can be guided to the tumor using the pretargeting strategy.
CONCLUSION
Despite the fact that each RCC tumor has distinct physiologic characteristics, with the pretargeting strategy very high uptake of the radiolabel in each of the tumors was observed. There was no relation between G250 antigen expression and uptake and retention of the radiolabel in the tumor. These studies show that the enhanced vascular permeability allowed efficient tumor targeting with the pretargeting strategy.
Acknowledgments
This work was supported by a research grant from the Dutch Cancer Society (KUN 2000-2306). We thank G. Grutters, H. Eijkholt, and B. de Weem (University of Nijmegen, Central Animal Laboratory) for technical assistance in the animal experiments.
Footnotes
Received Aug. 26, 2004; revision accepted Sep. 30, 2004.
For correspondence or reprints contact: Frank G. van Schaijk, MSc, Department of Nuclear Medicine, University Medical Center Nijmegen, P.O. Box 9101, 6500 HB Nijmegen, The Netherlands.
E-mail: F.vanSchaijk{at}nucmed.umcn.nl
REFERENCES
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Pretargeting: A Path Forward for Radioimmunotherapy
- Acetazolamide Serves as Selective Delivery Vehicle for Dipeptide-Linked Drugs to Renal Cell Carcinoma
- A 99mTc-Labeled Ligand of Carbonic Anhydrase IX Selectively Targets Renal Cell Carcinoma In Vivo
- A New Tri-Fab Bispecific Antibody for Pretargeting Trop-2-Expressing Epithelial Cancers
- Antibody-Radionuclide Conjugates for Cancer Therapy: Historical Considerations and New Trends
- Successful Radiotherapy of Tumor in Pretargeted Mice by 188Re-Radiolabeled Phosphorodiamidate Morpholino Oligomer, a Synthetic DNA Analogue.
- Pretargeting of Carcinoembryonic Antigen-Expressing Tumors with a Biologically Produced Bispecific Anticarcinoembryonic Antigen x Anti-Indium-Labeled Diethylenetriaminepentaacetic Acid Antibody
- Residualizing Iodine Markedly Improved Tumor Targeting Using Bispecific Antibody-Based Pretargeting
- The Direct Route May Not Be the Best Way to Home