


Abstract
Nicotine α4β2 receptor subtypes are implicated in the study of Alzheimer’s disease, schizophrenia, substance abuse, lung cancer, and other disorders. We report the development and evaluation of a putative antagonist, 5-(3′-fluoropropyl)-3-(2-(S)-pyrrolidinylmethoxy)pyridine (nifrolidine) as a PET agent for nicotine α4β2 receptors. Methods: In vitro binding affinity of nifrolidine was measured in rat brain slices labeled with 125I-iodoepibatidine or 125I-bungaratoxin. Selectivity of binding was measured in the presence of cytisine. 18F radiolabeling was performed by reacting the tosylate precursor with 18F-fluoride followed by deprotection. In vitro autoradiographic studies in rat brain slices with 5-(3′-18F-fluoropropyl)-3-(2-(S)-pyrrolidinylmethoxy)pyridine (18F-nifrolidine) were read on a phosphor imager. Rats were injected with 18F-nifrolidine (3.7 MBq each), and brain regions were counted at various times (2–120 min). Blocking studies were performed by subcutaneous injection of nicotine (10 mg/kg). A PET study of 18F-nifrolidine (approximately 148 MBq) was performed on an anesthetized rhesus monkey using a high-resolution scanner. Results: In vitro binding affinity of nifrolidine exhibited an inhibition constant of 2.89 nmol/L for the α4β2 sites. Radiosynthesis and high-performance liquid chromatography purifications yielded the product in approximately 20%–40% decay-corrected radiochemical yield to provide 18F-nifrolidine specific activities of approximately 111–185 GBq/μmol. In vitro autoradiography in rat brain slices revealed selective binding of 18F-nifrolidine to the anteroventral thalamic nucleus, ventral posteriomedial thalamus, dorsolateral geniculate, and, to a lesser extent, cortex and striata, which are known to contain α4β2 sites. This specific binding was completely abolished by 300 μmol/L nicotine. Ex vivo rat brain distribution studies indicated selective binding in the thalamus with a maximal thalamus-to-cerebellum ratio of approximately 3. The PET study revealed selective maximal uptake (0.01% injected dose/mL) in regions of the thalamus (anteroventral and anteromedial thalamus, ventrolateral thalamus) and extrathalamic regions such as cingulate gyrus, lateral geniculate, temporal cortex, and frontal cortex. Conclusion: Binding of 18F-nifrolidine to α4β2 receptor-rich regions in rats and monkeys indicates promise as a PET agent. Additionally, the thalamus-to-cerebellum ratio approached a plateau of 1.7 in 120 min, indicating relatively faster kinetics compared with previously reported imaging agents.
Nicotinic acetylcholine receptors (nAChRs) belong to the superfamily of ligand-gated ion channels and are distributed widely in the human and nonhuman brain (1). At least 17 different subunits are currently known, which can coassemble in several ways resulting in 3 distinct types of nAChRs: (a) heteromeric receptors found in neuromuscular junctions, (b) heteromeric receptors found in the neurons, and (c) homomeric receptors also found in the neurons. These receptors mediate some effects of the endogenous neurotransmitter acetylcholine (ACh) and are also the biologic target of the tobacco alkaloid nicotine, which is known to mimic the actions of ACh at these receptors. Subunit compositions determine functional properties such as ion selectivity, conductance, channel open times, rate of desensitization, and sensitivity to certain neurotoxins (2). Several nAChRs have been identified and characterized pharmacologically and have distinct patterns of distribution in the brain. The α4β2 nAChR heteromeric neuronal-subtype receptors are considered important for the study of Alzheimer’s disease (AD), schizophrenia, substance abuse, and other disorders (3,4). It has been suggested that the loss of the α4β2 receptors may be an early presymptomatic marker for AD (4). These neuronal receptors may be involved in the addiction to nicotine in chronic tobacco users and tobacco use may increase the number of the α4β2 receptor sites (5,6).
Clinical significance of the α4β2 nAChR subtype has resulted in the development of noninvasive imaging methods using PET and SPECT of this receptor system. PET studies have been performed with 11C-nicotine. However, the moderate affinity of nicotine for the α4β2 receptors resulted in rapid clearance and, therefore, precluded its usefulness as a radiotracer (7). Catalyzed by the discovery of epibatidine (8), various PET and SPECT radioligands have been discovered (9). In general, these include various epibatidine analogs and pyridylether analogs that have been radiolabeled with 11C, 18F, 76Br, or 123I (Fig. 1). Additional radioligands have been prepared in an effort to optimize in vivo imaging properties (10–12). Toxicity issues and appropriate kinetics have slowed the progression of these radiotracers for human studies. Nonetheless, human SPECT studies have now begun with 5-123I-A85380, and PET studies have begun with 2-18F-A85380 and, more recently, with 6-18F-A85380 (13–16).

Chemical structures of α4β2 radioligands. (A) Epibatidine analogs. (B) Pyridylether analogs. (C) Azetidinylether analogs in human studies. (D) Fluoroalkyl dervatives of pyrrolinylethers (nifrolidine, this work).
The PET and SPECT radioligands developed thus far for the α4β2 nAChR subtype have been agonists. It has been suggested that α4β2 nAChRs may occur in 4 possible conformations (17,18): (a) a resting state, when no agonist is present; (b) an activated state, when agonist is present and the ion channel is open; (c) a transiently desensitized state, when the ion channel is closed for small lengths of time (i.e., seconds); and (d) a desensitized state, when the ion channel is closed for longer periods of time. ACh is known to bind with different affinities to these different states (e.g., ACh has higher affinity for desensitized states). It is unclear at this time whether antagonists also would have different affinities for the various states or whether their in vivo binding pattern would be different compared with that of the agonists such as 2-18F-A85380. It is likely that development of an antagonist imaging agent for the α4β2 receptor subtype may provide additional information of this receptor system and may complement information that is obtained by agonist-based radiotracers.
We have thus embarked on the development of PET radiotracers for the α4β2 receptor based on antagonists. It has been shown that inclusion of alkyl groups at the 5-position in the pyridine ring of pyridyl ethers leads to a change from an agonist to an antagonist character (19,20). In a series of pyridylethers, inclusion of a propyl or butyl group in the 5-position instead of a hydrogen or a halogen leads to the inhibition of Rb2+ efflux, suggestive of the antagonistic character (19). We have used this approach and have included a 3′-fluoropropyl group at the 5-position in the pyridylether linked to a pyrrolidine ring. This 3′-fluoropropyl group is analogous to the propyl group and, therefore, it is anticipated that these compounds may be potential antagonists. The fluorine at the 3-position may only have a minimal effect on the pyridine nitrogen and is sufficiently away from the pyrrolidine nitrogen. The pyrrolidine ring was chosen (rather than the azetidine ring, which is known to result in higher affinity at the α4β2 receptor site) so that initially a moderate-affinity compound for this receptor could be prepared (21). The moderate affinity may help in accelerating in vivo binding kinetics, an issue that has been raised in the case of 2-18F-A-85380 and related compounds (22,23). We report here the synthesis of 5-(3′-fluoropropyl)-3-(2-(S)-pyrrolidinylmethoxy)pyridine (nifrolidine), in vitro pharmacology at the nAChRs, radiolabeling with 18F to provide 5-(3′-18F-fluoropropyl)-3-(2-(S)-pyrrolidinylmethoxy)pyridine (18F-nifrolidine), in vitro autoradiographic studies in rat brain slices with 18F-nifrolidine, in vivo biodistribution studies in rats, and a PET study in a rhesus monkey.
MATERIALS AND METHODS
All chemicals and solvents were of high grade from Aldrich Chemical Co. Electrospray mass spectra were obtained on a model 7250 mass spectrometer (Micromass LCT). Proton nuclear magnetic resonance (NMR) spectra were acquired on a General Electric NMR Omega 500 MHz. High-specific-activity 18F-fluoride was produced in the MC-17 cyclotron or the CTI RDS-112 cyclotron using an 18O-enriched water target (18O to 18F using p,n reaction). The high-specific-activity 18F-fluoride was used in subsequent reactions. Chromatographic separations were performed on semipreparative reverse-phase columns using the Gilson high-performance liquid chromatography (HPLC) systems. 18F radioactivity was counted in a Capintec dose calibrator, whereas low-level counting was performed in a well counter (Cobra Quantum; Packard Instruments Co.). Radioactive thin-layer chromatographs were obtained by scanning in a Bioscan System 200 Imaging scanner (Bioscan, Inc.). Rat brain slices were obtained on a Leica 1850 cryotome. 18F autoradiographic studies were performed by exposing tissue samples on storage phosphor screens. The apposed phosphor screens were read by a Cyclone Storage Phosphor System (Packard Instruments Co.). Monkey PET studies were performed using a high-resolution ECAT EXACT HR+ scanner (Siemens/CTI). All animal studies were approved by the Institutional Animal Care and Use Committee of University of California at Irvine and Wright State University.
Chemistry
5-Bromo-3-Hydroxypyridine.
Using reported procedures (24), 3,5-dibromopyridine 1 (5 g, 21 mmol) was dissolved in 100 mL anhydrous methanol (Fig. 2). The solution was cooled to 0°C and 16 g of sodium hydride (60% [w/w]) mineral oil suspension) were added in portions. This mixture was stirred for 1 h at room temperature and refluxed at 80°C for 4 h. The solvent was removed by rotary evaporation. The residue was taken in 100 mL of water. The mixture was extracted with dichloromethane (3 × 100 mL). The organic portions were combined, dried (anhydrous magnesium sulfate), filtered, and evaporated, and the residue (3.5 g) was characterized as 5-bromo-3-methoxypyridine 2 (25). The 5-bromo-3-methoxypyridine 2 was refluxed with 20 mL concentrated (57%) hydrogen bromide for 24 h. The reaction was quenched with saturated NaHCO3 solution, and the basic mixture was extracted with dichloromethane (3 × 50 mL). The organic portions were pooled together, dried (magnesium sulfate), filtered, evaporated to provide the 5-bromo-3-hydroxypyridine 3 (3 g, 82%) in >95% purity as ascertained by thin-layer chromatography (TLC) (Rf = 0.5, ethyl acetate/hexane,1:1). NMR (CDCl3, 500 MHz) δ-ppm: 8.28 (d, 1H, J = 1.8 Hz), 8.24 (d, 1H, J = 2.7 Hz), 7.38 (dd, 1H, J = 1.8, 2.7 Hz). MS, m/z, 173, 175 (25%, [M+H]+).
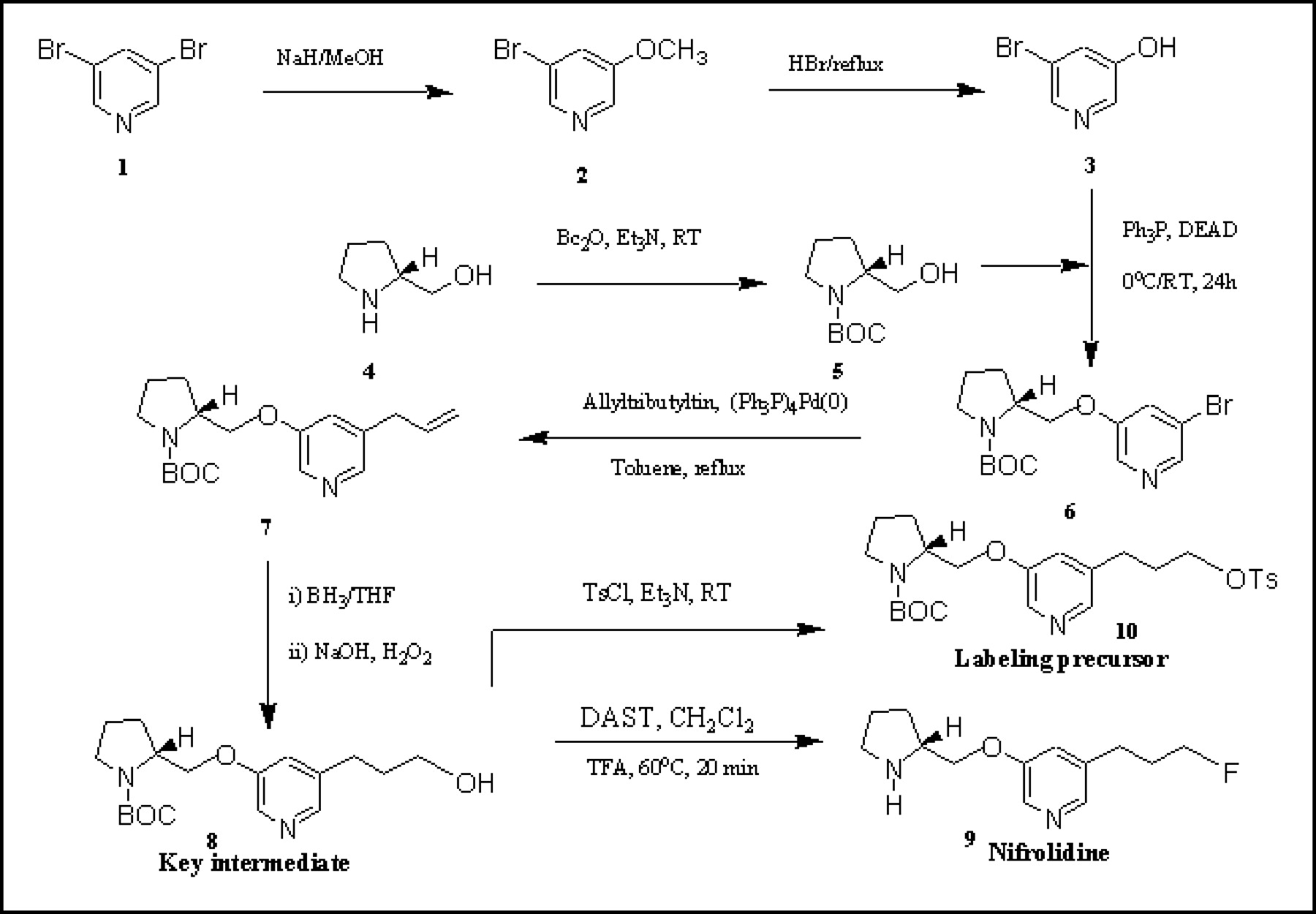
Synthesis scheme for 5-(3′-fluoropropyl)-3-(2-(S)-pyrrolidinylmethoxy)pyridine (9, nifrolidine) and tosylate precursor, 5-(3′-[[4-methylphenyl]sulfonyloxy]propyl)-3-(1-BOC-2-(S)-pyrrolidinylmethoxy)pyridine 10 for radiolabeling with 18F. BOC = butoxycarbonyl.
5-Bromo-3-(1-BOC-2-(S)-Pyrrolidinylmethoxy)Pyridine (BOC = Butoxycarbonyl).
Di-tert-butyl dicarbonate (4.3 g, 19.7 mmol) was added to a cold (0°C) mixture of 2-(S)-pyrrolidinemethanol 4 (2 g, 19.7 mmol) and triethylamine (2.8 mL, 20 mmol). After the mixture was stirred at room temperature for 30 min, 20 mL of dichloromethane were added and washed with saturated NaHCO3 (2 × 20 mL) and water (1 × 20 mL). The organic layer was dried (MgSO4), filtered, and concentrated to provide 1-BOC-2-(S)-pyrrolidinemethanol 5 (3.5 g, 88%) in >95% purity (MS, m/z, 224 (65%, [M+Na]+).
Diethyl azodicarboxlate (DEAD) (1.8 mL, 12 mmol) was added to a solution of triphenylphosphine (3.1 g, 12 mmol) in anhydrous tetrahydrofuran (THF) (30 mL) at 0°C, and the mixture was stirred for 30 min. A solution of 1-BOC-2-(S)-pyrrolidinemethanol (5) (2 g, 10 mmol) in 5 mL THF and 5-bromo-3-hydroxypyridine (3) (2 g, 11.4 mmol) in 5 mL THF was added at 0°C. The reaction mixture was allowed to stand at room temperature for 24 h. The solvent was removed by rotary evaporation and the residue was dissolved in dichloromethane (100 mL) and washed with saturated NaHCO3 (50 mL) and water (3 × 50 mL). The organic solution was dried over anhydrous MgSO4, filtered, and concentrated to oil. The crude oil was purified by silica column chromatography (hexane/ethyl acetate, 1:1) to afford the title compound 6 (1.56 g, 44%). NMR (CDCl3, 500 MHz) δ-ppm: 8.26 (dd, 2H, J = 15, 1.8 Hz), 7.37 (t, 1H, J = 2.4 Hz), 3.92 (dm, 2H, J = 34.7 Hz), 3.54 (m, 1H), 3.01 (m, 2H), 1.93 (m, 2H), 1.79 (m, 2H), 1.65 (s, 9H). MS, m/z, 379, 381 (5%, [M+Na]+).
5-Allyl-3-(1-BOC-2-(S)-Pyrrolidinylmethoxy)Pyridine.
A mixture of 5-bromo-3-(1-BOC-2-(S)-pyrrolidinylmethoxy)pyridine (6) (1.56 g, 4.4mmol), tetrakis(triphenylphosphine)palladium (0) (40 mg, 0.03 mmol), and allyltributyltin (1.5 mL, 4.4 mmol) in toluene (50 mL) was refluxed for 24 h. The mixture was filtered and the filtrate was evaporated. The residue so obtained was chromatographed (silica, ethyl acetate/hexane, 1:1) to afford the title compound 7 (800 mg, 57%). NMR (CDCl3, 500 MHz) δ-ppm: 8.16 (d, 1H, J = 2.5 Hz), 8.06 (d, 1H, J = 13.7 Hz), 7.05 (d, 1H, J = 27.7), 5.92 (m, 1H), 5.11 (m, 2H), 4.15 (m, 3H), 3.37 (d, 4H), 2.02 (m, 4H), 1.47 (s, 9H). MS, m/z, 319 (25%, [M+H]+), 341 (28%, [M+Na]+).
5-(3′-Hydroxypropyl)-3-(1-BOC-2-(S)-Pyrrolidinylmethoxy)Pyridine.
A solution of allyl derivative 7 (750 mg, 2.3mmol) in THF (5 mL) was cooled (0°C). To this solution was added diborane in THF (4.5 mL, 6.7 mmol), and the mixture was stirred at 0°C for 1 h followed by 1 h at room temperature. The reaction was cooled (0°C), and then 3N NaOH (5 mL) was added, followed by 30% aqueous hydrogen peroxide (200 μL). The reaction mixture was stirred at 0°C for 30 min and then at room temperature for 30 min. The THF was removed by rotary evaporation, and the resulting mixture was extracted with dichloromethane (3 × 5 mL). The combined organic extracts were dried (MgSO4), filtered, and concentrated to afford crude sticky product that was purified by preparative TLC (PTLC), to provide (ethyl acetate/hexane, 1:1) pure 8 (200 mg, 26%). NMR (CDCl3, 500 MHz) δ-ppm: 8.10 (s, 1H), 8.04 (s, 1H), 7.60 (s, 1H), 4.11 (m, 2H), 3.95 (m, 1H), 3.65 (m, 2H), 3.39 (m, 2H), 2.78 (m, 2H), 1.99 (m, 6H), 1.47 (s, 9H). MS, m/z, 337 (95%, [M+H]+), 359 (45%, [M+Na]+).
5-(3′-Fluoropropyl)-3-(2-(S)-Pyrrolidinylmethoxy)Pyridine.
The N-BOC alcohol 8 (40 mg, 0.12 mmol) in chloroform (200 μL) was treated with diethylaminosulfur trifluoride (DAST) (15.5 μL, 0.12 mmol) at 0°C. The reaction mixture was allowed to warm to room temperature and kept overnight. For work-up, 2 mL of chloroform and 2 mL of water were added to the reaction mixture, and the organic layer was separated and washed with 10% NaHCO3 (2 × 1 mL). The organic layer was dried (anhydrous sodium sulfate), filtered, and evaporated, and the residue was purified by PTLC (ethyl acetate/hexane, 1:1) to afford pure 5-(3′-fluoropropyl)-3-(1-BOC-2-(S)-pyrrolidinylmethoxy)pyridine (10 mg, 25%). NMR (CDCl3, 500 MHz) δ-ppm: 8.17 (d, 1H, J = 2.2 Hz), 8.08 (d, 1H, J = 15 Hz), 7.08 (d, 1H, J = 38.8 Hz), 4.47 (dt, 2H, J = 47.2, 5.8 Hz), 4.15 (m, 2H), 3.92 (m, 1H), 3.40 (m, 2H), 2.76 (m, 2H), 2.03 (m, 6H), 1.47 (s, 9H). MS, m/z, 339 (25%, [M+H]+), 361 (100%, [M+Na]+).
Deprotection of the N-BOC fluorinated derivative was performed by treatment with trifluoroacetic acid (TFA). The 5-(3′-fluoropropyl)-3-(1-BOC-2-(S)-pyrrolidinylmethoxy)pyridine (7 mg, 0.02 mmol) was dissolved in 0.8 mL dichloromethane and 0.2 mL TFA. The solution was heated at 80°C for 30 min. Solvents were removed in vacuo and the residue was neutralized to pH 9 with saturated sodium carbonate. The aqueous layer was extracted with dichloromethane, dried (MgSO4), filtered, and concentrated to afford crude product that was purified on PTLC (dichloromethane/methanol, 9:1) to provide pure 5-(3′-fluoropropyl)-3-(2-(S)-pyrrolidinylmethoxy)pyridine (4 mg, 81%). NMR (p-toluenesulfonate salt in CD3OH, 500 MHz) δ-ppm: 8.27 (d, 1H, J = 1.6 Hz), 8.19 (d, 1H, J = 6.8 Hz), 7.68 (dd, 2H, J = 6.5, 1.7 Hz), 7.63 (s, 1H), 7.22 (d, 2H, J = 7.9 Hz), 4.46 (dt, 2H, J = 47.4, 5.8 Hz), 4.38 (m, 2H), 4.08 (m, 1H), 3.40 (m, 2H), 2.84 (t, 2H, J = 7.6 Hz), 2.36 (s, 3H), 2.3–1.9 (m, 6H). MS, m/z, 239 (100%, [M+H]+).
5-(3′-[[(4-Methylphenyl)Sulfonyl]Oxy]Propyl)-3-(1-BOC-2-(S)-Pyrrolidinylmethoxy)Pyridine.
The alcohol 8 (75 mg, 0.22 mmol) was dissolved in CH2Cl2 (1 mL) and treated with Et3N (100 μL) and p-toluenesulfonyl chloride (42 mg, 0.22 mmol). The reaction mixture was stirred at room temperature for 24 h. The mixture was washed with water (2 × 1 mL), dried (Na2SO4), and separated on PTLC (ethyl acetate/hexane, 1:1) to afford 10 (50 mg, 46%). NMR (CDCl3, 500 Mz) δ-ppm: 8.10 (d, 1H, J = 2.8 Hz), 8.0 (s, 1H), 7.94 (s, 1H), 7.78 (d, 2H, J = 8.3 Hz), 7.22 (d, 2H, J = 7.9 Hz), 4.21 (m, 1H), 4.11 (m, 2H), 4.05 (t, 2H, J = 5.9 Hz), 3.60 (m, 2H), 2.72 (m, 2H), 2.47 (s, 3H), 1.99 (m, 6H), 1.55 (s, 9H). MS, m/z, 513 (100%, [M+Na]+).
In Vitro Binding Affinity
In vitro binding affinity of nifrolidine was measured in rat brain slices labeled with 125I-iodoepibatidine (125I-IEB) or 125I-α-bungarotoxin (26,27). The brains from male Sprague–Dawley rats (n = 4 per group) were extracted and frozen in isopentane at −20°C. Coronal sections (20-μm thick) were prepared from 4 brain levels on a cryostat at −20°C. These levels included cerebellum (Bregma −11.3), superior colliculus (Bregma −6.0 to −6.42), and 2 thalamic areas (Bregma −4.3 to −3.8). For 125I-IEB studies, slides were preincubated at room temperature for 10 min in buffer (50 mmol/L Tris HCl, 120 mmol/L NaCl, 5 mmol/L KCl, 2.5 mmol/L CaCl2, 1 mmol/L MgCl2, pH 7.4) and then incubated with 0.08 nmol/L 125I-IEB (specific activity, 81.4 TBq/mmol; Perkin Elmer) at room temperature for 90 min. Competition studies were performed by incubating alternate sections with 125I-IEB in the absence or presence of cytisine (200 nmol/L) and nicotine (300 μmol/L) and various concentrations of nifrolidine (0–30 nmol/L). Slides were then rinsed twice for 10 min in ice-cold buffer, dipped briefly in ice-cold water, blown dry, and laid out for autoradiography along with 125I plastic standards of known radioactivity (Fig. 3). For 125I-α-bungarotoxin binding competition studies, a similar method was used except that buffer consisted of 50 nmol/L Tris HCl, pH 7.4, with 120 nmol/L NaCl. Slides were incubated at room temperature for 2 h with 125I-α-bungarotoxin (5 nmol/L) in the absence and presence of various concentrations of nifrolidine (0–30 nmol/L) and α-cobratoxin (10 μmol/L) to define nonspecific binding. Slides were apposed to Kodak Biomax MR films for 2 or 48 h and then developed and fixed.
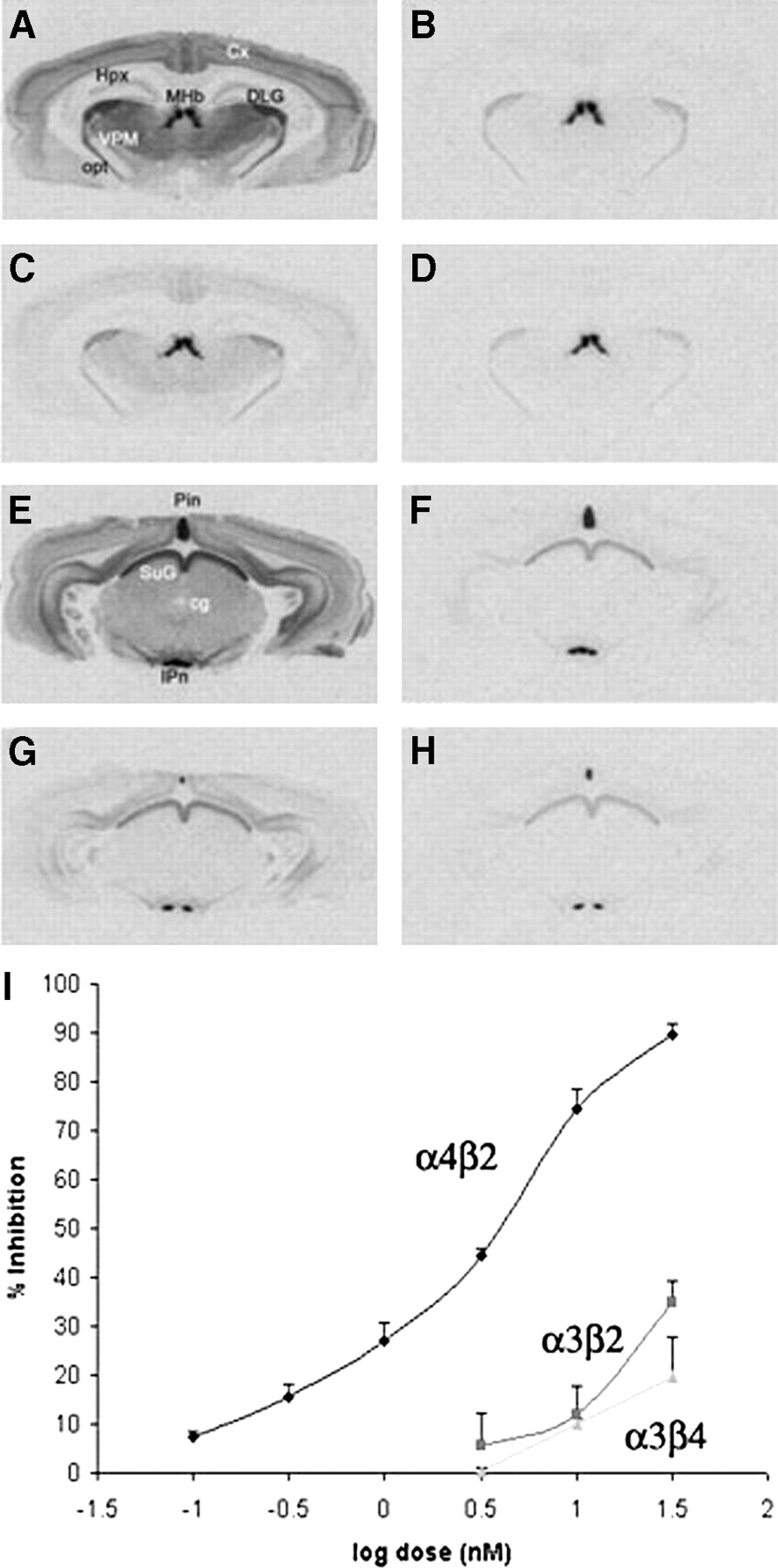
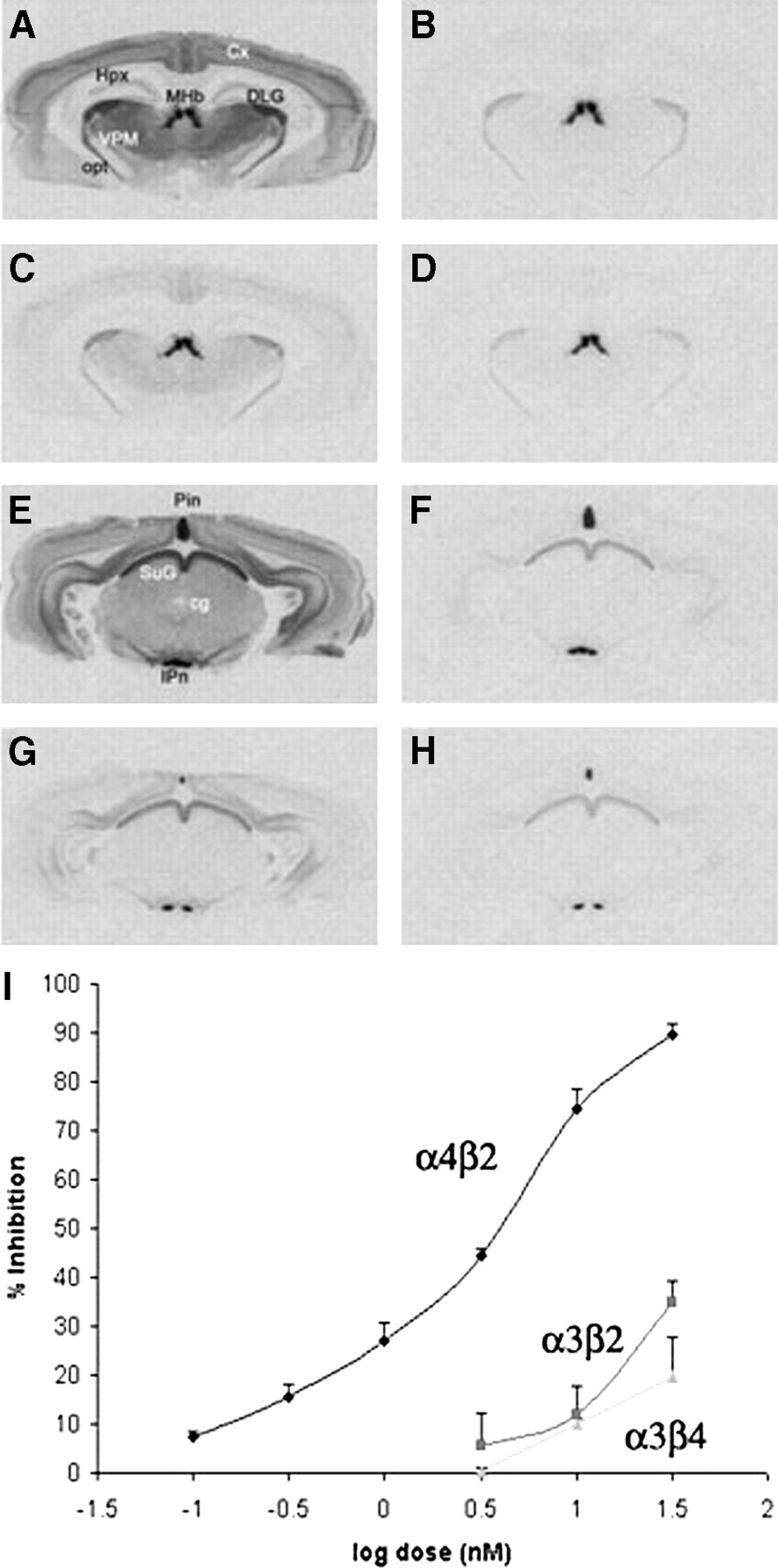
Inhibition of 125I-IEB in rat brain slices by nifrolidine. (A) Binding of 125I-IEB without nifrolidine and cytisine in cortex (Ctx), hippocampus (Hpx), medial habenula (MHb), dorsolateral geniculate (DLG), ventral posteriomedial nucleus of thalamus (VPM), and optic tract (opt). (B) Binding of 125I-IEB with 200 nmol/L cytisine. (C) Binding of 125I-IEB in presence of 30 nmol/L nifrolidine. (D) Binding of 125I-IEB in presence of 30 nmol/L nifrolidine and 200 nmol/L cytisine. (E) Binding of 125I-IEB without nifrolidine and cytisine in pineal (Pin), superior colliculus (SuG), central gray, and interpeduncular nucleus (IPn). (F) Binding of 125I-IEB with 200 nmol/L cytisine. (G) Binding of 125I-IEB in presence of 30 nmol/L nifrolidine. (H) Binding of 125I-IEB in presence of 30 nmol/L nifrolidine and 200 nmol/L cytisine. (I) Plot shows percent inhibition of 125I-IEB by nifrolidine at α4β2, α3β2, and α3β4 receptor subtypes obtained from autoradiographic experiments.
Autoradiograms were quantified with a computer-based image analysis system (MCID; Imaging Research) using calibrated standards of reference (27). A calibration curve of optical density against radioligand concentration (fmol/mg tissue) was constructed using values of known radioactivity. Optical densities in discrete regions of the autoradiographic images were measured, and corresponding values of radioactivity were determined by interpolation from the calibration curve. Specific binding values in each region were determined by subtracting binding in the presence of excess inhibitor from total binding values.
125I-IEB binding to heterogeneous sites can be discriminated by regional analysis of binding in the presence and absence of cytisine, which has modest selectivity for α4β2 nAChRs (26). Readings were obtained in various brain regions, including the pineal (Pin), interpeduncular nucleus (IP), medial habenula (MHb), optic tract (opt), superior colliculus (SuG), dorsolateral geniculate (DLG), central gray, cortex (Ctx), and ventral posteriomedial nucleus of the thalamus (VPM). Specific binding to α4β2 sites was measured in the VPM, Ctx and central gray, and was defined in the absence and presence of 200 nmol/L cytisine. Specific binding to α3β2 sites was measured in the DLG, SuG and opt, and was defined as 125I-IEB binding in the presence of 200 nmol/L cytisine minus nonspecific binding in the presence of added nicotine (300 μmol/L). Specific binding to α3β4 sites was measured in the Pin, IP, and MHb and was defined as 125I-IEB binding in the presence of 200 nmol/L cytisine minus nonspecific binding in the presence of added nicotine (300 μmol/L). Specific binding of 125I-α-bungaratoxin to α7 nAChRs was measured in the ventrolateral geniculate (VLG) and the SuG and was defined as total binding minus nonspecific binding in the presence of α-cobratoxin (10 μmol/L). Concentration–response curves for nifrolidine inhibition of specific binding to each nAChR were constructed for each brain region. For a given nAChR type, inhibition values did not differ across brain regions. Complete inhibition curves were obtained only for α4β2 sites, and resulting inhibition constant (Ki) values were derived by nonlinear computerized regression using Prizm (GraphPad, San Diego).
Radiochemistry
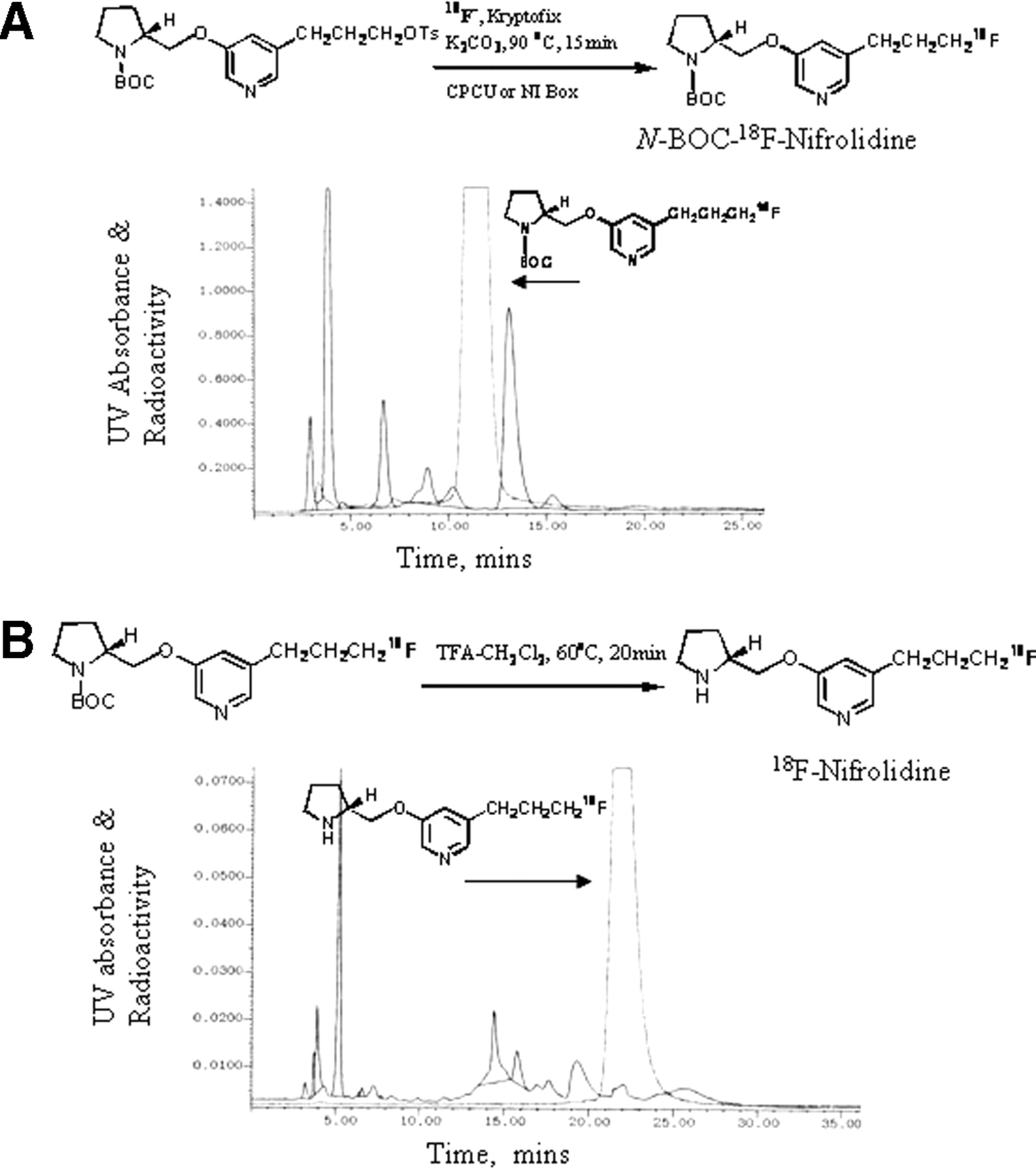
The radiosynthesis of 18F-nifrolidine was performed using an automated synthesis procedures that used a chemistry-processing computer unit (CPCU). 18F in H218O from the MC-17 cyclotron was passed through QMA-light Sep-Pak (Waters Corp.), preconditioned with 3 mL of K2CO3, 140 mg/mL, followed by 3 mL of anhydrous acetonitrile. The 18F trapped in QMA-light Sep-Pak was then eluted (using nitrogen gas) with 1 mL Kryptofix 2.2.2. (Aldrich)/K2CO3 (360 mg/75 mg in 1 mL of water and 24 mL of acetonitrile) and transferred to the CPCU reaction vessel. The SYNTH1 program in the CPCU was used for the synthesis that involved an initial drying step of the 18F-fluoride, Kryptofix 2.2.2., and K2CO3 mixture at 120°C for 10 min. Subsequently, acetonitrile (2 mL) from CPCU reagent vial 2 was added and evaporated at 120°C for 7 min to ensure dryness of this 18F-fluoride mixture. After this, the precursor, 5-(3′-[(4-methylphenyl)sulfonyloxy]propyl)-3-(1-BOC-2-(S)-pyrrolidinylmethoxy)pyridine 10 (3 mg in 0.5 mL of anhydrous acetonitrile contained in CPCU reagent vial 3) was added and the reaction proceeded for 15 min at 96°C. Subsequent to the reaction, CH2Cl2 (7 mL contained in CPCU reagent vial 4) was added to the mixture and the CH2Cl2 contents were passed through a neutral alumina Sep-Pak (prewashed with methanol) to remove any unreacted 18F-fluoride. The collected CH2Cl2 solution coming out of the CPCU now contained 5-(3-18F-fluoropropyl)-3-(1-BOC-2-(S)-pyrrolidinylmethoxy)pyridine (N-BOC-18F-nifrolidine). The CH2Cl2 was removed in vacuo and the residue was taken up for HPLC purification. The retention time of (N-BOC-18F-nifrolidine) was found to be 11–12 min using the solvent of 60% acetonitrile, 0.1 mol/L ammonium formate in water at a flow rate of 4.0 mL/min (Fig. 4A). The (N-BOC-18F-nifrolidine) fraction was collected into a flask and the solvent was removed in vacuo using a rotary evaporator. The residue was then dissolved in 1 mL CH2Cl2 and 0.2 mL of TFA. The solution was heated at 80°C for 20 min. The residue was cooled at room temperature, solvents were removed in vacuo, and the residue was neutralized to pH 7 with 10% NaHCO3. This mixture was then purified with a gradient of 0.1 mol/L ammonium formate (100%)/acetonitrile (0%) at time 1 min to 0.1 mol/L ammonium formate (40%)/acetonitrile (60%) at time 35 min with a flow rate of 4.0 mL/min. The retention time of 18F-nifrolidine was found to be 22 min. The radiosynthesis was accomplished in 2.5 h from the end of bombardment with an overall radiochemical yield ranging 20%–40% decay corrected.
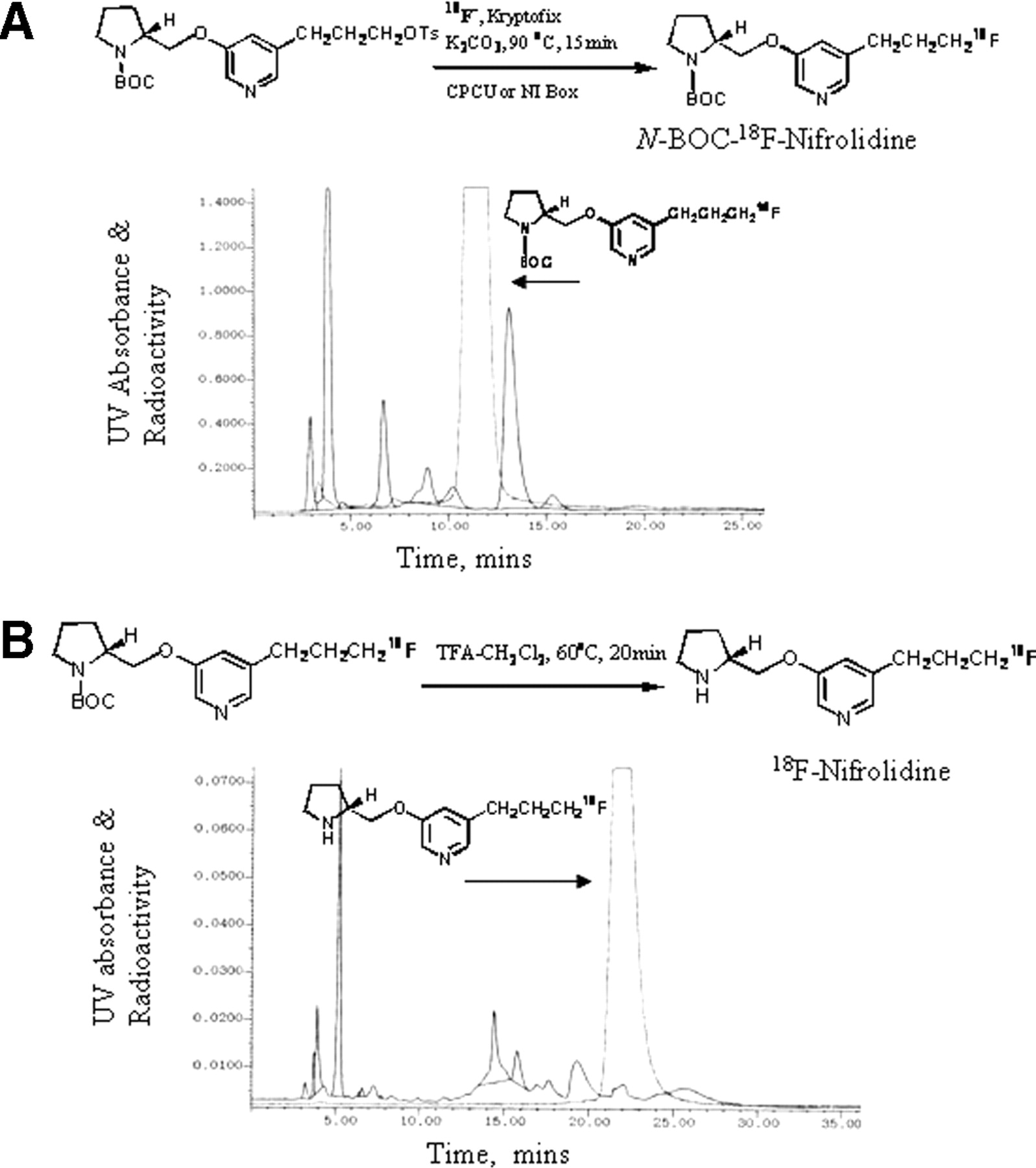
(A) Reaction scheme shows radiosynthesis of 18F-N-BOC-nifrolidine and HPLC purification of 18F-N-BOC-nifrolidine using C18 reverse-phase semipreparative column eluted with 60% acetonitrile/0.1 mol/L ammonium formate at flow rate of 4.0 mL/min. Retention time of 18F-N-BOC-nifrolidine was 11.5 min. (B) Reaction scheme shows radiosynthesis of 18F-nifrolidine and HPLC purification of 18F-nifrolidine using C18 reverse-phase semipreparative column eluted with gradient of 0.1 mol/L ammonium formate (100%)/acetonitrile (0%) at time 1 min to 0.1 mol/L ammonium formate (40%)/acetonitrile (60%) at time 35 min with flow rate of 4.0 mL/min. Retention time of 18F-nifrolidine was 22 min. UV = ultraviolet.
This collected fraction was taken to near dryness in vacuo. Approximately 5–8 mL of sterile saline (0.9% NaCl INJ [United States Pharmacopeia], 10-mL single dose; Abbott Laboratories) were added to the flask. The contents of the flask were then drawn into a 5- or 10-mL sterile syringe depending on volume. The contents of the syringe were then filtered through a 0.2-μm Millex-FG sterile filter (Millipore Corp.). This final dose was then used for in vitro and in vivo studies.
The final dose of 18F-nifrolidine was used to determine specific activity using nifrolidine standards. An aliquot of 18F-nifrolidine was injected on a C18 analytic column (250 × 4.6 mm) and eluted with 0.1 mol/L ammonium formate (40%)/acetonitrile (60%) at a flow rate of 1.0 mL/min. The radioactivity peak of 18F-nifrolidine appeared at approximately 21 min. The mass peak corresponding to this peak was compared with nifrolidine standards. The specific activity of the 18F-nifrolidine was found to be 140 ± 28 GBq/μmol (range, 111–185 GBq/μmol estimated in 10 radiosynthesis runs).
To reduce total radiosynthesis time, only 1 HPLC purification for the 2-step radiolabeling reaction was investigated. After the first radiolabeling step, the product was extracted with CH2Cl2 as described and the solvent volume was reduced to approximately 1 mL by a stream of nitrogen gas. Into this reaction vial, TFA (0.2 mL) was added and then reacted as described. The residue was cooled at room temperature, solvents were removed in vacuo, and the residue was neutralized to pH 7 with 10% NaHCO3 and with a gradient of 0.1 mol/L ammonium formate (100%)/acetonitrile (0%) at time 1 min to 0.1 mol/L ammonium formate (40%)/acetonitrile (60%) at time 35 min with a flow rate of 4.0 mL/min. The retention time of 18F-nifrolidine was found to be 22 min (Fig. 4B). Because the specific activity of 1 HPLC separation was comparable with that observed with 2 HPLC separations, all subsequent radiosyntheses of 18F-nifrolidine used 1 HPLC purification.
In Vitro 18F-Nifrolidine Autoradiographic Studies
Coronal and horizontal brain slices (10- to 20-μm thick) were obtained from male Sprague–Dawley rats. Sets of brain slices were preincubated in buffer (50 mmol/L Tris HCl containing 120 mmol/L NaCl, 5 mmol/L KCl, 2.5 mmol/L CaCl2, 1 mmol/L MgCl2, pH 7.4) for 10 min (26). Subsequently, the slices were incubated with 18F-nifrolidine (37 kBq/mL in fresh buffer) at 25°C for 60 min. Nonspecific binding was measured in the presence of 300 μmol/L nicotine. In some experiments, a lower concentration of nicotine (10 μmol/L) was also used. After incubation, slides were washed twice (1 min each) with ice-cold Tris HCl buffer, pH 7.4, followed by a quick rinse in cold deionized water. The slides were then air dried and apposed to phosphor screens overnight and read by the Cyclone Phosphor Imaging System (Packard Instruments Co). The amount of bound 18F-nifrolidine in the autoradiograms was evaluated in various brain regions (as digital light units [DLU]/mm2) using the OptiQuant acquisition and analysis program (Packard Instruments Co).
In Vivo 18F-Nifrolidine Rodent Biodistribution Studies
Male Sprague–Dawley rats (200–250 g) were anesthetized with halothane and injected via tail vein with 18F-nifrolidine (approximately 3.7 MBq each; mass injected <0.01 μg of nifrolidine). The animals were allowed to recover from anesthesia and had free access to food and water. They were sacrificed at various times (5, 60, and 120 min after injection of 18F-nifrolidine) and the brains were excised. Thalamus, Ctx, cerebellum, and blood were isolated from each rat and placed in preweighed tubes and were counted for 18F radioactivity. Using a standard of the injectate and the weight of the isolated regions, the amount of 18F-nifrolidine was calculated as percentage dose per gram for each of the regions. In one group of rats, nicotine, 10 mg/kg, was injected 5 min before administration of 18F-nifrolidine. Rats were sacrificed at 60 min after injection of 18F-nifrolidine and brain regions were evaluated as described.
Monkey PET Study
The male rhesus monkey (12 kg) was anesthetized using ketamine (10 mg/kg) and xylazine (0.5 mg/kg) and was subsequently maintained on 0.5%–1.5% isoflurane. Two intravenous catheters were placed, one on each arm, for purposes of administration of the radiopharmaceutical and for obtaining blood samples during the study. The head of the animal was placed in the gantry of an ECAT EXACT HR+ PET scanner and positioned in place with adhesive tape as described previously (28). A transmission scan using a 68Ge/68Ga rod source was acquired before administration of the radiopharmaceutical to correct for tissue attenuation of the coincident radiation. A dynamic sequence of scans for a total of approximately 150 min was acquired in the 3-dimensional mode immediately after administering approximately 148 MBq (mass injected, <0.3 μg of nifrolidine) of 18F-nifrolidine intravenously. Data in the final form are expressed in units of the percentage injected dose per milliliter (%ID/mL) or kilobecquerels per milliliter (kBq/mL). Areas showing maximal radioligand binding in the mediodorsal thalamus, ventrolateral thalamus, temporal Ctx, occipital Ctx, frontal Ctx, and cerebellum were delineated in the images. Time–activity curves were obtained for all these regions. To provide anatomic detail of the 18F-nifrolidine in the brain, the PET images were coregistered to an MR image of the rhesus brain. This MRI template of the rhesus (Macaca mulatta) brain is an average of 6 monkeys with T1-weighted MR scans. Postimage processing removed the skull and scalp from the template (courtesy of University of Wisconsin-Madison).
RESULTS
Synthesis
The synthesis route to prepare nifrolidine 9 is shown in Figure 2. Starting with 3,5-dibromopyridine 1, 5-bromo-3-methoxypyridine 2 was prepared in approximately 60%–89% yield by treatment with sodium hydride in methanol (24). Using an alternate procedure of sodium methoxide in N,N-dimethylformamide, more reproducible results were obtained in this reaction (25). Demethylation of 2 with refluxing hydrogen bromide provided 5-bromo-3-pyridinol 3 in 82% yield (24). The mixture generally had to be refluxed for longer times than reported (36 h rather than 16 h) for the high yields. Protection of (S)-pyrrolidinemethanol 4 was performed with di-tert-butyldicarbonate to provide 1-BOC-2-(S)-pyrrolidinemethanol 5 in 88% yield (alternatively, 5 is also available from Aldrich Chemical Co.). Mitsunobu reaction coupling of 3 and 5 was performed by diethyl azodicarboxylate in the presence of triphenylphosphine to provide the bromopyridyl ether 6 in 44% yield, which is somewhat lower than reported (24). To introduce a 3′-propanolic group at the 5-position, an allyl group was introduced. Using reported procedures (24), allylation of this bromo derivative 6 was first performed by allyltributyltin in the presence of catalytic amounts of tetrakis(triphenylphosphine)palladium to provide the 5-allylpyridyl ether derivative 7 in 57% yield. Hydroboration of the allyl group followed by alkaline hydrogen peroxide treatment using previously described conditions led to the important alcohol intermediate, 8 in 26% yield (29). The low yield in this step was probably a result of the formation of a BH3 complex of the alcohol 8. Some preliminary efforts to neutralize this BH3 complex were not successful. Similar BH3-complex formation has recently been reported for other amines (30). The substituted alcohol 8 was treated with DAST to convert the alcohol to the corresponding fluoride using methods that we have previously used (29). Removal of the N-BOC group was achieved by treating with TFA to provide 9 in 25% yield. Final product 9, nifrolidine, was used as a p-toluenesulfonate salt for in vitro binding assays. For radiolabeling with 18F, the tosylate 10 was prepared from the BOC-protected key alcohol intermediate 8 by reacting with toluenesulfonyl chloride in yields of 40%–50%. The tosylate 10 was found to be stable and suitable for 18F radiolabeling and was stored at 0°C to −20°C.
In Vitro Binding Affinity
Binding of 125I-IEB in rat brain slices followed a previously reported pattern (26). Specific binding to α4β2 subtypes such as VPM, central gray, and Ctx were defined in the absence and presence of cytisine (Fig. 3). Specific binding to α3β2 subtypes was measured in DLG, SuG, and opt in the presence of cytisine, and specific binding to α3β4 subtypes was identified in Pin, IP, and MHb in the presence of cytisine. As seen in Figure 3C, nifrolidine at a concentration of 30 nmol/L was able to displace a significant amount of 125I-IEB bound at the α4β2 subtypes. A significantly lower effect was observed on 125I-IEB bound to the α3β2 and α3β4 receptor subtypes (Figs. 3C and 3G). The dose–response curve of nifrolidine on the binding of 125I-IEB at the 3 different receptor sites is shown in Figure 3I. The binding affinity of nifrolidine, Ki = 2.89 nmol/L, was measured for the α4β2 sites, whereas weaker affinities (>30 nmol/L) were measured for the α3β2 and α3β4 sites.
Affinities for α7 sites using 125I-α-bungaratoxin in brain slices revealed no measureable displacement up to a concentration of 30 nmol/L (data not shown). These findings suggest that among the 4 nAChR receptor subtypes tested, nifrolidine is a relatively selective compound to study the α4β2 receptor subtypes.
Radiosynthesis
Radiosynthesis of 18F-nifrolidine required 2 steps. In the first step, 18F radiolabeling was performed by reacting the corresponding tosylate with 18F-fluoride (either from an MC-17 Scanditronix cyclotron or a RDS112 cyclotron) using Kryptofix 2.2.2. and K2CO3 in CH3CN at 96°C for 15 min. This product was purified in reverse-phase HPLC as shown in Figure 5. The radioactive peak was collected and the solvents were removed. The residue was dissolved in dichloromethane and used for the subsequent deprotection step. The purified radiolabeled BOC derivative was then deprotected (deprotection of the N-BOC–protecting group) with TFA at 80°C for 25 min. This product mixture was again purified by HPLC. Radiosynthesis and 2 HPLC purifications took approximately 2.5 h and yielded the product in approximately 20%–40% decay-corrected radiochemical yield to provide 18F-nifrolidine in specific activities of 111–185 GBq/μmol. The high specific activity for nAChR ligands is essential because of the low concentration of these receptors in the brain and also because of the associated toxicity of these agents. To reduce total production time, only 1 HPLC purification was performed after the completion of the deprotection. The HPLC purification profile of this process is shown in Figure 4B. Specific activities of 18F-nifrolidine were comparable with the 2-step purification and were produced in approximately 2 h in radiochemical yields of 20%–40%.
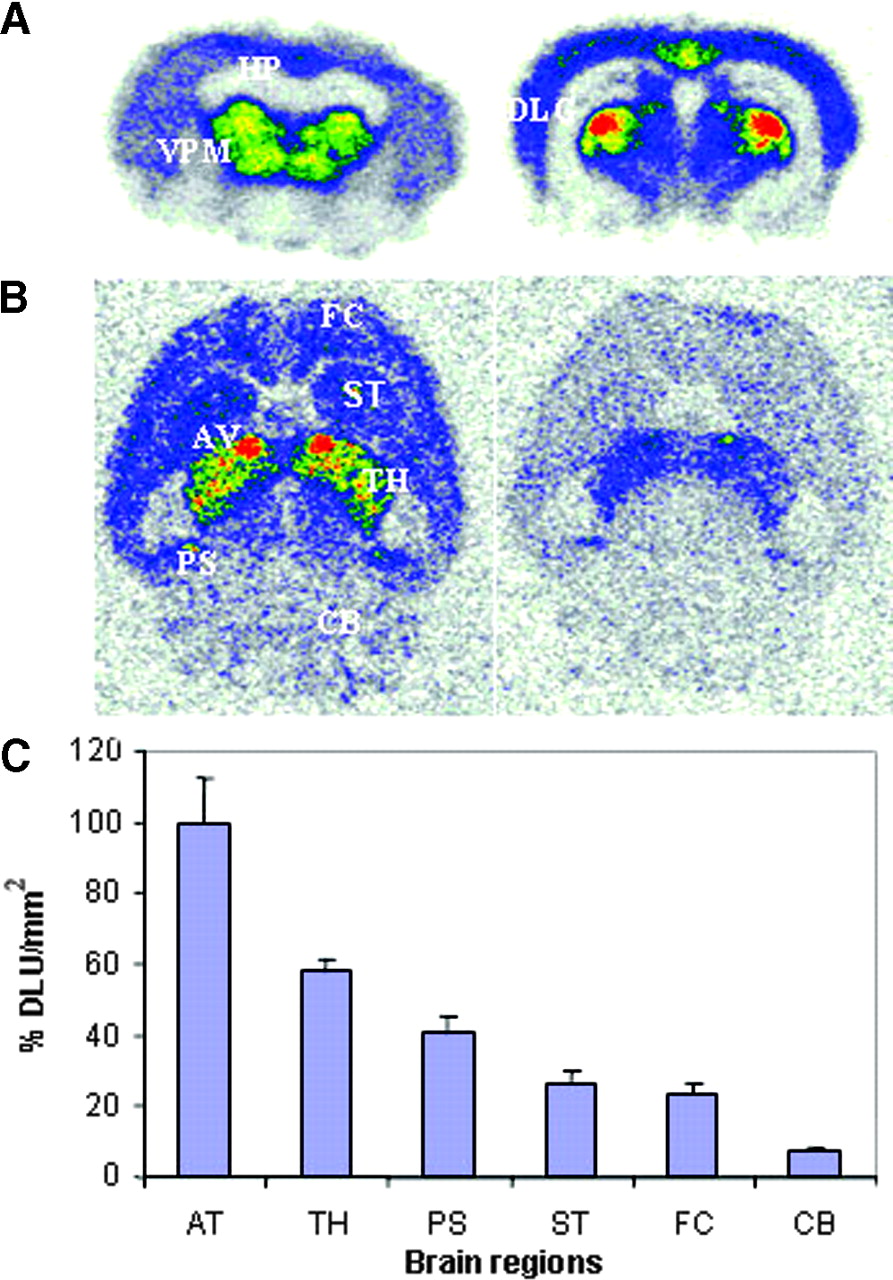
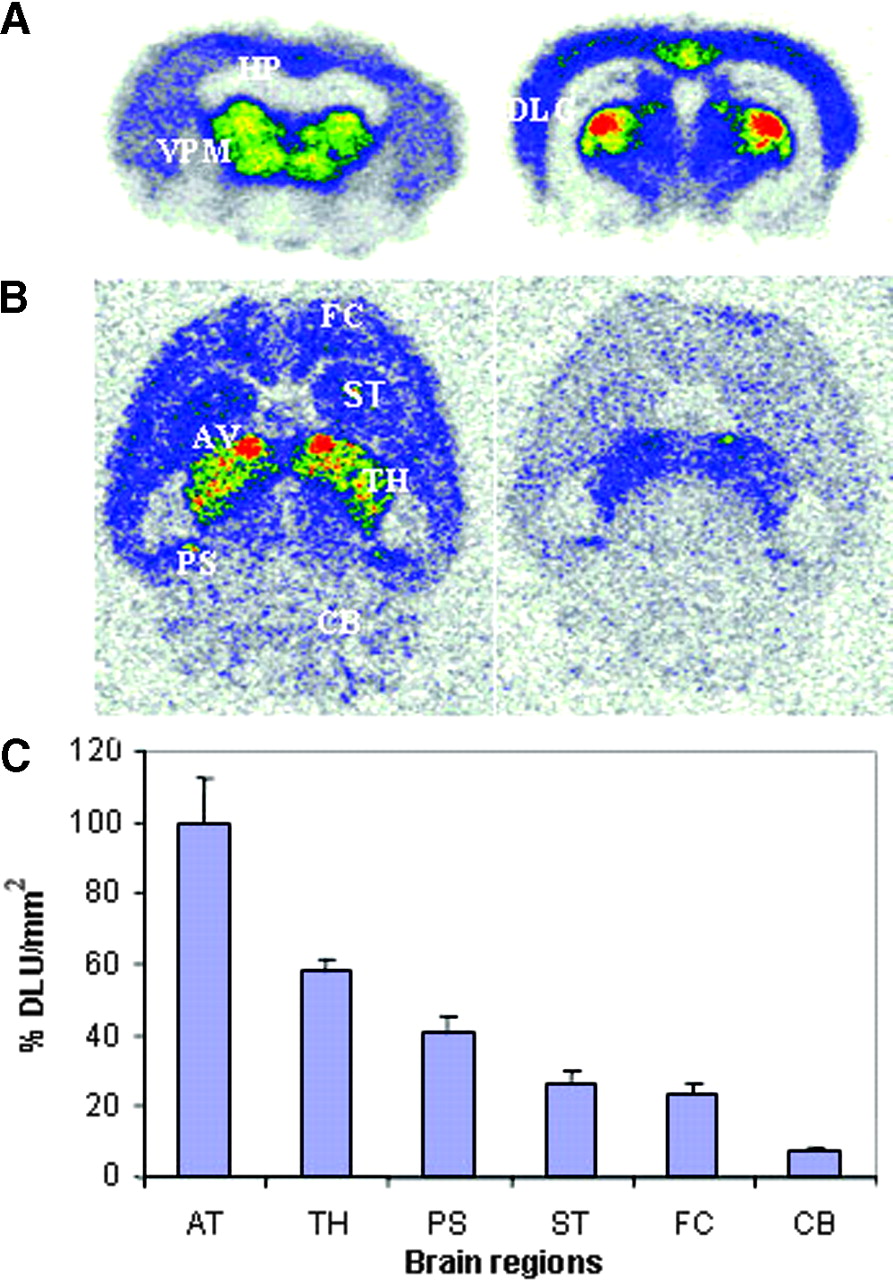
In vitro autoradiographic studies of 18F-nifrolidine in rat brain slices. (A) Binding of 18F-nifrolidine in 20-μm coronal slices (74 kBq/mL, 25°C; HP = hippocampus). (B) Total binding of 18F-nifrolidine in 10-μm horizontal slices (37 kBq/mL, 25°C) and binding in presence of 10 μmol/L nicotine. FC = frontal cortex; ST = striata; AV = anteroventral thalamic nucleus; TH = thalamus; PS = postsubiculum; CB = cerebellum). (C) Plot shows amount of 18F-nifrolidine binding (DLU/mm2) in brain regions shown in Figure 5B.
In Vitro Autoradiographic Studies
As seen in Figure 5A, in vitro autoradiography in coronal rat brain slices revealed selective binding of 18F-nifrolidine to thalamus, DLG, and other brain regions, such as the Ctx, known to contain α4β2 sites. The hippocampus did not reveal any selective binding. Specific binding of 18F-nifrolidine was completely abolished by 300 μmol/L nicotine in these brain regions. In horizontal slices, binding of 18F-nifrolidine was in the order thalamus > Ctx > striata > cerebellum, consistent with the known distribution of α4β2 receptor sites. The anteroventral thalamic nucleus (AV) exhibited the highest amount of binding, as shown in Figures 5B and 5C, and is consistent with previous autoradiographic studies with 125I-IEB (26). Portions of the subiculum also exhibited significant amounts of 18F-nifrolidine binding, which is known to contain α4β2 receptor sites (26). The cerebellum showed some binding (approximately 8% of that found in the AV). Nicotine at 10 μmol/L was able to partially displace the selective binding in various brain regions (Fig. 5B). These findings suggested the ability of 18F-nifrolidine to bind to α4β2 receptor–rich regions.
In Vivo Rat Biodistribution Studies
Table 1 shows the binding in the various brain regions. Levels in the blood decreased from 0.25 %ID/g at 2 min to 0.04 %ID/g at 120 min. Binding in the thalamus was highest (0.37 %ID/g) at 60 min, after which time it exhibited significant clearance. The thalamus-to-cerebellum ratio was about 3 at 60 min. Binding in the Ctx was greater than that in the cerebellum. Ratios of the thalamus to the cerebellum decreased at 120 min, indicative of clearance of the radiotracer from receptor sites. Blocking studies were performed by subcutaneous injection of nicotine (10 mg/kg), which decreased binding in the thalamus (Table 1). These results suggest that 18F-nifrolidine is stable in vivo in rodents, is able to cross the blood–brain barrier, and is able to bind preferentially to α4β2 receptor–rich regions, and the binding to receptor sites is reversible.
Biodistribution of 18F-Nifrolidine in Rats
Monkey PET Studies
Given the promising results in rodents, we then performed a PET study of 18F-nifrolidine on an anesthetized rhesus monkey using an ECAT EXACT HR+ scanner. The monkey was administered 155 MBq of high-specific-activity (185 GBq/μmol) 18F-nifrolidine. Vital signs were closely monitored; the monkey did not exhibit any unusual deviations from the baseline values.
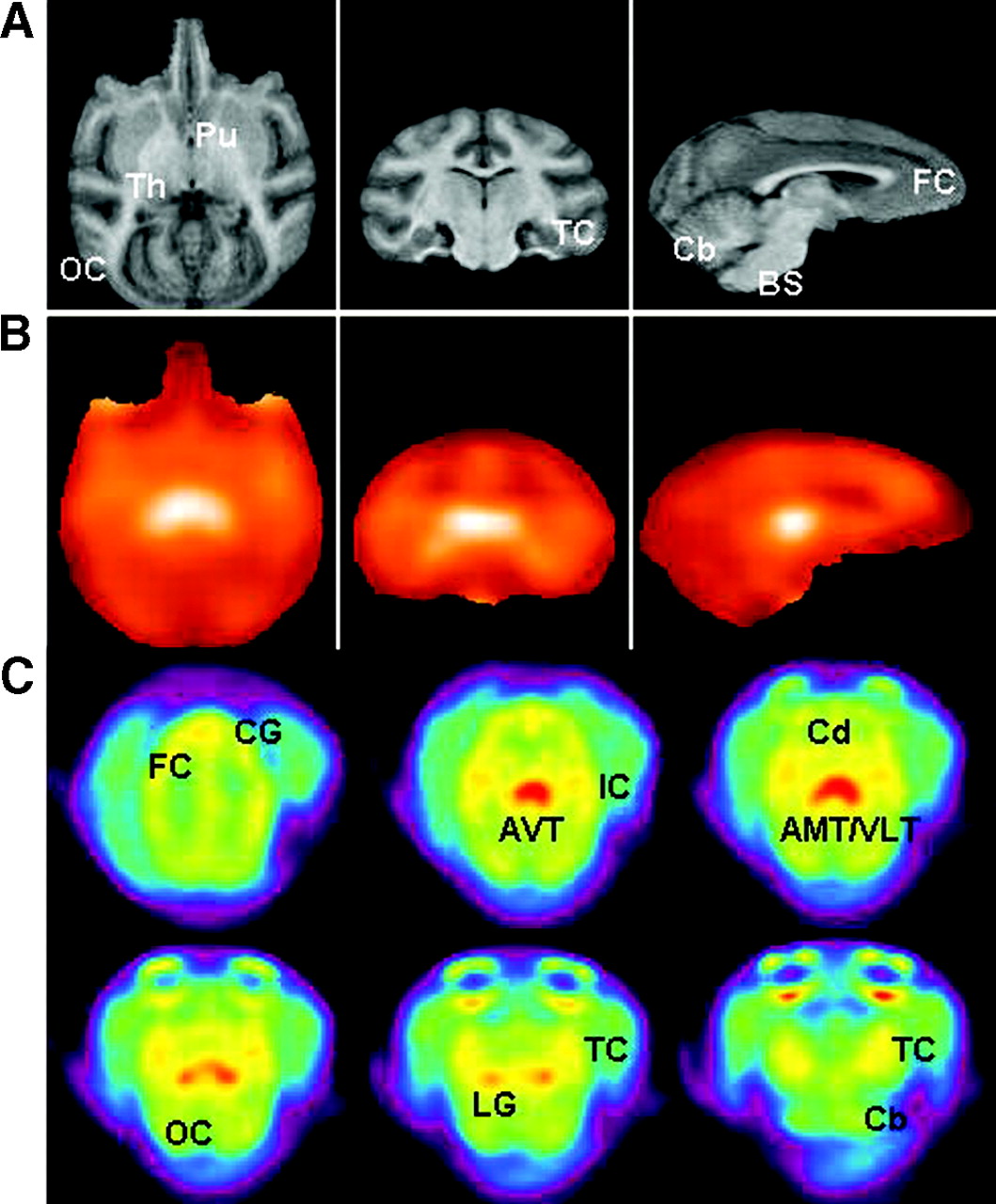
Correlation with rhesus brain MRI templates (Fig. 6A) indicated a significant amount of binding in the thalamic regions seen in axial, coronal, and sagittal slices (Fig. 6B). The temporal Ctx and frontal Ctx had significant amounts of uptake, whereas the occipital Ctx exhibited lower binding. The cerebellum and brain stem had the least amount of uptake. A closer look at the axial slices is shown in Figure 6C. The anteroventral thalamus (AVT), anteromedial thalamus (AMT), and ventrolateral thalamus (VLT) were regions that showed the highest uptake, which is consistent with the reported distribution of the α4β2 receptor subtype in the rhesus monkeys (31). Regions of moderate levels of binding included the lateral geniculate (LG; tentatively assigned) and cingulate gyrus (CG), both of which are known to contain significant proportions of the α4β2 receptor subtype (31). Cortical regions such as insular Ctx (IC; tentatively assigned) and regions of the temporal Ctx (TC) showed relatively more binding than other cortical regions. Some binding was also observed in other parts of the frontal Ctx (FC), occipital Ctx (OC), caudate (Cd), and cerebellum (Cb).

(A) MRI brain slice templates of rhesus monkey brain (axial, coronal, and sagittal sections) show putamen (Pu), thalamus (Th), occipital cortex (OC), temporal cortex (TC), frontal cortex (FC), cerebellum (Cb), and brain stem (BS). (B) Corresponding PET image slices acquired using 18F-nifrolidine. Images are shown in hot-metal color scale, where white indicates highest amount of radiotracer activity. (C) Distribution of 18F-nifrolidine in rhesus monkey brain. Summed PET images (100−150 min) show binding of 18F-nifrolidine in select brain slices. Brain regions include cingulate gyrus (CG), FC, anterioventral thalamus (AVT), insular cortex (IC), caudate (Cd), anteriomedial thalamus (AMT), ventrolateral thalamus (VLT), occipital cortex (OC), lateral geniculate (LG), temporal cortex (TC), and cerebellum (Cb).
Time–activity curves for the various brain regions are shown in Figure 7A. Selective maximal uptake occurred in regions of the AVT and AMT (0.011 %ID/mL), followed by the VLT (0.10 %ID/mL). Significant selective binding was also seen in the FC and TC. Regions such as the striatum—for example, the caudate—showed little binding. The Cb showed a significant amount of uptake that slowly cleared during the course of the scan. The thalamus-to-Cb ratio approached a plateau of 1.7 in 120 min, whereas the cortical regions exhibited ratios of 1.3 in 120 min (Fig. 7B). These findings suggest that 18F-nifrolidine may be able to approach transient equilibrium somewhat sooner than has been found with 2-18F-A85380 (22).

(A) Time–activity curves of 18F-nifrolidine binding in select areas of monkey brain corresponding to regions identified in Figure 6C (AMT, VLT, TC, OC, LG, and Cb). (B) Ratio plot of brain regions (in A) to Cb.
DISCUSSION
Agonist-based PET and SPECT agents have been reported previously for the α4β2 receptors. Our goal has been to develop antagonist-based imaging agents for this receptor subtype. Inclusion of alkyl groups (n-propyl and n-butyl) at the 5-pyridine position in pyridylethers reduces the agonist character of these compounds dramatically and turns them into antagonists (Fig. 1B) (19). Similarly, a change from the agonist character to the antagonist character has been reported for epibatidine analogs as well (32). The pyridylether skeleton seems to offer greater selectivity for the α4β2 receptor subtype than the epibatidine analogs (which bind to α3β2 and α3β4 as well) (26). We therefore selected the pyridylether backbone for development of the PET radiotracers. The initial design strategy for this series of compounds was to incorporate fluoroalkyl groups at the 5-position on the pyridine ring in the pyridylether compounds (Fig. 1D). The n-propyl and n-3′-fluoropropyl derivative maintain a high degree of structural similarity. Therefore, as a first derivative in our fluoroalkyl series, we developed the putative antagonist nifrolidine as a potential PET agent for this receptor. It must be noted that we have not performed in vitro assays that confirm the antagonist properties of nifrolidine. Radiolabeled 5-iodo-A85865 (Ki = 1.2 nmol/L), which is structurally similar to nifrolidine (an iodine instead of the 3′-fluoropropyl group), has shown some selective retention in the thalamus in SPECT studies (23).
The synthesis of nifrolidine was accomplished using modifications of reported procedures for the synthesis of the pyridylethers (24). The unique aspect of nifrolidine is inclusion of the 5-(3′-fluoropropyl) group on the pyridine ring (Fig. 2). This required functionalization of the alkyl group both for introducing fluorine and for eventual radiolabeling with 18F. As in the case of dopamine receptor agent fallypride, an allyl group was introduced at the 5-position to accomplish this task (29). The allyl group was converted to the corresponding alcohol. The alcohol thus served to introduce the fluorine or the 18F in the molecule. The yields of the various reaction steps were modest and are being currently optimized for the production of other derivatives. Nifrolidine was prepared as a p-toluenesulfonate salt for in vitro binding studies.
Taking advantage of the binding of 125I-IEB to α4β2, α3β2, and α3β4 receptor subtypes, binding affinities of these sites for nifrolidine were determined autoradiographically as reported (26). Nifrolidine exhibited high affinity (Ki = 2.89 nmol/L) in brain regions expressing the α4β2 sites. This is consistent with reported findings of the high affinity of 5-alkylated pyridylethers for this receptor subtype (19). Incorporation of the fluorine atom in the alkyl side chain did not have a detrimental effect on the binding of the compound. Weaker affinities (>30 nmol/L) were obtained for α3β2 and α3β4 receptor sites, thus demonstrating the good selectivity of nifrolidine for the α4β2 receptors. This is in contrast to the agonist A-85380, which has comparable affinities for α4β2 and α3β2 subtypes (26). Using 125I-bungaratoxin, affinities for α7 receptor sites were also found to be weak. Based on these initial binding studies, nifrolidine seems to have good selectivity for α4β2 sites over α3β2, α3β4, and α7 sites. In vitro binding affinities for other nonnicotinic receptor sites have yet to be performed.
The radiosynthesis of 18F-nifrolidine required a 2-step procedure because of a need to protect the pyrrolidine nitrogen using a BOC-protecting group. The N-BOC–protecting group has been used previously in the radiosynthesis of 18F-fluoro-A85380 (33). The first step, nucleophilic displacement of the tosylate group, proceeded in high radiochemical yields and was typically done either in a computer-controlled CPCU box or in a Nuclear Interface box. This product was purified in high specific activities and was deprotected using TFA. A second chromatographic purification resulted in the final purified product 18F-nifrolidine with specific activities of 111–185 GBq/μmol. We have now also purified the product in a single HPLC purification with high specific activities (>111 GBq/μmol). Specific activity is an important issue in imaging nAChRs. First, the receptors are in very small concentrations in the brain (1–6 fmol/mg in rat brains (26)), which requires a high specific activity to avoid saturating the receptor sites. Second, there may be issues of toxicity with the nAChR imaging agents requiring radioligands with higher specific activities.
Binding of 18F-nifrolidine in rat brain slices revealed a specificity toward brain regions rich in α4β2 receptor. Figures 5A and 5B show maximal binding in the AV, followed by other regions of the thalamus, including dorsal geniculate. This is consistent with the distribution observed for the α4β2 receptor subtype (26). Although the hippocampus has little α4β2, regions of the subiculum exhibited significant binding. The FC and striata had significant amounts of binding. The Cb exhibited the lowest amount of binding (Fig. 5C). The Cb, generally used as a reference region in some studies, does contain α4β2 receptors (26). Thus, care must be taken to identify a reference region in these studies. For example, in rodents an area within the hippocampus known to contain the lowest concentration of α4β2 receptors can be considered as a reference region (26). This low binding in the hippocampus is evident in Figures 5A and 5B. Nicotine (300 μmol/L) was able to completely block binding of 18F-nifrolidine to the receptor sites, whereas lower amounts of nicotine (10 μmol/L) revealed some residual specific binding. These findings suggested the ability of 18F-nifrolidine to localize in regions of α4β2 receptors in vitro and is consistent with previously reported radioligands.
The brain distribution in rats revealed the ability of 18F-nifrolidine to penetrate the blood–brain barrier and localize selectively in brain regions such as the thalamus. The maximal thalamus-to-Cb ratio was found to be 3 in about 60 min after injection. Although this uptake ratio is low compared with that of other radiotracers (9), it does indicate that 18F-nifrolidine may be able to achieve a transient equilibrium in vivo in 2–3 h. The selectivity of binding to the thalamus (and reduction of the thalamus-to-Cb ratio on nicotine pretreatment) indicated promising in vivo characteristics.
The imaging study in a rhesus monkey indicated good uptake of 18F-nifrolidine in the various regions of the brain. Coregistration with the MRI template confirmed localization of 18F-nifrolidine to the thalamus (Figs. 6A and 6B). The thalamus exhibited the highest amount of binding, which is consistent with previous reports using other radioligands, such as 2-18F-A85380 and 18F-epibatidine analogs. Binding was also seen in other parts of the brain, such as the TC and other cortical regions. The Cb and brain stem showed lower binding compared with that of the thalamus and Ctx. Several extrathalamic regions were identified in the serial axial slices. The CG, FC, areas in the TC, insular cortex, VLT, and LG were identifiable. These regions have not been identified previously with the agonist-based radiotracers.
Time–activity curves in Figure 7A reveal a plateauing effect in various regions, including the thalamus. Maximal binding in the thalamus occurs after 70 min following injection. Clearance from the Cb is slow; previous studies have shown a small amount of specific binding in the Cb to α4β2 receptors (34). In this study, the Cb was used as the reference region; other potential reference regions with lower α4β2 receptors in the monkey studies are being investigated because the Cb has been considered inadequate as a reference region (22). The ratio between the thalamus and the Cb was approximately 1.7 at about 140 min after injection (Fig. 7B). Thalamus-to-Cb ratios for the pyridylether PET radiotracers have ranged from 1.8 to 2.8 at 2 h after injection, whereas the epibatidine analogs have shown ratios of 2.9–4.2 at 2 h (9). Plateauing of the thalamus-to-Cb ratio in 150 min in the case of 18F-nifrolidine may reflect faster kinetics. In vitro and in vivo studies are currently underway to further validate the in vivo kinetic parameters of 18F-nifrolidine and other structural analogs.
CONCLUSION
18F-Nifrolidine is a new putative antagonist radiopharmaceutical for the α4β2 nAChR that shows promise as a PET agent. 18F-Nifrolidine shows better selectivity for this receptor subtype compared with the structurally related agonist A85380. In vitro studies indicate selective localization of the radiotracer in α4β2 receptor–rich regions. PET findings clearly indicate that binding of 18F-nifrolidine may reach a transient equilibrium during the course of a 2.5-h PET scan. The ability to detect α4β2 receptor sites in the thalamus and outside the thalamus (extrathalamic) are important attributes of 18F-nifrolidine.
Acknowledgments
This research was supported by the Biological and Environmental Program, U.S. Department of Energy, grant DE-FG02-03ER63598. Pilot project funds were also provided by the University of California-Irvine Transdisciplinary Tobacco Use Research Center (grant DA 13332) for support of this work. We thank Drs. Steve Shelton, Ned Kalin, and Terry Oakes (University of Wisconsin-Madison) for providing the rhesus MRI template and Yiling Chen for assistance with pharmacologic studies.
Footnotes
Received May 1, 2004; revision accepted Aug. 12, 2004.
For correspondence or reprints contact: Jogesh Mukherjee, PhD, Department of Psychiatry and Human Behavior, Brain Imaging Center, 162 Irvine Hall, University of California, Irvine, CA 92697-3960.
E-mail: mukherjj{at}uci.edu
REFERENCES
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Trapping of Nicotinic Acetylcholine Receptor Ligands Assayed by In Vitro Cellular Studies and In Vivo PET Imaging
- Trapping of Nicotinic Acetylcholine Receptor Ligands Assayed by in vitro Cellular Studies and in vivo PET Imaging
- 18F-ZW-104: A New Radioligand for Imaging Neuronal Nicotinic Acetylcholine Receptors--In Vitro Binding Properties and PET Studies in Baboons
- Synthesis and Biologic Evaluation of a Novel Serotonin 5-HT1A Receptor Radioligand, 18F-Labeled Mefway, in Rodents and Imaging by PET in a Nonhuman Primate