


Abstract
Regulatory peptides and their analogs are being extensively investigated as radiopharmaceuticals for cancer imaging and therapy. Receptors of the cholecystokinin family have been shown to be overexpressed in different types of neuroendocrine tumors. The purposes of this study were to evaluate the cholecystokinin octapeptide amide (CCK8) peptide tagged with a diethylenetriaminepentaacetic acid derivative (DTPAGlu) and to test whether a 111In-labeled conjugate (111In-DTPAGlu-G-CCK8, a derivative containing the chelating agent DTPAGlu bound through a glycine linker at the N-terminal end of the bioactive peptide CCK8) is suitable for cholecystokinin-B receptor (CCKBR) imaging. Methods: CCK8 was synthesized by solid-phase techniques and covalently coupled to DTPAGlu through a glycine linker at its amino terminus. The compound was labeled with 111In. The radiochemical purity and stability of the compound were assessed by chromatographic methods. NIH-3T3 and A431 cells overexpressing CCKBR were used to characterize the in vitro properties of the compound. Nude mice bearing control and CCKBR-overexpressing A431 xenografts were used as an in vivo model. Results: DTPAGlu-G-CCK8 showed rapid and efficient labeling with 111In. The radiolabeled conjugate showed specific binding to both cell lines overexpressing CCKBR. Binding was saturable, with a dissociation constant of ∼20 nmol/L in both cell systems. Both cell lines showed internalization of the ligand after interaction with the receptor. Biodistribution studies showed rapid localization of 111In-DTPAGlu-G-CCK8 on CCKBR-overexpressing A431 xenografts that was severalfold higher than that on control tumors at all time points tested. Unbound activity showed rapid clearance of over 80% through the kidneys by 30 min after injection. The labeled peptide conjugate was very stable in serum but showed a rapid breakdown after injection. Incubation with kidney homogenates suggested that most breakdown occurred in the kidneys, favoring the clearance of unbound activity. Conclusion: Our findings indicate that the in vitro and in vivo characteristics of 111In-DTPAGlu-G-CCK8 are favorable for CCKBR imaging, as the peptide shows high-affinity binding to the receptor, is internalized in CCKBR-expressing cells, and shows avid uptake in CCKBR-overexpressing xenografts, with rapid clearance of unbound radioactivity through the kidneys. Furthermore, the ease of synthesis, high labeling efficiency, and chemical stability of DTPAGlu make this chelating moiety an ideal candidate for widespread use in peptide radiolabeling for nuclear medicine applications.
Several receptor systems are being evaluated for the development of peptide-based radiopharmaceuticals for diagnostic and therapeutic purposes (1). This field is of particular interest for applications in oncology. Many of these receptor systems have been shown to be overexpressed in particular types of human cancers. The naturally occurring ligands for these receptors are, in many instances, small peptides. The sequences and interactions of these peptides with their specific receptors can be structurally characterized, and peptide derivatives can be designed and synthesized to bear a chelating group for a radiometal. The properties of the peptide derivative as a radiopharmaceutical then can be tested. An excellent example of the successful clinical application of a peptide–receptor system has been the use of the radiolabeled somatostatin analog octreotide, which has been used in several clinical studies to identify tumors and other pathologic conditions in which specific subtypes of the somatostatin receptor are overexpressed (2). The vasoactive intestinal peptide receptor (3) also has been evaluated for similar applications. Several potential receptor systems that can be used as molecular targets are under investigation (4)
We have focused on the cholecystokinin (CCK) family of receptors. Two major CCK receptor subtypes have been identified (CCKAR and CCKBR). CCKBR also is the gastrin receptor, which promotes acid secretion in the fundal mucosa of the stomach. These receptors show physiologic distributions in several different tissues, with rather selective expression of one or the other subtype. The CCKAR subtype is expressed in the pancreas, where it has been found to be involved in enzyme secretion in pancreatic acini and insulin secretion from islet cells. It also is found in the gallbladder and other areas of smooth muscle in the gastrointestinal tract, in the gastric mucosa, and in certain areas of the central nervous system, such as vagal afferent neurons. On the other hand, CCKBR is expressed throughout the central nervous system and in various cell types in the gastric mucosa, including parietal cells (responsible for acid production), chief cells (responsible for pepsinogen production), and enterochromaffin-like cells (producing histamine) (5). The overexpression of both of these receptor subtypes in certain human tumors has been demonstrated (6). CCKAR has been found to be overexpressed in several pancreatic adenocarcinomas and, to a lesser extent, in gastroenteropancreatic tumors. CCKBR has been found to be overexpressed in a large percentage of medullary thyroid cancers (MTCs), in other tumors of neuroendocrine origin (such as small-cell lung cancers), and gastroenteropancreatic tumors. It also has been found in stromal ovarian cancers, which are rather rare. Several cancer cell lines derived from human pancreatic adenocarcinomas have been found to express CCKAR. CCKBR has been found in the rat pancreatic cell line AR42J and in other cell lines, including those derived from medullary thyroid carcinoma, such as the human cell line TT.
Given the particularly high incidence of CCKBR overexpression in certain neuroendocrine tumors, such as MTCs (>90%) (6), CCKBR-directed radiopharmaceuticals may be more successful than currently available somatostatin analogs in the imaging and treatment of these cancer types. For this reason, several CCK and gastrin derivatives have been characterized over the past few years for the purpose of in vivo receptor targeting. Behr et al. initially evaluated radioiodinated gastrin I as a radiopharmaceutical (7); they could indeed specifically target CCKBR in a xenograft model of human medullary thyroid carcinoma (TT cells) and in a patient with metastatic MTC. The same animal model was used to screen several iodinated gastrin and CCK derivatives (8,9). This extensive screening led to the use of minigastrin for further preclinical and clinical development; diethylenetriaminepentaacetic acid (DTPA)–coupled derivatives of this peptide have been characterized (10,11). Reubi et al. described several DTPA and dodecanetetraacetic acid–coupled CCK analogs (12) with a high affinity for CCKBR. One of these analogs, DTPA(d-Asp26,Nle28,31)CCK(26–33), has been characterized in a rat tumor model (13) and in a clinical trial in patients with MTCs (14).
Among the several biologically active forms of CCK, the C-terminal CCK octapeptide amide (CCK8) is capable of binding both receptors, although the sulfated form of CCK8 (with a sulfate moiety on the Tyr27 side chain) is 1,000-fold more active than nonsulfated CCK8 in binding CCKAR (15). Nonsulfated CCK8 therefore displays higher affinity and selectivity for CCKBR. The bimolecular complexes of CCK8 with the N-terminal extracellular arm of CCKAR (16) and with the extracellular loops of CCKBR (17) have been structurally characterized by high-resolution nuclear magnetic resonance analysis and computational refinement. The structures of both complexes indicate that modifications at the N-terminal end of CCK8, obtained by introducing chelating agents and their metal complexes, should not affect the interaction of the peptide with CCKAR or CCKBR.
Although most ligands that have been evaluated have been demonstrated to specifically target CCKBR, there are still some issues that can benefit from further study; therefore, it is worthwhile to assess the efficacies of different compounds and different labeling approaches. Kidney uptake and retention, for example, have been shown to be extremely high, a situation that may cause problems for kidney toxicity, particularly in therapeutic applications. This problem appears to be significant with DTPA-coupled minigastrin (8,9) but less evident with DTPA-coupled CCK derivatives, such as sulfated CCK8 (8) and DTPA(d-Asp26,Nle28,31)CCK(26–33) (13). Another issue being addressed is how to improve the kinetic stability of DTPA conjugates to prevent transchelation of the radiometal after injection. In particular, one objective is to allow more secure coordination of therapeutic nuclides, such as 90Y (11). This radionuclide displays a high level of accumulation in bone and bone marrow toxicity when dissociated from its chelating group.
On these bases, we have taken a different approach to targeting CCKBR. We have used a native nonsulfated CCK8 peptide sequence bearing a recently described DTPA derivative, the chelating agent DTPAGlu (18), coupled to the N terminus of the peptide during solid-phase synthesis. The use of this novel chelating moiety has allowed us to obtain a peptide conjugate having 5 carboxyl groups (as opposed to the 4 normally available with conventional DTPA labeling) and 3 amino groups available for radiometal coordination. A glycine residue has been introduced as a spacer between the chelating agent and the biologically active CCK8 peptide. The combination of the CCK8 peptide sequence and the DTPAGlu moiety should allow us to obtain a lower level of retention of radioactivity in the kidneys, as shown for other CCK8 peptides (8,13), and to make use of a more stable chelating group. We have characterized the properties of this compound as a radiopharmaceutical by evaluating its binding specificity and affinity, cellular internalization, and retention in 2 cell culture systems of CCKBR overexpression as well as its in vivo biodistribution properties in xenograft-bearing nude mice. We have also performed a detailed analysis of compound stability.
MATERIALS AND METHODS
Peptide Synthesis
DTPAGlu-G-CCK8 peptide conjugate synthesis was performed by solid-phase techniques under standard conditions with a 9-fluorenylmethoxy carbonyl (Fmoc) strategy. The DTPAGlu pentaester, the chelating agent fully protected by tert-butyl (tBu) groups on its carboxyl groups, with the exception of the carboxyl group on the side chain of glutamic acid, N,N-bis[2-[bis[2-(1,1-dimethylethoxy)-2-oxoethyl]-amino]ethyl]-l-glutamic acid 1-(1,1-dimethylethyl)ester, was provided by Bracco Imaging SpA and was used without further purification. Its synthesis is described elsewhere (18). When the sequence Fmoc-Gly-Asp(OtBu)-Tyr(tBu)-Met-Gly-Trp(Boc)-Met-Asp(OtBu)-Phe-resin was completed (OtBu is tert-butoxy; Boc is tert-butoxycarbonyl), the Fmoc N-terminal protecting group was removed and the peptide resin was transferred to a manual vessel for coupling of the DTPAGlu pentaester.
The DTPAGlu pentaester was coupled to the Gly α-amino group by use of 2 equivalents of DTPAGlu pentaester, benzotriazole-1-yl-oxy-tris-pyrrolidinophosphonium hexafluorophosphate (PyBoP) and 1-hydroxybenzotriazole (HOBt), and 4 equivalents of N,N-diisopropylethylamine (DIEA) in a single coupling. The reaction was performed by stirring the mixture in dimethylformamide (DMF) for 3 h. For deprotection and cleavage, the fully protected DTPAGlu peptide resin was treated with trifluoroacetic acid (TFA) containing triisopropylsilane (TIS) (2.0%), ethanedithiol (EDT) (2.5%), and water (1.5%). DTPAGlu-G-CCK8 was precipitated at 0°C by the dropwise addition of diethyl ether. Purification of the crude mixture was performed by reversed-phase (RP) high-pressure liquid chromatography (HPLC) with a C18 column; elution was done by use of H2O:0.1% TFA (A) and CH3CN:0.1% TFA (B) linear gradients with 5%–95% B over 45 min at a flow rate of 1 mL/min. This step was followed by analytic HPLC, which showed >97% purity (retention time, 24.6 min). Mass spectra (matrix-assisted laser desorption ionization–time of flight) confirmed the product identity (molecular weight, 1,566.8; calculated, 1,566.0).
Radiolabeling
111In was purchased from Nycomed Amersham Sorin Srl in the chemical form 111In-Cl3 (740 MBq/mL). The DTPAGlu-G-CCK8 peptide was dissolved in CH3CN:20% H2O at concentrations of 0.1–1 mmol/L. Before labeling was done, 111In-Cl3 was buffered by the addition of 10 volumes of sodium citrate (0.3 mol/L). One volume of the peptide solution then was mixed with 10 volumes of 111In-citrate, and the mixture was incubated for at least 1 h at room temperature. Labeling efficiency was determined by silica gel thin-layer chromatography with sodium citrate (0.3 mol/L) as the eluent or RP HPLC performed with a model 305 system (Gilson), an analytic C18 column (Vydac) connected to a model 170 radiodetector (Beckman), and H2O:0.1% TFA and CH3CN:0.1% TFA gradients that were slightly different from those described above (5%–70% CH3CN:0.1% TFA over 31 min). The retention time of 111In-DTPAGlu-G-CCK8 was 22.0 min.
Cell Lines and Nude Mouse Xenograft Models
NIH-3T3 cells overexpressing human CCKBR (NIH-3T3-CCKBR cells) through stable transfection were obtained from Dr. Sue Watson (University of Nottingham, Nottingham, U.K.). These cells originally were developed by Dr. Y. Matsui (University of Kobe Medical School, Kobe, Japan) (19) and used with his permission. A431 cells were obtained from the American Type Culture Collection. A431 is a human epidermoid carcinoma cell line that has been shown to yield subcutaneous tumors with a high efficiency in immunologically incompetent mice (20). All cells were grown in a humidified atmosphere containing 5% CO2 and 95% air at 37°C in Dulbecco modified Eagle medium containing glucose (4.5 g/L) and supplemented with 10% heat-inactivated fetal bovine serum (GibcoBRL, Life Technologies). To obtain A431 cells overexpressing CCKBR (A431-CCKBR cells), complementary DNA for the receptor was generated by reverse transcriptase PCR of total RNA obtained from NIH-3T3-CCKBR cells. Primers were designed on the basis of the published sequence of human CCKBR (GenBank accession number L07746) to amplify the region between nucleotides 163 and 1,509, containing the full coding sequence. The PCR product then was cloned into vector pCR3.1 (Invitrogen) by use of a eukaryotic TA cloning kit (Invitrogen). The plasmid was sequenced, linearized, and transfected into A431 cells by use of dioleoyltrimethylammonium propane liposomal transfection reagent (Roche Molecular Biochemicals). Transfectants were obtained after 2 wk of selection in medium containing the neomycin analog G418 (500 μg/mL; GibcoBRL). One positive transfectant and one control transfectant (transfected with vector pCR3.1 alone) were isolated for further analysis.
In Vitro Cellular Assays
All in vitro cellular assays were performed with cells that had been plated at a density of 100,000–200,000 cells per well in 12-well plates 2 or 3 d before the experiments. These conditions allowed for the cells to be almost confluent at the time of the assays. Experiments aimed at assessing the specificity of the interaction were performed by comparing cell-associated activity after incubation of the radiolabeled peptide with A431-CCKBR cells and A431 control cells and with A431-CCKBR cells in the presence of excess unlabeled peptide. Triplicate wells were incubated with known amounts of radiolabeled peptide for different times at 37°C. After incubation, cells were washed with phosphate-buffered saline (pH 7.4) (PBS) and then recovered by trypsinization. Cell-associated radioactivity and total radioactivity were determined with a Wizard γ-counter (Wallac Oy). The amount of bound compound was normalized for protein content, which was determined by use of a commercially available kit (Bio-Rad Laboratories) after cells were solubilized in 1N NaOH.
To assess the binding affinity of the compound, duplicate wells were incubated with serial dilutions of 111In-DTPAGlu-G-CCK8 in culture medium at 4°C for 1 h; concentrations ranged from 0.05 to 50 nmol/L. To assess nonspecific binding, a 100- to 1,000-fold excess of unlabeled peptide was added to some of the wells. For all experiments, cell-associated radioactivity was recovered from the wells by trypsinization of cells after 2 rapid washes in ice-cold PBS. Radioactivity then was counted and normalized for the number of cells, and nonspecific binding was subtracted. Binding curves were analyzed with a Macintosh computer and Kaleidagraph software (Abelbeck Software, version 3.0.5; distributed by Synergy Software). Dissociation constants (Kds) and the apparent numbers of binding sites per cell (Bmax) were derived by fitting the data to the following equation: bound = (Bmax × [111In-DTPAGlu-G-CCK8])/(Kd + [111In-DTPAGlu-G-CCK8]).
Cellular internalization was determined by comparing differences in the accumulation of the compound incubated with A431-CCKBR cells at 4°C, the temperature at which most metabolic processes are blocked, and at 37°C, at which all metabolic processes, including receptor internalization, are active. Cells were incubated with 111In-DTPAGlu-G-CCK8 (20 nmol/L) at the 2 temperatures for 60 and 120 min. After 120 min of incubation, some wells were rinsed with PBS to remove unbound radioactivity and then were incubated for an additional 60 min at 4°C with unlabeled peptide (30 μmol/L) to displace any surface-bound radioactivity. Radioactivity associated with cells was determined after unbound radioactivity was washed away and then was normalized for protein content.
Retention of the cell-associated label was assessed by allowing A431-CCKBR cells to accumulate 111In-DTPAGlu-G-CCK8 at 37°C for 2 h, washing away unbound radioactivity, and replacing the cells in the incubator with fresh culture medium. After different times, cell-associated radioactivity was determined as described above.
Biodistribution Studies
Mice bearing xenografts of A431-CCKBR cells and A431 control cells were generated by injection of 100 μL of the respective cell suspensions at a density of 2 × 107 cells per milliliter in PBS into opposite flanks of 6-wk-old CD-1 nude mice (weight, 17–23 g). Tumors were allowed to grow for 10–14 d. The final tumor weights were between 0.5 and 1 g at the time of the biodistribution and imaging experiments. The time courses for tissue and tumor distributions of 111In-DTPAGlu-G-CCK8 were determined after injection into the lateral tail vein of ∼1.85 MBq (50 μCi, ∼0.4–1 μg or 0.2–0.5 nmol of peptide) of labeled peptide per mouse. At least 5 animals per time point were used. The animals were killed at 30, 120, and 240 min after injection. Blood, lung, liver, spleen, kidney, stomach, intestine, muscle, and xenograft samples were weighed, and radioactivity was determined with the γ-counter. Counts for dilutions of the injected compound were obtained simultaneously for accurate determination of the injected dose. The relative amounts of radioactivity in the organs were calculated and expressed as the percentage injected dose (%ID) per gram of tissue normalized for a 20-g mouse. For some animals, pinhole γ-camera imaging was performed after the mice were sacrificed to visualize CCKBR-positive tissues.
Compound Stability
The stability of 111In-DTPAGlu-G-CCK8 was tested by incubating the compound with mouse serum for 1 h at 37°C or by injecting the compound into a nude mouse and recovering blood samples at 30 and 120 min after injection. The blood samples were centrifuged to recover serum, and all samples were extracted with an equal volume of CH3CN to precipitate high-molecular-weight proteins. The resulting soluble fraction was subjected to RP HPLC, and radioactivity was assessed directly with the radiodetector; alternatively, when count rates were too low, 1-min fractions were collected and assayed in the γ-counter. Similarly, experiments aimed at determining the tissue metabolism of the compound were performed with homogenates obtained from freshly prepared mouse tissue samples. Aliquots of 111In-DTPAGlu-G-CCK8 were incubated with the homogenates for times ranging from 0 to 60 min, extracted with CH3CN, and analyzed by RP HPLC as described above.
To assess the transchelation of 111In to iron-binding proteins in the serum, such as transferrin, the labeled peptide was incubated with mouse or fetal bovine serum for up to 24 h at 37°C or recovered after injection as described above. Serum and plasma from blood samples were subjected to size exclusion chromatography with Sephadex G-50 spin columns (Roche). Controls for these experiments consisted of freshly labeled 111In-DTPAGlu-G-CCK8 and fetal bovine serum (GibcoBRL) incubated with small amounts of 111In-Cl3 for 2 h at 37°C.
RESULTS
Synthesis of DTPAGlu-G-CCK8
After standard solid-phase peptide synthesis of the CCK8 peptide, DTPAGlu was coupled to the N terminus of the resin-bound peptide according to the schematic representation shown in Figure 1. This coupling step was very efficient; however, the subsequent deprotection reaction required a prolonged time to ensure complete removal of the tBu protecting groups. Final HPLC purification resulted in high purity and yields of the DTPAGlu-G-CCK8 conjugate.

Schematic representation of coupling of the DTPAGlu moiety to the resin-bound G-CCK8 peptide. Subsequent deprotection yields the final compound, which is ready for radiolabeling.
Radiolabeling and Quality Control
Rapid and efficient labeling was obtained as early as 30 min after the addition of 111In to the compound. Typical yields were >97%, as determined by RP HPLC, at specific activities ranging from 4 to 9 GBq/μmol (100–250 Ci/mmol). The elution pattern was identical to that observed with the conjugate in the presence of stable indium and monitored by UV absorption. No purification was needed after radiolabeling for all subsequent experiments (Fig. 2A).
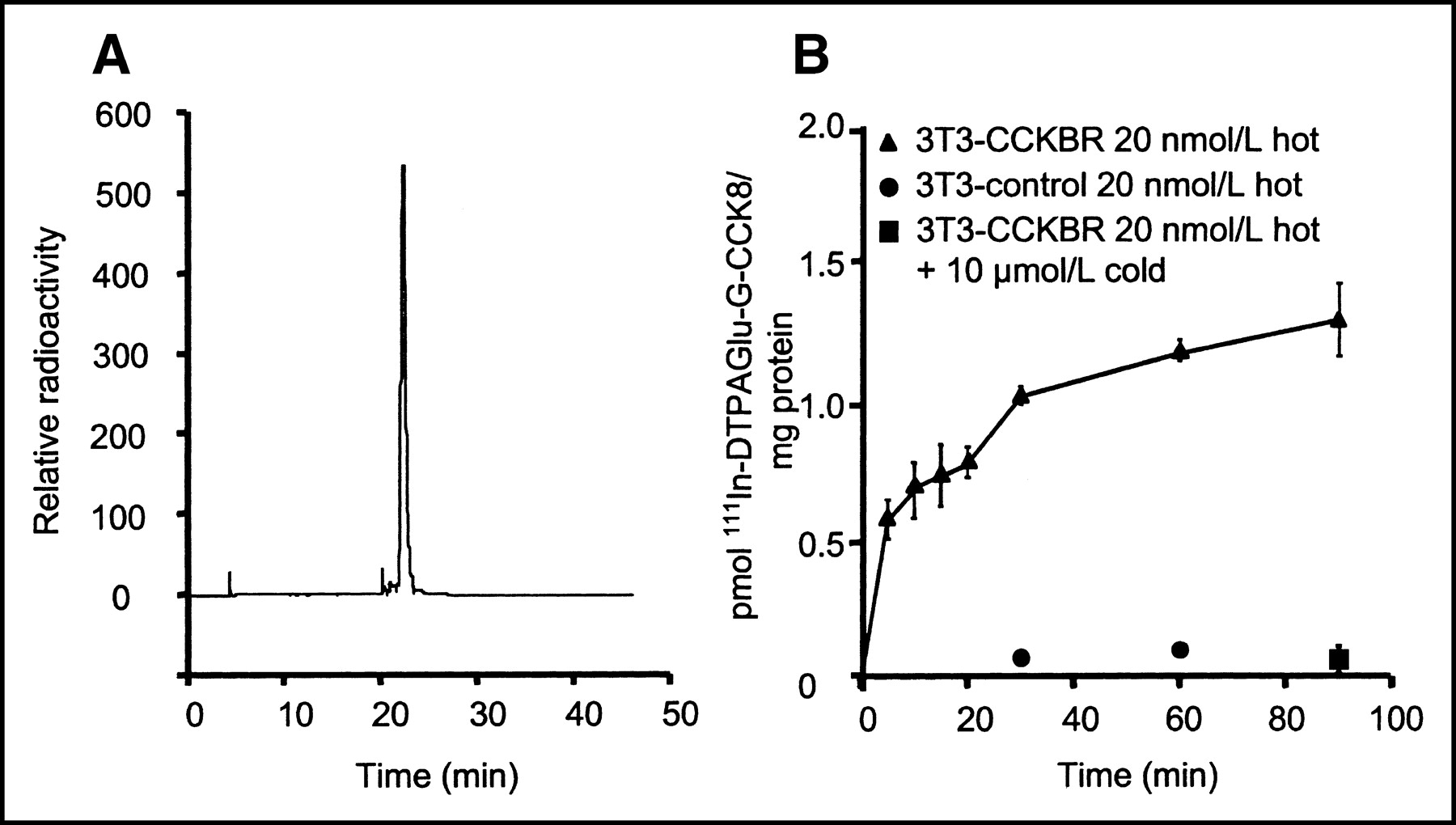
(A) Radioactive trace of RP HPLC analysis after labeling for 60 min at room temperature. 111In-DTPAGlu-G-CCK8 shows a retention time of approximately 22 min on the CH3CN:H2O gradient. Unincorporated 111In, which has a retention time of approximately 3 min, is barely detectable. (B) Specificity of interactions of 111In-DTPAGlu-G-CCK8 with NIH-3T3-CCKBR cells and control cells. There is a progressive increase of cell-associated activity in receptor-positive cells incubated with the radioactive peptide alone (▴). Receptor-negative cells (•) and receptor-positive cells incubated with excess unlabeled peptide (▪) show very little interaction with the radiolabeled peptide. There were 3 samples per time point; error bars indicate SDs.
Cell Binding and Internalization
111In-DTPAGlu-G-CCK8 showed rapid specific binding to both NIH-3T3-CCKBR cells and A431-CCKBR cells. Figure 2B shows a time course experiment with NIH-3T3-CCKBR and NIH-3T3 control cells incubated with nonsaturating concentrations of 111In-DTPAGlu-G-CCK8. The experiment showed rapid initial binding of the compound to CCKBR-expressing cells and slowly increasing cell-associated amounts over time. The level of nonspecific binding, as assessed by incubation of the compound with non–receptor-expressing cells or with receptor-expressing cells in the presence of a 100-fold excess of unlabeled peptide, was very low. Similar results were obtained with A431-CCKBR cells and A431 control cells (data not shown). Figure 3 shows saturation curves for the compound on 2 CCKBR-overexpressing cell lines. These experiments were performed at 4°C to block receptor internalization and thus measure only ligand–receptor interactions. In both cell systems, binding was saturable, with Kds on the order of 20 nmol/L. A431-CCKBR cells had a larger number of receptors (approximately 4.7 × 106 sites per cell compared with 1.6 × 106 sites per cell for NIH-3T3-CCKBR cells).

Binding of 111In-DTPAGlu-G-CCK8 on NIH-3T3-CCKBR cells (A) and A431-CCKBR cells (B) at 4°C. Both cell lines showed saturable binding of the peptide, with equivalent Kds (22 ± 11 nmol/L [mean ± SD] for NIH-3T3-CCKBR cells; 23 ± 4 nmol/L for A431-CCKBR cells). A431-CCKBR cells had higher Bmax values (1.6 × 106 ± 0.4 × 106 sites per cell [mean ± SD] for NIH-3T3-CCKBR cells; 4.7 × 106 ± 0.4 × 106 sites per cell for A431-CCKBR cells). There were 2 samples per concentration; error bars indicate SDs.
To evaluate whether the peptide was internalized by the cells, we performed parallel experiments in which A431-CCKBR cells were incubated with 111In-DTPAGlu-G-CCK8 (20 nmol/L) at 4°C (internalization blocked) or 37°C (internalization active) (Fig. 4A). There was a progressive accumulation of radiolabel in cells incubated at 37°C with labeled peptide alone, whereas the levels of cell-associated radioactivity were constant in cells incubated at 4°C for 60 and 120 min, suggesting that internalization occurred to some extent at 37°C. Coincubation with excess cold peptide produced very low levels of cell-associated activity, confirming the specificity of the interaction. After incubation for 120 min with labeled peptide alone, cells were incubated with excess cold peptide for 60 min at 37°C. A large portion of the radioactivity (∼85%) was displaced by the cold peptide in cells incubated at 4°C, whereas very little (∼10%) was displaced in cells previously incubated at 37°C; these results indicated that the compound indeed had been internalized to a large extent in cells incubated at 37°C. Similar results were obtained with NIH-3T3-CCKBR cells (data not shown). The release of cell-associated radioactivity (Fig. 4B) was biexponential, with an early, rapid component likely attributable to the effect of dilution of the ligand–receptor complex after the addition of fresh medium and a later, slower component likely attributable to the release of radiolabel internalized in cells.

Cellular internalization and release of 111In-DTPAGlu-G-CCK8 for A431-CCKBR cells. (A) Internalization. Cells were incubated at 4°C (□) and 37°C (▪) for 60 and 120 min with radiolabeled peptide (20 nmol/L). Excess unlabeled peptide was added during the 2-h incubation (third set of bars from left) or after the 2-h incubation and after washes in PBS to displace label on the cell surface (fourth set of bars from left). There is a progressive increase of cell-bound activity at 37°C, whereas a constant level is observed at 4°C. Coincubation with cold ligand produced a very low level of binding under both conditions. The addition of cold ligand after the 2-h incubation displaced most of the radioactivity from cells incubated at 4°C, consistent with the ligand being on the cell surface, whereas very little displacement was observed for cells incubated at 37°C, indicating that the label was in an intracellular compartment in these cells. (B) Release. Cells incubated at 37°C for 2 h were rinsed with PBS. Fresh medium was added, and cell-associated radioactivity was determined at the indicated times. There were 3 samples per time point; error bars indicate SDs.
Biodistribution and Imaging
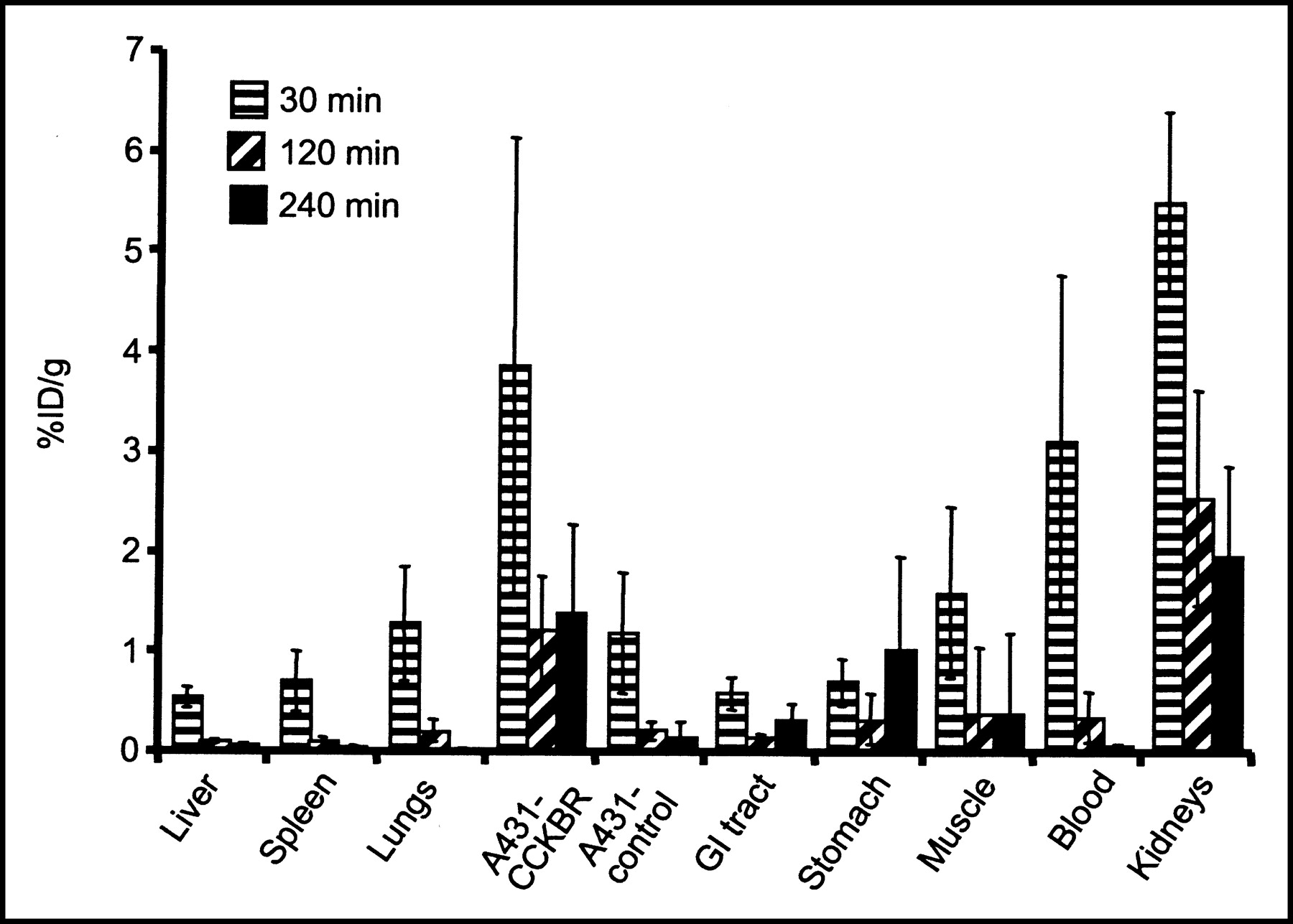
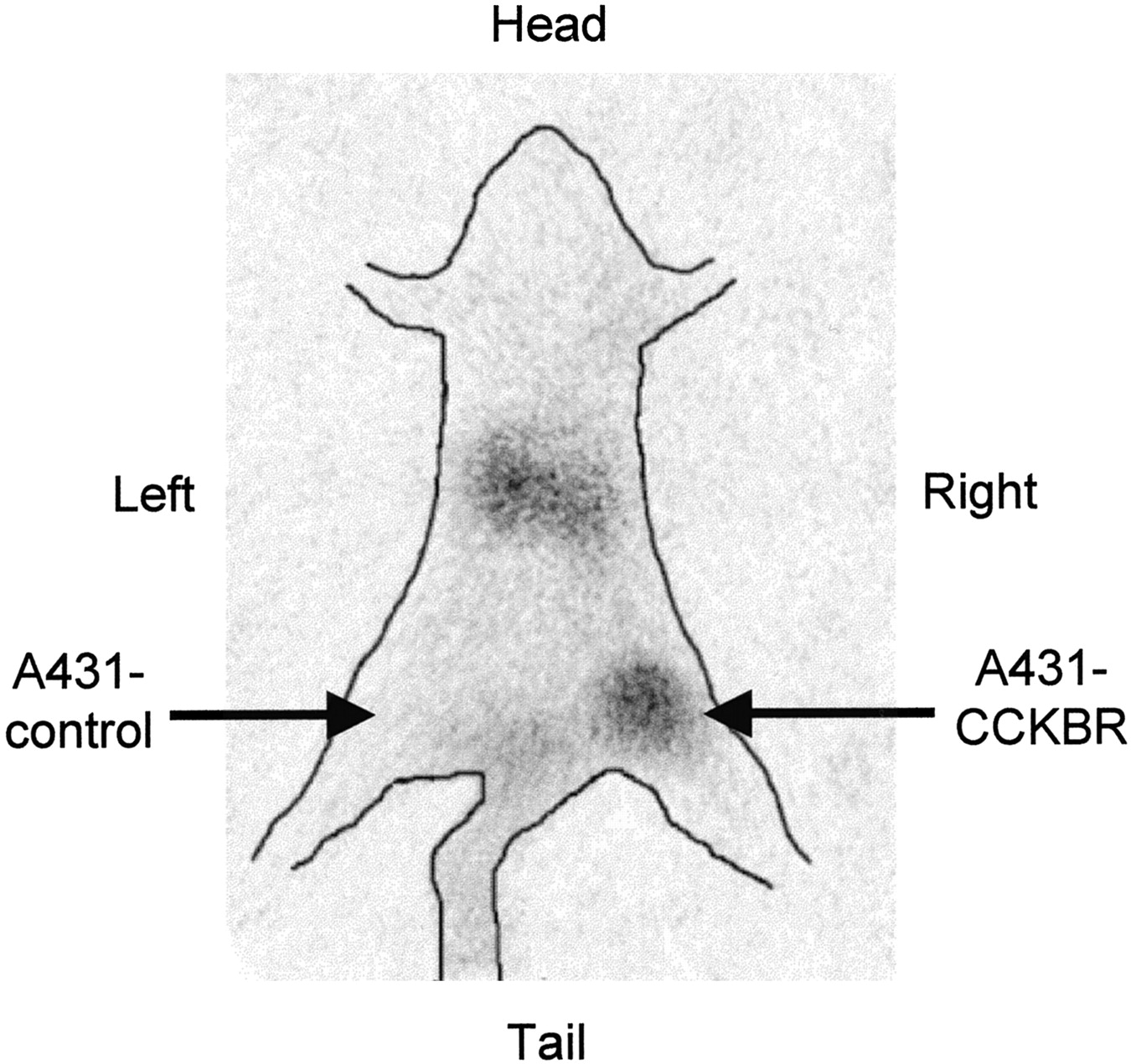
The data obtained in the biodistribution experiments are displayed in Figure 5. The uptake of 111In-DTPAGlu-G-CCK8 into A431-CCKBR xenografts was higher than that into A431 control tumors at all time points tested, with a progressive increase in the target-to-nontarget ratio (receptor-positive vs. receptor-negative tumors) from 3.3:1 at 30 min after injection to 5.5:1 at 120 min and 10:1 at 240 min. The only organs showing uptake higher than that in CCKBR-positive xenografts were the kidneys, which also showed a progressive release of radioactivity over time. Uptake in the stomach was slightly higher than that in the rest of the gastrointestinal tract. The accumulation of 111In-DTPAGlu-G-CCK8 in the remaining organs, including the liver, was negligible and appeared to decrease in parallel to blood clearance of the compound. More than 80% of the injected activity was cleared through the kidneys by 30 min. The pinhole γ-camera image obtained at 2 h after injection (Fig. 6) showed marked accumulation of the compound in the CCKBR-positive tumor and in the kidneys and very low uptake in the control tumor.

Biodistribution of 111In-DTPAGlu-G-CCK8 after intravenous injection. Organ-associated radioactivity is expressed as the %ID per gram of tissue normalized for a 20-g mouse. There were at least 5 samples per time point; error bars indicate SDs. GI = gastrointestinal.

Pinhole γ-camera image obtained at 2 h after injection of 111In-DTPAGlu-G-CCK8. Avid accumulation of the peptide was seen in a CCKBR-positive xenograft (right thigh) but not in a control tumor (left thigh). Hot spots in the abdomen are consistent with kidney accumulation.
Stability and Metabolites
Because the overall clearance of radioactivity was extremely rapid, we evaluated the stability of the peptide conjugate under different conditions. Incubation of the peptide in mouse serum for 1 h produced almost no effect on the compound (Fig. 7A, top trace). On the other hand, radioactivity recovered from the blood at 30 and 120 min after injection showed a marked breakdown of the starting compound to hydrophilic by-products, and the starting material was no longer detectable (Fig. 7A, bottom 2 traces). Gel filtration analysis of blood samples obtained at the same time points showed that >99% of the radioactivity was retained in the included volume of the spin columns. This result suggests that among the metabolites generated after injection, there is no unbound 111In. In fact, in control experiments, incubation of 111In-Cl3 in serum for the same times caused most (>60%) of the radioactivity to be excluded from the resin, consistent with the transchelation of the radiometal to transferrin or other metal-binding proteins. Radiolabeled peptide incubated at 37°C in fetal bovine serum for up to 24 h showed very little transchelation (6% ± 0.7% [mean ± SD] at 24 h) of 111In to high-molecular-weight proteins, indicating very high chelate stability. Because metabolism in the organs rather than in serum appears to play a major role, the compound was incubated with tissue homogenates obtained from the liver and kidneys (Figs. 7B and 7C). Incubation with homogenates from both of these organs produced degradation of the compound that appeared to be more rapid and complete in kidney extracts.
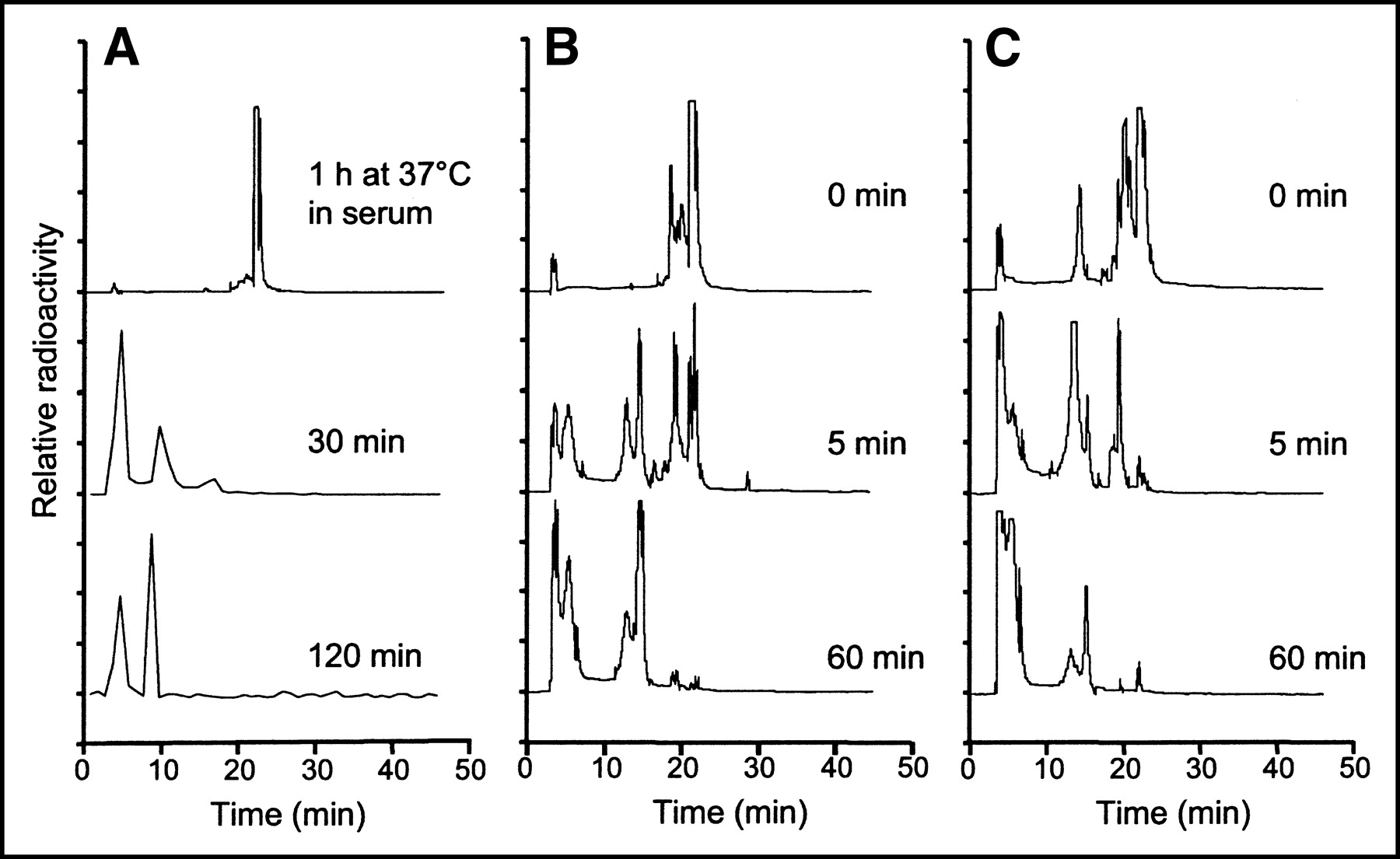
(A) RP HPLC analysis of 111In-DTPAGlu-G-CCK8 incubated for 1 h in serum (top trace) and radioactivity recovered from blood samples taken at different times after injection into a nude mouse (bottom 2 traces). The compound is very stable in serum but appears to be rapidly metabolized after injection to more hydrophilic compounds. The small amount of radioactivity in the blood samples taken after injection required the use of a fraction collector and γ-counting. (B and C) RP HPLC analysis of 111In-DTPAGlu-G-CCK8 incubated with tissue homogenates of liver (B) and kidneys (C). Homogenates were freshly prepared and incubated on ice. Radiolabeled peptide was added to the homogenates and either immediately extracted with CH3CN (0 min) or incubated at 37°C for the indicated times. A breakdown of the peptide to more hydrophilic forms appears to be more rapid and prominent in the kidneys.
DISCUSSION
We have characterized the in vitro and in vivo properties of 111In-DTPAGlu-G-CCK8, a derivative containing the chelating agent DTPAGlu bound through a glycine linker at the N-terminal end of the bioactive peptide CCK8. Other investigators already have demonstrated the feasibility of using CCK8-based peptides to target CCKBR in vivo (8,13). The novelty of our work involves the use of a new DTPA derivative. There are 2 advantages to this approach.
First, the use of the chelating agent DTPAGlu allows a peptide conjugate having 5 carboxyl groups and 3 amino groups available for radiometal coordination to be obtained. The 8 chelating functions constitute a coordinated set able to yield stable metal complexes, in particular, with metal ions in the +3 oxidation state. There is one more chelating function beyond the coordinated set displayed by DTPA directly bound to a peptide. The 8 functions displayed in the coordinated set by our peptide conjugate are the same as those used by DTPA alone to yield very stable complexes. The additional coordinating moiety may provide better stability for chelating other radiometals of interest, such as 90Y, which is used in therapeutic applications and is known to form rather unstable complexes with conventional DTPA-coupled peptides (21).
Second, the entire peptide conjugate is synthesized in the solid phase, allowing high yields of the compound and high purity of the raw product to be obtained. The final HPLC purification step yields a well-characterized compound that corresponds to the desired molecule. Up to now, most peptide–chelate conjugates, as well as some DTPA-coupled derivatives of CCK8 and gastrin and their analogs, have been prepared by solution coupling of the chelating moiety to the bioactive peptide (8). This procedure produces potentially undesirable by-products and lower yields than does chelator coupling during solid-phase synthesis, which also has been described for coupling CCK8 analogs to DTPA (12).
Radiolabeling of the compound was rapid and yielded a high radiochemical purity (Fig. 2A), consistent with the predicted high efficiency of the DTPAGlu moiety for coordinating indium. The labeled compound displayed a specific interaction with CCKBR in both of the cell lines that we tested. Radiolabeled peptide bound to CCKBR-overexpressing cells was displaced easily by the addition of unlabeled peptide. Binding to non–receptor-expressing cells was negligible (Fig. 2B). The affinity of the interaction between 111In-DTPAGlu-G-CCK8 and CCKBR was adequate for use in clinical applications. Kds on the order of 10−8 mol/L are well within the acceptable range for receptor-binding radiopharmaceuticals (22).
One of the biologic features of G-protein–coupled receptors, such as CCKBR, is that the receptor and the ligand are internalized to intracellular compartments after interaction. This mechanism constitutes the principal means by which the cell limits the molecular signal provided by receptor activation. For radiopharmaceutical application of peptides, this mechanism provides higher levels of labeled material to be associated with target cells through at least 2 mechanisms. First, the ability to internalize and continuously turn over the receptor population that is available on the cell surface guarantees that there are always receptors on the cell surface capable of interacting with the ligand present in the extracellular space. Second, because the radiolabeled ligand is internalized, it is removed from the equilibrium between unbound compound and bound compound, thus increasing target-associated radioactivity. Internalization also contributes to prolongation of the retention times of radiopharmaceuticals in the target organ, which are of particular interest in therapeutic applications with radionuclides. Our compound appears to be internalized to a large extent when incubated with cells at 37°C (Fig. 4A) in both cell systems tested. Furthermore, the retention time of the radiolabel in cells appears to be sufficient for use of the compound in clinical applications, and this feature likely is aided by the fact that the compound is internalized (Fig. 4B).
In vivo evaluation of 111In-DTPAGlu-G-CCK8 showed rapid and specific targeting of CCKBR-expressing xenografts (Fig. 5). At all time points evaluated, there was a severalfold-higher accumulation of radioactivity in these xenografts than in control xenografts. Clearance of unbound radioactivity was extremely rapid and occurred exclusively via the urinary tract, with retention of a portion of the injected radioactivity in the kidneys. There appears to be virtually no clearance through the hepatobiliary system, a great advantage, because such clearance produces low background activity in the gastrointestinal tract that may impair the detection of CCKBR-overexpressing tumors in the abdomen. Nonspecific retention of radioactivity in other organs is low. There is a slightly higher level of retention of the compound in the stomach than in the rest of the gastrointestinal tract, consistent with higher concentrations of CCKBR in specific areas of this organ. Imaging experiments performed with 111In-DTPAGlu-G-CCK8 (Fig. 6) confirm the suitability of the peptide conjugate for in vivo applications. The low level of background activity and high level of specific targeting of CCKBR-positive tumors are evident in the scintigraphic image.
The in vitro stability of 111In-DTPAGlu-G-CCK8 in serum is high, and the compound remains intact for prolonged periods of incubation at 37°C. Blood samples drawn from mice after injection of the compound, however, show the presence of only circulating metabolites as early as 30 min after injection (Fig. 7A). These observations suggest that although the compound is stable enough in serum to reach receptor-expressing tissues, it is also actively metabolized at some level. We presume that such breakdown occurs in the kidneys because, first, there is a high level of accumulation of label in the kidneys in the biodistribution experiments (Fig. 5) and, second, the experiments performed with kidney homogenates ex vivo show that enzymatic activities present in the kidneys are capable of rapidly and thoroughly metabolizing the peptide conjugate (Fig. 7C). Such rapid in vivo metabolism of the radiolabeled peptide likely is a major contributing factor for high target-to-nontarget ratios in A431 xenografts, as it favors the rapid clearance of unbound radioactivity. The hydrophilic breakdown products generated are cleared rapidly through the urine, keeping background radioactivity levels low. During this process, there appears to be no breakdown of the 111In-DTPA complex. In fact, the 111In appears to remain bound to the chelating agent, as no generation of free 111In ions occurs. The stability of the chelating moiety also contributes to keeping background radioactivity levels low, as free 111In would be retained on metal-binding proteins present in the serum and would show slower clearance. Overall, the rapid metabolism of the peptide conjugate and the stability of the chelating moiety contribute to the achievement of low background radioactivity levels but to no hindrance of the ability of the compound to bind specifically to CCKBR-expressing tumors.
Kidney uptake of small peptides is a major concern, as the kidneys are typically the critical organs for these types of radiopharmaceuticals and for CCKBR-targeting peptides as well. DTPA-coupled minigastrin derivatives (9,11) have kidney retention values of 40–50 %ID/g in tumor-bearing nude mice. These values are rather constant from 10 min after injection up to 24 h. Reducing kidney accumulation while maintaining good specific targeting would lower the radiation dose absorbed by these organs. This situation would be a major improvement, as concerns for nephrotoxicity already have been raised in preliminary therapeutic applications with 90Y (10). The retention in kidneys of our compound progressively decreases over time (at 4 h, kidney retention is 2.0 ± 0.9 %ID/g [mean ± SD]). However, no definitive conclusions can be gathered by comparing tumor uptake values, because different models were used and there were differences in the numbers of receptors expressed and other variables, such as vascular permeability and blood flow. Our compound shows better relative CCKBR targeting; A431-CCKBR xenografts at 4 h show uptake of 1.4 ± 0.9 %ID/g and a tumor-to-kidney ratio of 1:1.5. For 111In-DTPA-minigastrin, uptake in TT xenografts has been reported to be ∼4 %ID/g at 3 h after injection (9), with a tumor-to-kidney ratio of ∼1:10, and 0.31 ± 0.06 %ID/g at 4 h (11), with a tumor-to-kidney ratio of ∼1:160. 111In-DTPA-sulfated CCK8 (8) has a much lower level of kidney retention, 6 %ID/g at 4 h, and an uptake value of ∼6 %ID/g at 4 h in TT xenografts (∼1:1 tumor-to-kidney ratio); however, this compound has similar retention levels in other organs, such as the lungs, liver, and spleen, and therefore shows poor specific targeting and elevated background activity levels. DTPA(d-Asp26,Nle28,31)CCK(26–33) (13) was evaluated in a rat tumor model and showed good relative targeting of CA20948 tumors (0.09 % ID/g at 24 h, compared with 0.3 %ID/g in the kidneys); however, a later evaluation in humans (14) showed high background activity levels in scintigraphic images, with evidence of hepatobiliary clearance and relatively poor targeting of CCKBR-positive structures, such as the stomach.
Improvement of the kinetic stability of DTPA-coupled peptides for chelating indium and yttrium also has been addressed as a way to advance this class of radiopharmaceuticals. Behe et al. recently showed that the stability of 111In-DTPA-minigastrin is improved greatly by the addition of a d-Glu moiety adjacent to the chelating group (11). This structure results in lower rates of indium transchelation to proteins, although the exact mechanism by which this process occurs still is not fully understood. DTPA-d-Glu-minigastrin shows transchelation rates on the order of 7%–8% after incubation for 24 h in serum; the rate for DTPA-minigastrin is ∼25%. The transchelation rate that we obtained (6% at 24 h in fetal bovine serum) is in the same range as those reported for DTPA-d-Glu-minigastrin, indicating that the DTPAGlu chelating moiety provides an advantage in stability. Future studies will address the use of this approach for the labeling, stability, and therapeutic efficacy of 90Y-DTPAGlu-G-CCK8.
CONCLUSION
We have characterized the chemical, radiochemical, in vitro, and in vivo biologic properties of 111In-DTPAGlu-G-CCK8. The compound shows specific, high-affinity binding and is internalized by CCKBR-expressing cells in vitro. The specificity of the interaction of the compound with its receptor is maintained in applications in nude mice, showing a high level of accumulation in CCKBR-positive xenografts, rapid renal clearance, and a low level of background activity. Our findings indicate that 111In-DTPAGlu-G-CCK8 shows promise for clinical use in CCK receptor imaging and has biodistribution characteristics more favorable than those of previously described CCKBR-binding ligands. Furthermore, the ease of synthesis, high labeling efficiency, and chemical stability of the 111In-DTPAGlu complex make this an ideal candidate for use in other peptide-radiolabeling applications.
Acknowledgments
This work was funded by Ministero Universitá e Ricersca Scientifica e Tecnologica and Consiglio Nazionale delle Ricerche, Progetto Strategico: Oncologia. The technical assistance of Carmine Di Nuzzo is greatly appreciated. We are grateful to Bracco Imaging SpA for providing the DTPAGlu compound. We thank Silvio Aime for critical assessment of the results obtained.
Footnotes
Received Apr. 14, 2003; revision accepted Nov. 24, 2003.
For correspondence or reprints contact: Luigi Aloj, MD, Istituto di Biostrutture e Bioimmagini, CNR, Edificio 10, Via S. Pansini 5, I80131 Napoli, Italy.
E-mail: luigi.aloj{at}mailcmn.area.na.cnr.it
REFERENCES
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Signaling Network Response to {alpha}-Particle-Targeted Therapy with the 225Ac-Labeled Minigastrin Analog 225Ac-PP-F11N Reveals the Radiosensitizing Potential of Histone Deacetylase Inhibitors
- DOTA-MGS5, a New Cholecystokinin-2 Receptor-Targeting Peptide Analog with an Optimized Targeting Profile for Theranostic Use
- "To Serve and Protect": Enzyme Inhibitors as Radiopeptide Escorts Promote Tumor Targeting
- Evaluation of an 111In-Radiolabeled Peptide as a Targeting and Imaging Agent for ErbB-2 Receptor Expressing Breast Carcinomas
- Selection of Radiolabeled Gastrin Analogs for Peptide Receptor-Targeted Radionuclide Therapy
- CCK-2/Gastrin Receptor-Targeted Tumor Imaging with 99mTc-Labeled Minigastrin Analogs