


Abstract
Imaging techniques have demonstrated in various cardiomyopathies that an altered uptake of radiolabeled norepinephrine (NE) analogs may coexist with β-adrenergic receptor downregulation, but the relationships between these 2 alterations and their mechanisms remain unclear. The aim of this study was to evaluate the hypothesis of a chronic elevation of circulating NE levels as a mechanism of decreased uptake of radiolabeled NE analogs and reduced β-adrenergic receptor sites in the heart. Methods: Osmotic minipumps containing either NE or NaCl were implanted intravenously in rats for 5 d. The uptake-1 function was assessed in vitro by measuring in excised hearts 3H-NE and 123I-metaiodobenzylguanidine (123I-MIBG) uptakes and uptake-1 carrier density using 3H-mazindol binding assay. The myocardial β-adrenergic receptor pathway was assessed in vitro by 3H-CGP 12177 binding and measurement of adenylyl cyclase activity at baseline and under stimulation. Results: A 34% decrease in 3H-NE uptake and a 35% decrease in 123I-MIBG uptake were found in the hearts of rats infused with the NE pump compared with that of rats infused with saline solution (P < 0.05 for both). Moreover, the uptake-1 carrier protein density was decreased by 29% (P < 0.05) and 33% (P < 0.05) in right and left ventricles, respectively, of rats infused with NE compared with those infused with saline solution. Myocardial β-adrenergic receptor desensitization was associated with a 36% reduction in receptor density in the right ventricle (P < 0.05) and a 31% reduction in the left ventricle (P < 0.05) of rats infused with NE compared with those infused with saline solution. Conclusion: The decrease in myocardial β-adrenergic receptors and radiolabeled NE analog uptake found in different cardiomyopathies using neuroimaging techniques may be related to a functional mechanism of NE-induced downregulation of both β-adrenergic receptor and uptake-1 carrier sites.
- norepinephrine
- uptake-1
- β-adrenergic receptor
- radiolabeled norepinephrine analogs
- MIBG
- hydroxyephedrine
- single-photon imaging
- PET
Contraction of cardiac muscle is regulated by both neuronally derived and circulatory catecholamines. Carrier-mediated uptake transport of norepinephrine (NE) plays a key role in the regulation of sympathetic neurotransmission. The introduction of radiolabeled catecholamine analogs such as radioiodinated metaiodobenzylguanidine (MIBG) and 11C-hydroxyephedrine (11C-HED) has allowed the noninvasive assessment of the NE reuptake (uptake-1) and storage system, whereas the use 11C-CGP 12177 with PET has permitted the measurement of β-adrenergic receptor density (1). Decreased myocardial uptake and kinetics of MIBG or 11C-HED have been reported in many experimental and clinical conditions in which the adrenergic system was deeply altered (2–11). Although the exact mechanisms of decreased uptake of radiolabeled NE analogs remain putative, many complications have been related to this alteration. Clinical and animal studies have demonstrated a correlation between the heterogeneity of sympathetic innervation and arrhythmias in ischemic cardiomyopathy (5,12). The role of the autonomic nervous system in the pathophysiology of congestive heart failure and arrhythmias is now established. In congestive heart failure, many studies have shown a direct relationship between the importance of neurohormonal disorders and mortality and there is evidence that pharmacologic inhibition of the adrenergic pathway by angiotensin-converting enzyme inhibitors or β-blockers improves cardiac function and survival by protecting the heart from overexposure to catecholamines.
Diminution of vesicular storage, reduction in uptake-1 carrier sites, or competitive inhibition at the binding sites by elevated synaptic NE concentration could explain decreased NE analog uptake. Some reports in animal models have demonstrated that an exposure to high catecholamine levels could cause a loss of uptake-1 carrier sites in a manner that suggests a potential downregulation of the NE transporter (13–15) similar to that observed for β-adrenergic receptors. The purpose of this study was to evaluate the role of a chronic increase in circulating NE level as a mechanism for both decreased NE analog uptake and β-adrenergic receptor downregulation. To evaluate the effects of an overexposure to the neurotransmitter, miniosmotic pumps delivering intravenously either NaCl or NE were implanted for 5 d in rats. The adrenergic innervation function was assessed by measuring in vitro cardiac fixations of intravenously injected 3H-NE and 123I-MIBG and uptake-1 carrier protein density. To evaluate the postsynaptic modifications, the density of cardiac β-adrenergic receptors was determined using in vitro binding assays with 3H-CGP 12177 and the adenylate cyclase activity was measured either in basal state or after stimulation.
MATERIALS AND METHODS
All animal procedures were in accordance with the recommendations of the European Economic Community (86/609) and the French Committee (87/848) for the care and use of laboratory animals.
Minipump Implantation
Male Wistar rats (200–250 g) were kept in individual cages, received food and water ad libitum, and were submitted to a 12-h:12-h dark:light cycle.
Rats were anesthetized with ketamine before minipump implantation. Alzet model 2001 osmotic minipumps were used for chronic intravenous infusion of NE (Sigma Chemical Co.) at a rate of 0.02 μg/min or for NaCl infusion. A small neck incision was made to expose the right jugular vein. The silastic catheter was inserted into the jugular vein to place its tips in the right atrium. The pump was fixed subcutaneously over the right scapula and then the incision was closed.
At the end of the 5 d, rats were anesthetized with ketamine and a small neck incision was made. The intravenous NE infusion was stopped by closing the catheter of the pump with a thread, and then the incision was closed. The animals were kept in their cage for 2 h to permit the elimination of the extrinsic NE and to allow recovery from the effects of anesthesia.
Uptake Measurements of NE Analogs and Perfusion Tracer
Two series of rats were injected with either 3H-NE or 123I-MIBG. For the first series of rats, animals were removed from their cage and injected in the tail vein with 37 MBq 99mTc-sestamibi to assess the myocardial perfusion. One hour later, rats were injected in the tail vein with 17 kBq 3H-NE. The animals were killed 10 min later, because the myocardial 3H-NE concentrations peak during the first 10 min after the administration and the tracer metabolization is fast (16). Heart, lungs, liver, and muscles (soleus and extensor digitorum longum) were excised and a blood sample was drawn. The right and left ventricles were separated and all samples were weighed. The 99mTc activity in the right and left ventricles was measured in a γ-counter (Kontron 2000). One sample of each organ was immersed in a liquid (Beckman tissue solubilizer-450) to dissolve tissues overnight at 55°C. After dissolution, a β-scintillator cocktail (Ready solv MP; Beckman) was added to each sample. After the decay of 99mTc activity (3 d), the 3H-NE activity was measured in a β-counter (80% efficiency; Packard SL 2000). For the second series of rats, animals were killed 4 h after 123I-MIBG injection, because myocardial MIBG uptake measured after this delay has been shown to be the best index of neuronal accumulation (17). Heart, lungs, liver, and muscles were excised and blood was drawn. Right and left ventricles were separated. All samples were weighed (wet weight). Their 123I tissue activities were determined in the γ-counter.
For the different radiopharmaceuticals, the tissue concentrations were expressed in % kg dose/g to normalize for differences in animal weights (7).
Preparation of Rats for In Vitro Binding Assays
Another series of experiments was done on 20 rats (10 control and 10 NE rats). After 5 d, the pump infusion was stopped as previously described, and the rats were killed. The hearts were excised, the right and left ventricles were separated, and blood was drawn to determine the catecholamine concentrations. The myocardial samples were immediately immersed in liquid nitrogen and kept in a −80°C freezer until measurements of β-adrenergic receptor density, uptake-1 carrier density, and adenylate cyclase activity.
Preparation of Cardiac Membranes for Binding and Adenylyl Cyclase Assays
Preparation of Cardiac Membranes.
For the uptake-1 carrier protein binding assay, the frozen ventricles were cut and homogenized in 10 mL of ice-cold buffer (50 mmol/L Tris-HCl, pH 7.7) with a Polytron (PCU Kinematica, Gmbh- Bioblock) with one 5-s burst at a setting of 7 and centrifuged at 40,000g for 15 min at 4°C. The pellet was resuspended in 50 mmol/L Tris-HCl, 120 mmol/L NaCl, 5 mmol/L KCl, pH 7.4, to obtain a protein concentration of 0.3 mg/mL. Protein measurements were made using serum albumin as the standard (18).
For the β-adrenergic receptor binding assay, the frozen ventricles were cut and homogenized in 10 mL of ice-cold buffer (20 mmol/L Tris-HCl, 1 mmol/L EDTA [ethylenediaminetetraacetic acid], pH 8) using the Polytron with one 5-s burst at a setting of 7; 1 mL of ice-cold 2.5 mol/L KCl was added to extract contractile proteins followed by stirring at 4°C for 15 min. The suspension was then centrifuged at 40,000g for 20 min. The pellet was resuspended in 20 mmol/L Tris-HCl, 1 mmol/L EDTA ice-cold buffer with a Dounce homogenizer (H&R Co.) to achieve a protein concentration of 0.3 mg/mL.
Radioligand Binding for Uptake-1.
Mazindol was used to bind uptake-1 carrier protein (19). The incubation buffer contained 0.1 mL of 50 mmol/L Tris-HCl, 120 mmol/L NaCl, 5 mmol/L KCl, pH 7.4, or 0.1 mL of 100 μmol/L desipramine (uptake-1 inhibitor), 0.1 mL 3H-mazindol (concentration, 3–60 nmol/L), and 0.3 mL of membrane preparation. The assay was incubated 20 min at 4°C.
Radioligand Binding for β-Adrenergic Receptors.
The radioligand 3H-CGP 12177 was used to label the receptors. The incubation buffer contained 0.1 mL of 50 mmol/L Tris-HCl, 10 mmol/L MgCl2, pH 7.4, or 0.1 mL of 10 μmol/L l-propranolol (nonspecific binding), 0.1 mL of 3H-CGP 12177 (0.1–8 nmol/L), and 0.3 mL of membrane preparation. The assay was incubated 60 min at 37°C.
Termination of the Binding Assay.
The reaction was terminated with filtration through Whatman GF/C filters (Millipore) and washing with ice-cold incubation buffer. Filter radioactivity was determined by liquid scintillation counting. Uptake-1 carrier or β-adrenergic receptor density and affinity were determined from Scatchard plots of the counting data.
Adenylyl Cyclase Assay.
Adenylyl cyclase activity was measured in cardiac homogenate membranes according to the method described by Salomon et al. (20). Briefly, this method is based on the measurement of the level of 32P-cyclic adenosine monophosphate (cAMP) production after incubation with 32P-ATP-Mg2+ (where ATP is adenosine triphosphate), which is proportional to adenylyl cyclase activity. The membrane preparation was similar to that described for the receptor binding assay but we used another ice-cold buffer (50 mmol/L Tris-HCl, 10 mmol/L MgCl2, pH 7.4). After homogenization and centrifugation, the pellet was resuspended in the same buffer with a Dounce homogenizer to obtain a protein concentration of 2.5–5 mg/mL. The reaction was performed in an assay medium (final volume, 60 μL) containing 50 mmol/L Tris-HCl, 5.0 mmol/L MgCl2, 1.0 mmol/L 3-isobutyl-1-methylxanthine (phosphodiesterase inhibitor), 1.0 mmol/L EDTA, an ATP-regenerating system (1 mg/mL creatine kinase and 1 mmol/L phosphocreatine), 1 mmol/L 32P-ATP (106 counts per minute [cpm]), 1 mmol/L cAMP to inhibit endogenous phosphodiesterase activity, and ∼104 cpm of 3H-cAMP were also added to calculate the efficiency of the alumina column (column recovery, 70%–90%). The incubation was started by addition of membrane fractions (20 μL, 50–100 μg protein). After a 10-min incubation period in a shaking water bath at 37°C, the reaction was terminated by adding 200 μL of 0.5 mol/L HCl followed by immediate boiling for 7 min. The pH of the assay mixture was adjusted to 7.6 with 250 μL of 1.5 mol/L imidazole. Samples were then eluted with 3 mL of 10 mmol/L imidazole-HCl, pH 7.6, through an alumina column that retains 32P-ATP but allows 32P-cAMP to be eluted. The 3H and 32P activities of the eluate were then counted, after addition of scintillation cocktail, in a β-scintillation counter. All assay points were the means of triplicate values. Adenylyl cyclase activity was determined with and without addition of stimulants. Adenylyl cyclase activity was stimulated using isoproterenol, NaF, forskolin, 5′guanylylimidodiphosphate (Gpp(NH)p), and MnCl2: 5 mmol/L NaF, 0.1 mmol/L l-isoproterenol + 0.1 mmol/L guanosine triphosphate (GTP), 100 μmol/L forskolin, 0.1 mmol/L Gpp(NH)p, or 5 mmol/L MnCl2. GTP was added to optimize the coupling of agonist to receptor by acting on regulatory G proteins. Isoproterenol was used to stimulate the enzyme by activating β-adrenergic receptors, NaF was used to activate the system at the level of G proteins, and forskolin was used to produce direct stimulation of the catalytic moiety of the enzyme. All determinations were performed in triplicate.
Plasma Catecholamine Concentration
Blood was placed in ice-cold tubes containing heparin and then centrifuged; the plasma was stored at −80°C until the assay. Plasma NE and epinephrine levels were measured by high-performance liquid chromatography in 4 groups of rats: 6 control rats, 6 rats killed immediately after stopping saline solution pumps, 6 rats killed immediately after stopping NE pumps, and 6 rats killed 1 h after stopping NE pumps.
Statistical Analysis
All parameters are expressed as mean ± SD. Two-way ANOVAs were used to compare parameters between groups, followed when appropriate, by nonparametric tests (Kruskall–Wallis). The level of statistical significance was set at 0.05.
RESULTS
Body and Cardiac Ventricle Weights
Animals infused with saline solution weighed 274 ± 43 g (n = 8), and animals infused with NE weighed 271 ± 36.6 g (n = 9). The ratios of left ventricle to body weight were not statistically different when comparing animals infused with saline solution with those infused with NE (1.95 ± 0.22 mg/g vs. 1.89 ± 0.28 mg/g, respectively; P = not significant).
Catecholamine Levels
Results are listed in Table 1. Plasma NE levels were higher in the NE pump group than in the group of rats (control rats) without pump implantation (7.01 ± 0.61 vs. 0.6 ± 0.2 ng/mL, respectively; P < 0.05). Both pump implantation and anesthesia were factors of stress for the rats, as suggested by the increase in plasma NE levels found in the group of rats implanted with saline solution pumps compared with that of control rats (3.22 ± 0.57 vs. 0.60 ± 0.2 ng/mL, respectively; P < 0.05). In another group of NE-infused rats (n = 6), plasma catecholamine levels were measured 1 h after stopping the NE infusion. For this group, the plasma NE level decreased but stayed at a level higher than that measured in saline solution group (4.63 ± 0.70 vs. 3.22 ± 0.57 ng/mL, respectively; P < 0.05).
Circulating Catecholamine Levels
Cardiac Perfusion Assessed by 99mTc-Sestamibi Uptake
When comparing NE-infused rats with saline solution-infused animals, there was no difference in 99mTc-sestamibi concentrations measured in the whole heart (69.8% ± 11.6% vs. 68% ± 7.6% kg dose/g, respectively; P = not significant) as well as in either the right ventricle (66.2% ± 12.2% vs. 64.4% ± 6.8% kg dose/g, respectively; P = not significant) or the left ventricle (65.2% ± 8.8% vs. 64.8% ± 6.8% kg dose/g, respectively; P = not significant).
Myocardial 3H-NE and 123I-MIBG Uptakes
When comparing NE-infused rats with saline solution-infused animals (Table 2), the myocardial 3H-NE uptake was decreased (P < 0.05) by 34.8% in the whole heart (0.75 ± 0.06 vs. 1.15% ± 0.16% kg dose/g, respectively), by 37% in the right ventricle (0.390 ± 0.025 vs. 0.620% ± 0.100% kg dose/g, respectively), and by 32% in the left ventricle (0.360 ± 0.060 vs. 0.530% ± 0.061% kg dose/g, respectively).
3H-NE and 123I-MIBG Tissue Concentrations
When comparing NE-infused rats with saline solution-infused animals (Table 2), the 123I-MIBG myocardial fixation was decreased by 35.2% in the whole heart (0.590% ± 0.065% vs. 0.910% ± 0.073% kg dose/g, respectively; P < 0.05).
There was no statistical difference in 3H-NE or 123I-MIBG uptake among the others organs (lungs, liver, muscles) when comparing rats infused with either NE or saline solution.
Uptake-1 Carrier Protein
When comparing NE-infused rats with saline solution-infused animals (Table 3), a 30% decrease in uptake-1 carrier protein density was found in the right ventricle (295 ± 55 vs. 416 ± 73.4 fmol/mg protein, respectively; P < 0.05) and a 33% decrease was found in the left ventricle (327 ± 75 vs. 488 ± 40.3 fmol/mg protein, respectively; P < 0.01). The affinity constant (Kd) of 3H-mazindol for the uptake-1 carrier protein was not different between the saline solution pump group and NE pump group.
Effects of NE Infusion on Uptake-1 Carrier Protein Density
β-Adrenergic Receptors
When comparing the NE pump group with the saline solution pump group (Table 4), β-adrenergic receptor density decreased by 35.7% in the right ventricle (17.6 ± 2.97 vs. 27.4 ± 1.07 fmol/mg, respectively; P < 0.001) and decreased by 31.3% in the left ventricle (25.8 ± 2.77 vs. 37.6 ± 1.52 fmol/mg, respectively; P < 0.0001). The Kd for 3H-CGP 12177 was not significantly different between the 2 groups.
β-Adrenergic Receptor Density
Adenylyl Cyclase Activity
When comparing the NE pump group with the saline solution pump group (Table 5), the right ventricle showed a 37.5% decrease in basal adenylyl cyclase activity (P < 0.001), a 37% decrease in isoproterenol- or Gpp(NH)p-stimulated adenylyl cyclase activity, and a 42% decrease in forskolin-stimulated adenylyl cyclase (P < 0.01 for the 3 stimuli), whereas the left ventricle showed a 33.5% decrease in basal adenylyl cyclase activity (P < 0.001), a 26% decrease in isoproterenol-stimulated adenylyl cyclase activity, a 45% decrease in Gpp(NH)p-stimulated adenylyl cyclase activity, and a 37% decrease in forskolin-stimulated adenylyl cyclase (P < 0.01 for the 3 stimuli). In all experiments, NaF- and MnCl2-sensitive adenylyl cyclase activity was unchanged.
Effects of NE Infusion on Adenylyl Cyclase Activity
DISCUSSION
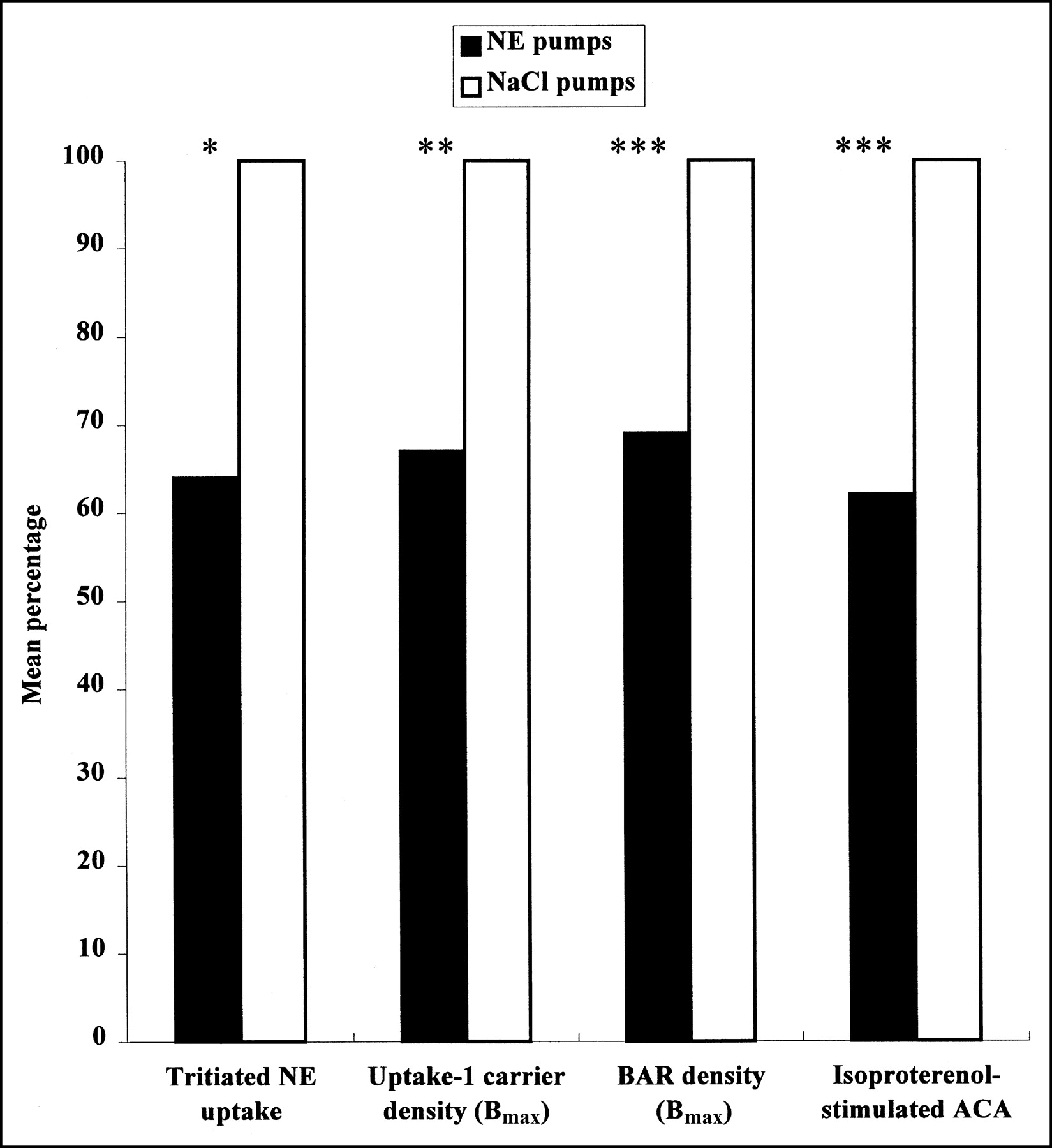
This study demonstrates that a chronic elevation of circulating NE causes a reduction in myocardial uptake of radiolabeled NE analogs, in proportion to the loss of uptake-1 carrier sites that parallels desensitization and downregulation of β-adrenergic receptors (Fig. 1). Our data point out the interest of studying both the uptake of radiolabeled NE analogs and the β-adrenergic receptor density by neuroimaging techniques to assess the pathophysiologic mechanisms of disorders involving hyperactivity of the sympathetic system and to evaluate their potential clinical implications.

Effect of chronic infusion of NE on myocardial noradrenergic neurotransmission. After 5-d NE infusion, similar reduction in NE uptake, uptake-1 carrier density, β-adrenergic receptor (BAR) density, and responsiveness to agonists was observed in left ventricle of rats. ACA = adenylyl cyclase activity; Bmax = carrier or receptor protein density. *P < 0.05. **P < 0.01. ***P < 0.001.
Uptake-1 is the main mechanism for removal of NE from the presynaptic cleft and termination of action of NE on the myocardial β-receptors, because as much as 90% of the released NE is taken back into presynaptic nerve and is returned to storage granules from which it can be released again. In nonneuronal tissue, uptake-2 is another transport mechanism that removes NE from the extracellular space through a transport protein but that is quantitatively much less important than uptake-1 (21). Because neuronal reuptake of NE is the major mechanism for terminating the action of the neurotransmitter, decreased uptake-1 increases interstitial NE and causes myocyte hyperstimulation. Indeed, in experimental models, the cardiac response to exogenous NE was prolonged by the blockade of uptake-1 but was unchanged by the blockade of uptake-2 (22,23). Therefore, elevation of the NE concentration at the synaptic level generated by impaired uptake-1 can induce deleterious effects such as hypertrophy, myocyte calcium overload, arrhythmias, increase in myocardial oxygen consumption, alteration in coronary artery tone, or β-adrenergic desensitization.
The use of radiolabeled NE analogs allows the noninvasive exploration of uptake-1 function using single-photon imaging techniques or PET. MIBG is unmetabolized by catechol-O-methyltransferase or monoamine oxidase (24) and this property favors a slow tracer accumulation in adrenergic tissue high enough to be measurable by single-photon imaging (17). The MIBG uptake is similar to that of NE and can be largely reduced with pharmacologic inhibitors of uptake-1 such as desipramine and cocaine or with anatomic disruption of the sympathetic innervation (25,26). Conversely, PET radiolabeled NE analogs seems to display a higher level of specific uptake in the myocardium of rats (27).
A decrease in uptake-1 and NE storage has been repeatedly found using either single-photon imaging with MIBG or PET with 11C-HED in various heart diseases (1–4,8). This finding has been confirmed in patients with ischemic and idiopathic end-stage cardiomyopathies by sophisticated invasive measurements or by in vitro assays (8,28,29). The clinical implications of reduced radiolabeled NE analog uptake may vary considerably in different cardiac diseases. Impaired uptake-1 function seems to contribute to β-adrenergic desensitization only when it is associated with sympathetic hyperactivity (13,30). In heart failure, evidence has accumulated that sympathetic activation plays an important role in the progression of cardiac dysfunction. An inverse relationship between myocardial radiolabeled NE analog uptake and β-receptor density has been found in the failing heart. In idiopathic dilated cardiomyopathy, decreased scintigraphic MIBG uptake was found to be correlated with blunted response to myocardial β-adrenergic stimulation using intracoronary dobutamine infusion (31). In vitro analyses of left ventricles of patients with end-stage dilated cardiomyopathy demonstrated that reduced 3H-NE uptake was directly proportional to decreased β-adrenergic receptor density (32). The concept that the hyperactivity of the adrenergic nervous system plays a dysregulatory role in chronic heart failure was confirmed when the beneficial effects of β-blockade were shown not only on both hemodynamic condition and clinical outcome (33–35) but also on uptake-1 function (36).
Similarly, data from 123I-MIBG or 11C-HED imaging studies have suggested that impaired adrenergic nerve function was involved in mechanisms of primary hypertrophic cardiomyopathy (6,9) and in idiopathic right ventricular outflow tract tachycardia (37). As in heart failure, a concomitant decrease in both β-adrenergic receptor sites and 11C-HED uptake has been demonstrated (9,37).
On the other hand, in the case of full anatomic denervation in cardiac transplantation, leading to dramatically decreased NE analog uptake in the heart, the only apparent physiologic consequence appears to be a prolongation of the action of β-receptor agonists, which are usually taken up through the uptake-1 carrier system (30). This fact implies that NE concentration does not increase at the receptor site in the transplanted heart, as suggested by the lack of changes in myocardial β-adrenergic receptor density during the first 18 mo after transplantation (38). Moreover, in a dog model associating experimental heart failure and ventricular denervation, significantly less catecholamine-induced desensitization was found in comparison with that of dogs having the same heart failure but intact myocardial adrenergic innervation (39). These data indicate that the presence of normal ventricular innervation is required for physiologic expression of catecholamine overexposure.
As a potential methodologic limitation of our study, species differences exist regarding the respective importance of uptake-1 and uptake-2 systems in the myocardial fixation of MIBG. That is the reason why, in this study, some rats were injected intravenously with 3H-NE, which has been shown to enter the cell at 90% through the uptake-1 pathway whatever the species studied. A comparable decrease in tracer uptake was found when using the 2 tracers, suggesting that our results may be extended to other NE analogs and to other species.
Another potential confounding factor in this study may be an interference of changes in flow related to NE administration. Cardiac uptake of MIBG has been shown to be closely related to blood flow in pig hearts (40). Therefore, we checked that the blood flow was not altered after a 5-d NE infusion. The decreased number of uptake-1 carrier sites found in rats exposed to NE confirms that the decreased uptake of radiolabeled NE analogs was not due to any alteration in flow delivery.
The value of the model we used to generate an extrinsic elevation of circulating NE may be limited because rats implanted with saline solution pumps showed elevated circulating NE concentrations in comparison with those of control rats. This finding may be due to an increased propensity to stress as a consequence of the prior minipump implantation. However, this stress was not sufficient to induce either uptake-1 carrier downregulation or β-adrenergic receptor desensitization. Nevertheless, a potentiation of the effect of extrinsic NE by endogenous NE cannot be ruled out.
CONCLUSION
This study indicates a potential mechanism of the decrease in the myocardial fixation of radiolabeled NE analogs in patients with cardiomyopathies. This mechanism involves a downregulation of uptake-1 carrier sites that parallels β-adrenergic receptor downregulation in response to chronic elevation of neurotransmitter concentration. This phenomenon probably explains why the anatomic sympathetic denervation created by the cardiac transplantation is much better tolerated than the functional denervation present in cardiomyopathies—such as idiopathic, arrhythmic, or hypertrophic cardiomyopathy, in which uptake-1 deficiency is associated with sympathetic hyperactivation.
Footnotes
Received Oct. 22, 2002; revision accepted Mar. 24, 2003.
For correspondence contact: Pascal Merlet, MD, PhD, Service de Médecine Nucléaire, Centre Hospitalo-Universitaire Henri Mondor, 51 Avenue du Maréchal de Lattre de Tassigny, 94010 Créteil Cedex, France.
E-mail: pascal.merlet{at}hmn.ap-hop-paris.fr
REFERENCES
In this issue




{kind=link}
Jump to section
Related Articles
Cited By...
- Nuclear Imaging of the Cardiac Sympathetic Nervous System: A Disease-Specific Interpretation in Heart Failure
- Increased Epicardial Adipose Tissue Volume Correlates With Cardiac Sympathetic Denervation in Patients With Heart Failure
- Prognostic Value of Lymphocyte G Protein-Coupled Receptor Kinase-2 Protein Levels in Patients With Heart Failure
- Myocardial Iodine-123 Meta-Iodobenzylguanidine Imaging and Cardiac Events in Heart Failure: Results of the Prospective ADMIRE-HF (AdreView Myocardial Imaging for Risk Evaluation in Heart Failure) Study
- Evidence for Pre- to Postsynaptic Mismatch of the Cardiac Sympathetic Nervous System in Ischemic Congestive Heart Failure
- Decreased Myocardial {beta}-Adrenergic Receptor Density in Relation to Increased Sympathetic Tone in Patients with Nonischemic Cardiomyopathy
- Presence of Specific 11C-meta-Hydroxyephedrine Retention in Heart, Lung, Pancreas, and Brown Adipose Tissue
- Cardiac Sympathetic Dysfunction Correlates With Abnormal Myocardial Contractile Reserve in Dilated Cardiomyopathy Patients
- Effects of Carvedilol on Myocardial Sympathetic Innervation in Patients with Chronic Heart Failure
- What Is the Clinical Role of Neuronal Imaging?