Article Text

Abstract
The majority of deletions of the short arm of chromosome 5 are associated with cri du chat syndrome (CdCS) and patients show phenotypic and cytogenetic variability. To perform a genotype-phenotype correlation, 80 patients from the Italian CdCS Register were analysed. Molecular cytogenetic analysis showed that 62 patients (77.50%) had a 5p terminal deletion characterised by breakpoint intervals ranging from p13 (D5S763) to p15.2 (D5S18). Seven patients (8.75%) had a 5p interstitial deletion, four (5%) a de novo translocation, and three (3.75%) a familial translocation. Of the remaining four patients, three (3.75%) had de novo 5p anomalies involving two rearranged cell lines and one (1.25%) had a 5p deletion originating from a paternal inversion. The origin of the deleted chromosome 5 was paternal in 55 out of 61 patients (90.2%). Genotype-phenotype correlation in 62 patients with terminal deletions highlighted a progressive severity of clinical manifestation and psychomotor retardation related to the size of the deletion. The analysis of seven patients with interstitial deletions and one with a small terminal deletion confirmed the existence of two critical regions, one for dysmorphism and mental retardation in p15.2 and the other for the cat cry in p15.3. Results from one patient permitted the cat cry region to be distally narrowed from D5S13 to D5S731. Furthermore, this study lends support to the hypothesis of a separate region in p15.3 for the speech delay.
- cri du chat syndrome
- 5p deletion
- phenotype-genotype correlation
- FISH
Statistics from Altmetric.com
The majority of the deletions in the short arm of chromosome 5 (5p−) are associated with cri du chat syndrome (CdCS).1The size of the deletion ranges from the entire short arm to only 5p15.3.2 Hallmark clinical features of CdCS include a high pitched, monotonous cry, microcephaly, a round face, hypertelorism, epicanthic folds, micrognathia, abnormal dermatoglyphics, growth delay, and severe psychomotor and mental retardation. Characteristic phenotypic changes are seen in adolescent and adult patients.3-10
CdCS has an estimated incidence of between 1:15 000 and 1:50 000 live births.7 11 Among the mentally retarded population, the prevalence may be as high as 1:350.7 Most CdCS patients have a de novo deletion (about 80%), parental translocations account for slightly more than 10%, and rare cytogenetic aberrations cause less than 10%.7 Parent of origin studies show a majority of paternally derived deletions: 20/2512 and 10/12.13
Although CdCS is a well defined clinical entity, subjects with 5p deletion show phenotypic and cytogenetic variability. The existence of a critical region responsible for the CdCS phenotype was suggested from the karyotype analysis of 35 Danish patients with 5p deletion, indicating that a region in 5p15.2-15.3 must be deleted for the typical phenotype of the syndrome to be present.7 14 This hypothesis was supported by the identification of subjects with deletions not encompassing 5p15.2 and who either did not display the classical CdCS phenotype15-18 or were completely normal.19 Recent molecular cytogenetic analyses2 20 have led to the definition of two distinct regions, one for the cat-like cry in 5p15.3 between loci D5S13 and D5S760 and the other for dysmorphism, microcephaly, and mental retardation in 5p15.2 between loci D5S23 and D5S791 (CdCCR). The semaphorin F (SEMAF) gene, covering at least 10% of this region, was recently cloned.21 Because of its role in guiding axons or migrating neuronal precursors during cortical development in mice, it has been suggested thatSEMAF may be responsible for some of the features of CdCS.
Church et al,13 22 in studies involving patients with atypical CdCS features or no symptoms, mapped the cat-like cry to the proximal part of 5p15.3 and to the distal region of 5p15.2. Two further separate regions were mapped to 5p15.2, one for childhood facial dysmorphism and moderate mental retardation and the other for adult facial dysmorphism and severe mental retardation. Recently, the δ-catenin gene (CTNND2), a neuronal specific protein involved in cell motility and potentially associated with mental retardation in CdCS, was mapped within this region.23 24The presence of a region for speech delay in the distal portion of 5p15.3 was also hypothesised.13
Few studies have attempted to correlate genotype and phenotype in CdCS7 8 25-30 and the results differ. To investigate possible correlations and to define a molecular and phenotypic map of the short arm of chromosome 5, molecular and phenotypic analyses of 80 patients from the Italian CdCS Register were performed.
Patients and methods
PATIENTS
Eighty patients (35 males and 45 females) with 5p deletion were recruited from the Italian CdCS Register, which, with the help of various cytogenetic laboratories, genetic counselling services, and paediatric units as well as the Italian Cri du Chat Children's Association, has managed to collect personal, cytogenetic, and clinical data for 198 patients since it was set up in 1980. These include anthropometric data at birth and in subsequent follow ups, dysmorphism of childhood and adult age, major and minor malformations, and other medical problems.6 9 31 Parents of subjects participating in the study were personally informed and signed an informed consent form.
All patients underwent a detailed clinical examination by the same clinician (PCM). Their age at the time of molecular-cytogenetic analyses ranged between 4 months and 32 years (median 8 years 7 months). Evaluation of dysmorphism was performed both by pattern recognition (Gestalt)32 and in an analytical manner.9 31 Eight types of dysmorphism (broad nasal bridge, epicanthic folds, strabismus, downturned corners of the mouth, short philtrum, low set ears, microretrognathia, transverse flexion creases) which remain stable throughout life7 9 were evaluated.
Evaluation of psychomotor development was performed using the Denver Developmental Screening Tests (DDST).33 34 The main milestones from each of the four areas of development (gross motor, fine motor-adaptive, personal-social, and language) were considered. The ages at which the patients mastered each skill were retrospectively obtained by having a questionnaire filled in by parents and carers and centiles (25th, 50th, 75th, 95th) were derived based on the total number of subjects recorded as having completed the skill. Skills with less than 23 responses were not included.
All clinical, genetic, and developmental data were collected in a database and the statistical analysis was performed using the χ2 test with Yates's correction, χ2 test for trend, and Student's t test. Bonferroni correction for multiple comparison was used.
CYTOGENETIC ANALYSIS
Peripheral blood cultures of patients and their parents were set up according to a standard protocol. Staining was performed by QFQ banding. Slides were analysed by fluorescent microscope (Olympus Provis AX70) equipped with a CCD camera (Photometrics Sensys). Image analysis was carried out with PSI MacType software. An aliquot was used to obtain IL-2 cell lines. All cell lines were stored in the Galliera Genetic Bank.
MOLECULAR-CYTOGENETIC ANALYSIS
In order to rule out cryptic translocations or other chromosomal rearrangements in the parents, fluorescence in situ hybridisation (FISH) was performed with a biotinylated “painting” probe specific for 5p (LiStar FISH). The chromosome “painting” probe was used according to the manufacturer's protocols.
To find out the extent of the deletions in the patients and to detect interstitial deletions, FISH experiments were performed using 136 single locus DNA lambda phage probes spanning the 5p arm.35 Phage clones were propagated by infection and concentrated by polyethylene glycol precipitation; DNA was extracted by two phenol/chloroform extractions and concentrated by ethanol precipitation. Phage DNA was labelled by nick translation with biotin-16-dUTP (Boehringer-Mannheim), coprecipitated with human Cot1 DNA (Gibco BRL, 1:40) by ethanol, and resuspended in 50% formamide/2 × SSC/10% dextran sulphate at a final concentration of 40 ng/μl.
FISH analysis was carried out as described by Lichter and Cremer.36 In short, slides were denatured in 70% formamide/2 × SSC for four minutes at 75°C in an oven, and dehydrated with ethanol. Phage probes were denatured at 70°C for 10 minutes. Repetitive sequences were suppressed by Cot1 DNA at 37°C for 15 minutes. Hybridisations were carried out at 37°C for 20 hours. Post hybridisation washes were performed in 50% formamide/2 × SSC at 43°C for 15 minutes and in 2 × SSC at 37°C for 15 minutes. Hybridisation was detected by avidin-Cy3 (Amersham). Slides were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (200 ng/ml) and analysed by fluorescent microscope (Olympus Provis AX70) equipped with a CCD camera (Photometrics Sensys). Image analysis was carried out with PSI MacProbe software.
MOLECULAR ANALYSIS
DNA from the patients and their parents was extracted from peripheral blood using conventional techniques and were typed with highly polymorphic PCR based microsatellite markers (Research Genetics)37 spanning the distal chromosome 5p in order to determine the parental origin of the rearranged chromosome. A total of 100 ng of genomic DNA from each subject were amplified using 1 UTaq polymerase in a cocktail containing 0.1 μmol/l of the cold forward primer, 0.4 μmol/l of the reverse primer, and 0.3 μmol/l of 32P labelled forward primer, 0.25 mmol/l of each deoxynucleotide triphosphate, and 1 × PCR buffer in a total volume of 10 μl. PCR amplification was performed using conditions specific for each marker as reported by the manufacturers. After PCR, 5 μl of the sample were denatured for five minutes at 95°C and immediately electrophoresed on a 6% polyacrylamide sequencing gel. Autoradiography was performed after drying the gel in a vacuum.
Results
CYTOGENETIC AND MOLECULAR ANALYSIS
Eighty patients and 148 parents were analysed. Sixty two patients (77.50%) showed a 5p terminal deletion with a wide variability of breakpoints ranging from p13 (locus D5S763) to p15.2 (locus D5S18). Seven patients (8.75%) had a 5p interstitial deletion, four (5%) a de novo translocation involving 5p, and three (3.75%) a 5;autosome translocation inherited from a parent. Three others (3.75%) had de novo 5p anomalies involving two rearranged cell lines and one (1.25%) a 5p deletion inherited from a paternal inversion. Karyotypes of the seven patients with unbalanced translocations and of the three with mosaicism are summarised in tables 1 and 2. Out of 148 parents, four showed an abnormal karyotype with a rearranged chromosome 5, indicating that the deletion in their child was the result of adjacent segregation of a heterozygous parental translocation and in one case a paternal inversion.
Karyotypes of seven cases with unbalanced translocation
Karyotypes of three cases with mosaicism
The parental origin of the deleted chromosome 5 was defined in 61 families (all sporadic cases) and it was paternal in 55 patients (90.2%). The same percentage was found for patients with interstitial deletions.
GENOTYPE-PHENOTYPE CORRELATION
Genotype-phenotype correlation was done separately for 62 patients with terminal deletions including the critical region for dysmorphism and mental retardation in 5p15.2 between the loci D5S23 and D5S791 (CdCCR),2 for the seven patients with interstitial deletions and for the patient whose terminal deletion does not include the CdCCR. The seven patients with a translocation and three with mosaicism38 were not included in the correlation analysis because of the misleading effect of the partial trisomies in the former cases and of the different cell lines in the latter.
Sixty two patients with terminal 5p deletions
The typical CdCS phenotype including the cat-like cry was observed in all 62 patients. In order to analyse if the severity of the syndrome was related to the deletion size, the patients were subdivided into four groups: group A (seven patients, mean age 14.3 years, range 5.9-25.11) with deletion breakpoints proximal to the CdCCR2 down to p14 (from D5S755 to D5S796); group B (22 patients, mean age 11.1 years, range 2.0-26.11) with deletion breakpoints in distal p14 corresponding to the region which was reported to have no phenotypic effect19 (from D5S699 to D5S711); group C (20 patients, mean age 13.4 years, range 4.0-25.4) with deletion breakpoints in proximal p14 (from D5S769 to D5S776); group D (13 patients, mean age 15.5 years, range 3.11-34.11) with deletion breakpoints in p13 (from D5S692 to D5S734). Distribution of the dysmorphism showed an increasing frequency from group A to group D (A=35.7%, B=54%, C=66.4%, D=82%) which reaches statistical significance (χ2 test) between group A and group B (p<0.05), C (p<0.05), and D (p<0.05) (χ2 for trend p<0.05) (fig1).

Molecular analysis of 62 patients with 5p terminal deletions. Black bars represent the deleted chromosome 5 of each patient. On the left is a diagram of chromosome 5p and the matching phage probes. Patients were subdivided into four groups according to the deletion size (A, B, C, D). The percentages refer to the frequency of dysmorphism in the patients of different groups (χ2for trend p<0.05).
The frequency of microcephaly (cranial circumference <3rd centile) at birth (data available for 44 patients) compared to breakpoint distribution is shown in fig 2. No patient in group A had microcephaly and the frequency increased from group B to group D (χ2for trend p<0.05). Forty two out of 44 patients were microcephalic at the most recent check up (age 11 months-24 years). However, the most pronounced microcephaly was found in the patients of group D (>−4 SD) compared to those of group A (−2.9 SD).
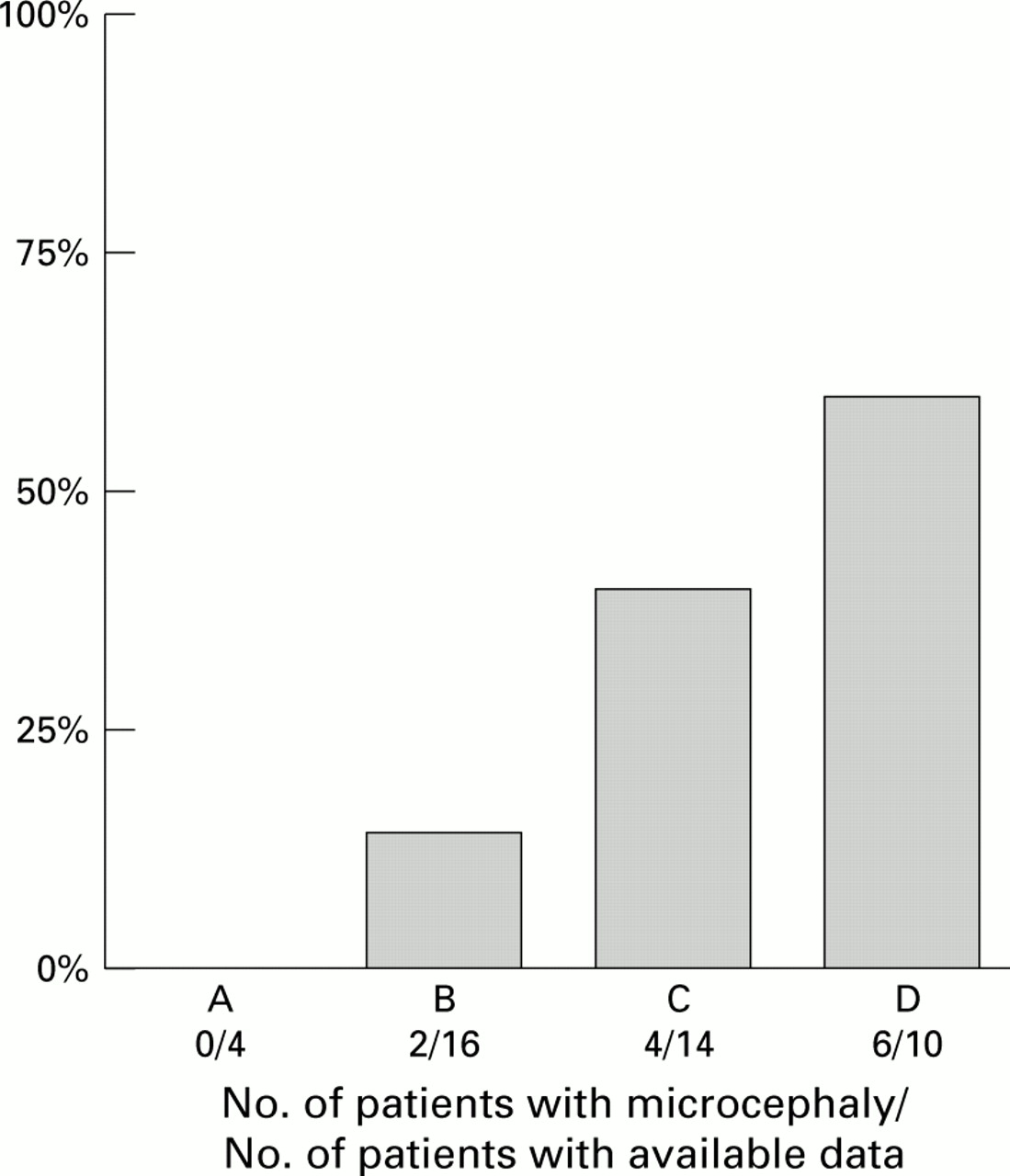
Distribution of microcephaly (head circumference <3rd centile) at birth in 44 patients with 5p terminal deletion (χ2 for trend p<0.05).
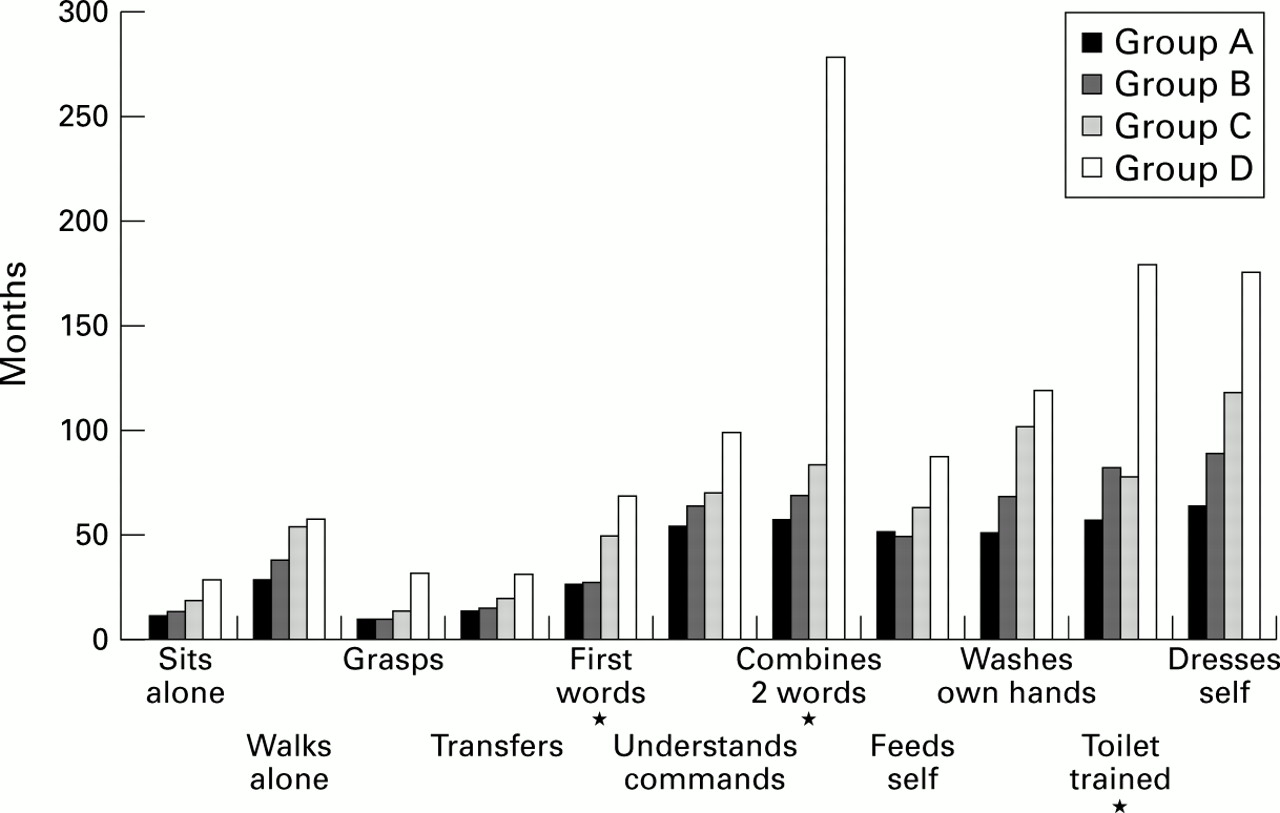
The evaluation of psychomotor development is shown in fig3.33 34 39 40 Acquisition of new skills occurred from the age of 2 months to 19 years without any loss of previously acquired skills. Patients who reached the milestones of psychomotor development below the 25th centile were more frequent in group A than in groups B, C, and D for nine of 11 milestones (fig 4). The difference reached statistical significance (χ2 test) between groups A and D for “walks alone” (p<0.05) and between groups A and C for “walks alone” (p<0.05) and “feeds self” (p<0.05). The statistical trend was observed for three skills: “first words”, “combines two words”, and “toilet trained” (χ2 for trend p<0.05) and was borderline for “walks alone”, “grasps”, and “feeds self”.

Developmental chart for 62 patients with CdCS comparing ages of achievement of milestones. The number of subjects screened for every skill is shown in brackets.

Psychomotor development in the CdCS patients who reached the milestones <25th centile. *A/D p<0.05, **A/C p<0.05, ***χ2 for trend p<0.05.
The mean age at which each group mastered different skills increases from group A to group D. Statistical significance (Student'st test) was reached for “first words” (p<0.05), “combines two different words” (p<0.05), and “toilet trained” (p<0.05) (fig 5).

Psychomotor development in 62 CdCS patients. Mean ages at which each group mastered different skills. *A/D p< 0.05.
No phenotypic differences owing to imprinting effects were noted in this group of patients for any of the parameters considered (dysmorphism, microcephaly, psychomotor developmental delay).
Seven patients with interstitial deletions and one patient with a terminal deletion not including the CdCCR
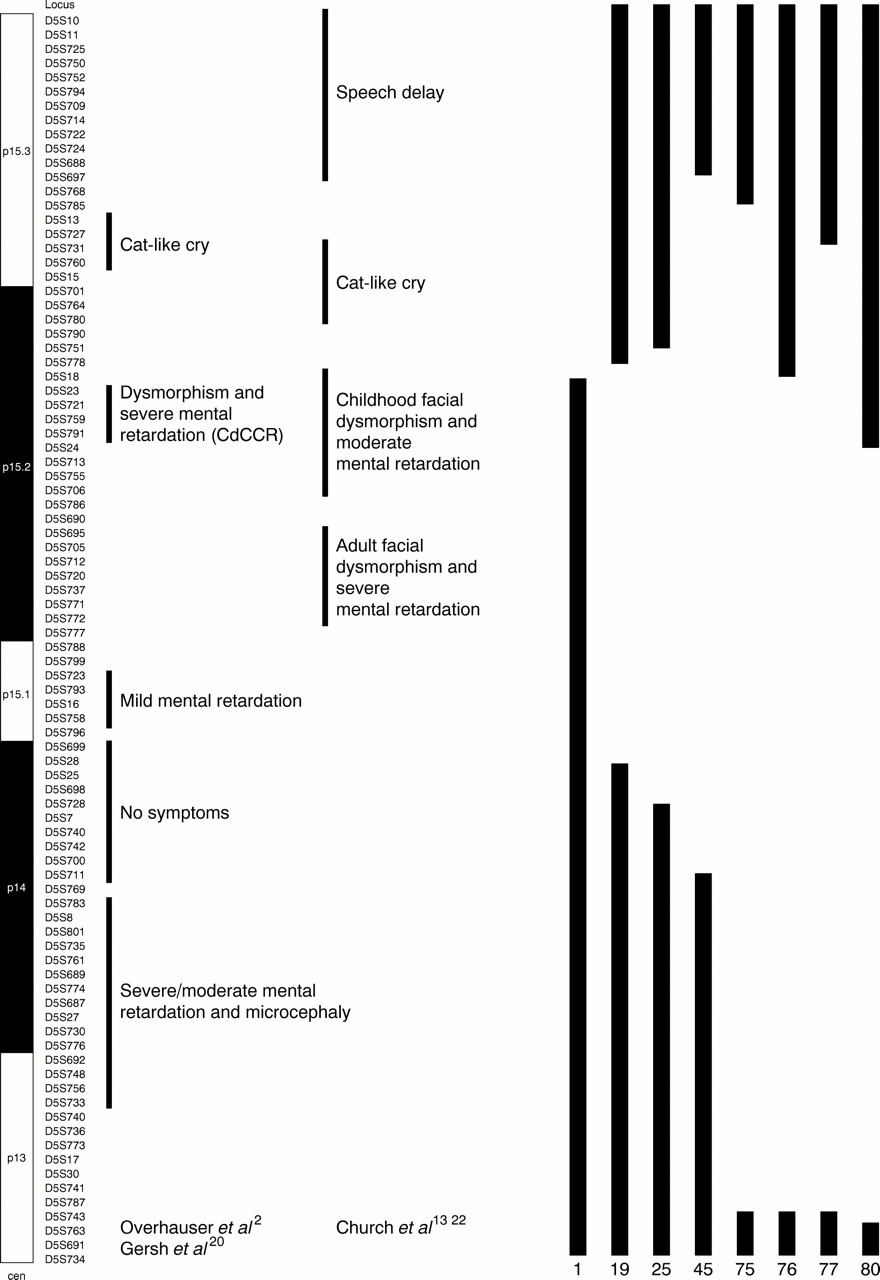
Six out of seven patients with interstitial deletions (19, 25, 45, 75, 76, 77) lacked both the CdCCR2 and the regions for dysmorphism and mental retardation defined by Churchet al 13 22 (fig 6) and showed the typical dysmorphism, microcephaly, and psychomotor retardation. Clinical data of all these patients are summarised in table3.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Seven patients with 5p interstitial deletions and one patient with a terminal deletion. On the left is a diagram of chromosome 5p and the matching phage probes.
Clinical data of the seven patients with interstitial deletions and the patient with the terminal deletion not including the CdCCR
Patient 80, with a deletion that did not encompass the CdCCR, but included the region for adult facial dysmorphism and severe mental retardation,13 22 (fig 6) did not show the classical CdCS phenotype nor the cat cry, but had microcephaly and severe speech and psychomotor developmental delay.
Patient No 1, with a terminal deletion (D5S23+/D5S18−), which does not include the CdCCR2 nor the regions for dysmorphism and mental retardation defined by Church et al 13 22 (fig 6), did not show the CdCS dysmorphism or microcephaly in infancy or adulthood, but he had a high pitched cry and slight psychomotor and language retardation in childhood.
Discussion
A cytogenetic, molecular, and phenotypic correlation study on a large number of patients with 5p deletions was carried out. Firstly, the importance of FISH analysis for a precise diagnosis of 5p deletions must be stressed.30 In the present study, seven patients (five with interstitial deletions, one with a small terminal deletion, and one with mosaicism) had not been correctly diagnosed by previous routine cytogenetics.
The range of breakpoints from 5p15.2 (locus D5S18) to 5p13 (locus D5S763) indicates that the short arm of chromosome 5 does not have a breakage hot spot. This phenomenon may be attributable to the unusual richness of low or medium copy number repetitive sequences, such as LINE-1 elements and chromosome 5 specific repeats causing unequal recombination.41-44
Few studies have attempted to correlate the deletion size of the short arm of chromosome 5 to the CdCS phenotype. Niebuhr,7 25in his study of 35 Danish patients, found that the anthropometric data and dermatoglyphic anomalies of the probands with the largest deletions showed a general tendency to deviate more from normal than those with smaller deletions. Cephalic breadth and bi-iliac width were significantly lower (0.05>p>0.025) in the group with the largest deletions. Kjaer and Niebuhr45 suggested that the CdCS manifestations varied in severity depending on the breakpoint. In 48 patients with terminal deletions, Wilkins et al 8 found a significant negative correlation (p=0.02) between IQ and the size of the deletion. Cornishet al,26-28 in their studies on 26 typical and four atypical CdCS patients, found that subjects with deletion breakpoints in 5p15.3 had a milder degree of cognitive impairment and fewer behavioural problems than those with deletion breakpoints in p15.2. Marinescu et al 29 did not find any relationship between the size of the deletion and the level of developmental delay in 50 subjects with CdCS.
The difficulties of genotype-phenotype correlation in the evaluation of complex (quantitative) traits (dysmorphism, psychomotor development) must be considered. In fact, the genetic background or environmental factors or both may affect the clinical phenotype.46Nonetheless, a study with such a large number of patients makes it possible to make some interesting observations.
The 62 patients with terminal deletions that include the CdCCR in 5p15.2 (D5S23+/D5S791+)2 presented the classical CdCS phenotype. However, the clinical evaluation showed a difference of severity for microcephaly, dysmorphism, and psychomotor retardation between the patients with small deletions in 5p15.1 and those with larger ones. The statistical analysis of the 62 patients subdivided into four groups (A, B, C, D) according to the deletion size confirmed a significant trend from group A to group D for the expression of the most important dysmorphism and for the delay in acquisition of some important milestones of psychomotor development. Microcephaly at birth proved to be closely correlated to deletion size. Almost all patients became microcephalic as they grew, but the most severe cases were those with larger deletions.
The lack of the distal region of 5p14, which had no phenotypic effect in the family described by Overhauser et al,19 seems to have influence on dysmorphism and psychomotor retardation in group B, on the basis of both clinical and statistical evaluations. Recently, Johnson et al 47 reported a healthy father and an affected son (microcephaly, seizures, and global developmental delay) with an identical interstitial deletion of 5p14. Hand et al 48 also reported a family in which a patient with a peroxisomal disorder and his normal mother had the same interstitial deletion in 5p14. It is possible that the phenotypic effect of the haploinsufficiency may depend on the presence of recessive alleles on the homologous chromosome or genetic background or both. The fact that Marinescu et al 29 did not find any relationship between deletion size and developmental delay may be because of different characteristics of the two populations (age and deletion size).
The analysis of the parental origin of the deleted chromosome showed a prevalence of paternal origin. No phenotypic differences resulting from imprinting effects as suggested by Bengtsson et al 18 were observed in this group of patients.
The study of key patients with interstitial and small terminal deletions was particularly interesting for the study of the critical regions. The analysis of patients 1 and 80, who showed deletions that did not include CdCCR and did not show CdCS dysmorphism, and patient 76, who had the typical clinical features of CdCS, would suggest a correlation between dysmorphism and CdCCR2 rather than the region proposed by Church et al.13 22
The existence of a region for severe mental retardation in proximal 5p15.224 might be confirmed by the data provided by patients 2 and 80. In fact, the former maintained it and showed mild mental retardation, while the latter lacked it and was severely mentally retarded. However, the definition of specific regions associated with mental retardation appears particularly complex because of the presence of different genes (SEMAF,CTNND2) involved in brain development and function along the 5p arm.
The existence of a separate region for the cat cry in p15.32 20 was confirmed by patients 19, 25, 76, and 80, who maintained it and did not have the cry, and by patient 75, who lacked it and had the typical cry. Patient 77 lacks only a part of the suggested cat cry region,2 yet he had the typical cry at birth and even now, at the age of 25, has a typical plaintive voice. This fact distally narrows the critical region for the cat cry from D5S13 to D5S731 and may move it down as suggested by Churchet al 13 (fig 6).
The hypothesis of a separate region for speech delay in distal p15.3 was suggested by Church et al 13and is confirmed in the data provided by Baccichettiet al 17 and Cornishet al.28 In the present study, the existence of this region would be supported by patient 19, who had moderate psychomotor retardation, but developed unusually good speaking skills. This skill was not present in the other six patients probably because of a more serious delay in psychomotor development owing to a larger deletion. In fact, the severity of the impairment in language skills could be a consequence of significant intellectual delay which may have had a profound influence on early vocal development.28
This study has enabled the following: (1) to define the karyotypes responsible for CdCS phenotype and their frequencies better; (2) to stress the difficulty of defining specific critical regions for mental retardation; (3) to highlight a progressive severity of clinical manifestations and psychomotor retardation related to the increasing size of deletion; (4) to confirm the presence of two distinct regions, for dysmorphism and mental retardation in p15.2 and the other for the cat cry in p15.3; (5) to narrow the cat cry region distally from D5S13 to D5S731; (6) to support the hypothesis of a separate region for the speech delay in p15.3; and (7) to confirm that not all 5p deletions result in the CdCS phenotype.
The identification of phenotypic subsets associated with specific deletions may be of great diagnostic and prognostic relevance. Furthermore, clinical examination combined with the molecular analysis of the deletion results in a more personalised evaluation of the patient.
Acknowledgments
The first two authors contributed equally to this work. This study was supported by Telethon Italian (grant E.511) and the Italian Cri du Chat Children Association. The cell lines of the CdCS patients are stored at the Galliera Genetic Bank supported by Telethon Italian (grant C.23). The following colleagues collaborated in providing patients, material, and clinical information: G Andria (Napoli), A Baraldi (Brescia), L Boggi (Massa Carrara), C Borrone (Genova), M Cammarata (Palermo), D Caufin (Pordenone), M L Cavaliere (Napoli), L Chessa (Roma), A Di Comite (Taranto), B Dallapiccola (Roma), M Farina (Lamezia Terme), P Franceschini (Torino), A Garau (Cagliari), L Garavelli (Reggio Emilia), G Gemme (Genova), A Giannotti (Roma), M L Giovannucci (Firenze), L Giuffrè (Palermo), R Lingeri (Como), A Lomangino (Bari), A Lumini (Pistoia), R Magistrelli (Ancona), M Martinazzi (Gallarate), T Mattina (Catania), F Mollica (Catania), G Pagano (Como), M Pagano (Roma), G Palka (Chieti), M Pergola (Roma), M G Pirastu (Sassari), G Presta (Brindisi), M M Rinaldi (Napoli), G Rovetta (Manerbio), B Sacher (S Daniele del Friuli), M Stabile (Napoli), A Selicorni (Milano), L Tarani (Roma), R Tenconi (Padova), E Valletta (Verona), V Ventruto (Napoli), M G Vianello (Genova), P Vignetti (Roma), N Weber (Trieste), L Zelante (S Giovanni Rotondo).