


Abstract
The bicyclam AMD3100 is known as a small synthetic inhibitor of the CXCL12-binding chemokine receptor CXCR4. Here, we show that AMD3100 also binds to the alternative CXCL12 receptor CXCR7. CXCL12 or AMD3100 alone activate β-arrestin recruitment to CXCR7, which we identify as a previously unreported signaling pathway of CXCR7. In addition, AMD3100 increases CXCL12 binding to CXCR7 and CXCL12-induced conformational rearrangements in the receptor dimer as measured by bioluminescence resonance energy transfer. Moreover, small but reproducible increases in the potency of CXCL12-induced arrestin recruitment to CXCR7 by AMD3100 are observed. Taken together, our data suggest that AMD3100 is an allosteric agonist of CXCR7. The finding that AMD3100 not only binds CXCR4, but also to CXCR7, with opposite effects on the two receptors, calls for caution in the use of the compound as a tool to dissect CXCL12 effects on the respective receptors in vitro and in vivo.
Chemokine receptors belong to the G-protein coupled receptor (GPCR) family. GPCR binding by ligands is believed to alter receptor conformation in a way that is transmitted to the cytoplasmic face of the receptor and triggers the activation of the heterotrimeric G-proteins as well as that of other G-protein-independent effectors. Although ligand-induced conformational change of the receptor has for long been deduced from functional data, the advent of new biophysical methods has eventually permitted direct measurement of such changes in native receptors present in the plasma membrane of live cells. bioluminescence resonance energy transfer (BRET) is one of the resonance energy transfer techniques used to show constitutive dimerization of chemokine receptors and other GPCRs (Terrillon and Bouvier, 2004). Constitutively dimeric GPCR BRET couples can also be used to probe receptor conformation, because BRET depends on the distance between the luminescence donor [Renilla reniformis luciferase (RLuc)] and the acceptor [the yellow fluorescent protein (YFP)]. For instance, the use of CXCR4-RLuc/CXCR4-YFP dimers as sensors permitted the detection of different conformations in a panel of CXCR4 mutants (Berchiche et al., 2007). Moreover, dimeric BRET sensors permit the measurement of ligand-induced conformational changes in the receptor dimer (Ayoub et al., 2004; Percherancier et al., 2005). These effects are not only observed for cognate agonists but also for small synthetic ligands that may be orthosteric or allosteric modulators.
Allosteric modulation of receptor-ligand interactions results from binding of a second (allosteric) ligand to a distinct site on the receptor, in a way that does not directly compete with binding of the cognate (orthosteric) ligand. Binding of the allosteric ligand may decrease the affinity of the cognate ligand, resulting in negative allosteric modulation. Conversely, the presence of the allosteric modulator may increase binding of the cognate ligand, called positive allosteric modulation (May et al., 2007). Allosteric ligands may also have intrinsic activity on one or several signaling pathways. These are referred to as allosteric agonists (Schwartz and Holst, 2006).
Chemokines are small secreted proteins. The chemokine receptor CXCR4 was first identified as one of the coreceptors that permit entry of the HIV-1 into target cells (Berger et al., 1999). Small antiviral molecules targeting CXCR4 have shown side effects because they inhibit other essential functions of the receptor (Hendrix et al., 2004). Indeed, CXCR4 has turned out to be involved in physiological and pathological processes, such as stem cell homing and cancer metastasis dissemination (Zlotnik, 2004; Ratajczak et al., 2006). In contrast to other chemokines, which often bind to more than one receptor, it was initially believed that the chemokine CXCL12 (also called SDF-1, for stromal cell-derived factor) binds only CXCR4. However, two recent reports showed that CXCL12 also binds to CXCR7 (Balabanian et al., 2005; Burns et al., 2006). To date, the CXCL12-CXCR7 interaction has not been shown to activate G-protein-mediated signal transduction, and it has been speculated that the receptor may clear secreted CXCL12 from the extracellular environment and thereby help maintain defined CXCL12 gradients (Thelen and Thelen, 2008) and references herein). The expression of CXCR7 on cancer cells suggests that CXCR7 might be responsible for some of the effects of CXCL12 on these cells, an issue that warrants re-examination.
AMD3100 is a small molecule of the bicyclam family, first identified by virtue of its antiretroviral effects and subsequently shown to bind CXCR4 (De Clercq, 2005). Because of its established selectivity as an antagonist at the CXCR4 receptor AMD3100 is widely used as a pharmacologic tool to demonstrate the involvement of CXCR4 in the action of CXCL12 in various settings. In line with the role of CXCR4 in hematopoietic stem cell homing, single administration of high doses of AMD3100 leads to massive release of such cells into the peripheral blood, and the compound now undergoes testing as a drug facilitating access to the hematopoietic stem cell compartment. Derivatives of AMD3100 are also under investigation for their effect on cancer cells (De Clercq, 2005).
This study set out to analyze the effects of CXCL12 and AMD3100 on CXCR7 using a homodimeric receptor BRET sensor. We find that AMD3100 is a ligand not only for CXCR4, but also for CXCR7, at concentrations that are used by investigators in in vitro studies. In contrast to its negative effect on the CXCR4/CXCL12 interaction, AMD3100 positively modulates CXCL12 effects and binding on CXCR7. In addition, AMD3100 alone recruits β-arrestin to CXCR7 but blocks β-arrestin recruitment to CXCR4. AMD3100 therefore qualifies as an allosteric agonist of CXCR7.
Materials and Methods
Materials.125I-CXCL12 (SDF-1α) tracer was from GE Healthcare (Chalfont St. Giles, Buckinghamshire, UK). AMD3100 was synthesized in-house or from Sigma Corp. (St. Louis. MO), and CXCL12 (SDF-1β) was acquired from R&D Systems (Minneapolis, MN).
BRET Measurements. Each receptor was cloned into pCDNA3.1/Zeo+ (Invitrogen, Carlsbad, CA), modified with an N-terminal HA-Flag sequence. Tagged receptor sequences were subcloned to yield-YFP and -Rluc fusion proteins as described previously (Berchiche et al., 2007). HEK293T cells (American Type Culture Collection, Manassass, VA) were transiently transfected in six-well plates with HA-Flag-CXCR4-YFP and HA-Flag-CXCR4-RLuc or HA-Flag-CXCR7 cDNA using the polyethylenimine method; the total amount of transfected DNA was kept constant for all transfections by adding empty vector. Transfected cells were seeded in 96-well white clear-bottomed plates treated with poly-d-lysine and left in culture for 24 h. At 48 h after transfection, the culture medium was replaced by PBS supplemented with 0.1% bovine serum albumin (Sigma) at room temperature. Coelenterazine H (Nanolight technology) was added at a final concentration of 5 μM, and fluorescence and luminescence readings were collected, with the use of a Mithras LB940 plate reader (Berthold Technologies, Bad Wildbad, Germany), as described previously (Percherancier et al., 2005; Berchiche et al., 2007). All BRET experiments were performed while the cells remained attached to the plastic surface.
For BRET titration experiments, HEK293T cells were cotransfected with a constant amount of receptor-Rluc and increasing concentration of plasmids encoding eYFP-tagged receptors. The BRET signal is determined by calculating the ratio of the receptor-eYFP over the receptor-RLuc emission. The values were corrected to net BRET by subtracting the background BRET signal detected when the receptor-RLuc construct was expressed alone. Saturation curves were obtained by plotting net BRET values as a function of the [acceptor]/[donor] ratio. Total fluorescence and luminescence were used as a relative measure of total expression of the acceptor and donor proteins, respectively. Experiments were carried out in triplicate.
To assess ligand-induced changes of the BRET signal, HEK293T cells expressing-eYFP and -Rluc fusion proteins at a ratio at BRETmax were stimulated with the indicated ligands for 5 min at 37°C before the addition of the substrate. In dose-response experiments, BRET ratio was measured in cells after stimulation with increasing concentrations of AMD3100 or CXCL12 in the presence or absence of 10 μM AMD3100.
Competition Binding Studies. HEK293T cells (American Type Culture Collection) were transiently transfected with HA-Flag-hCXCR4 or HA-Flag-CXCR7. In brief, cells were transfected with Lipofectamine-2000 (Invitrogen) using 5 μg of HA-Flag-hCXCR4 or 5 μg of HA-Flag-CXCR7 cDNA per 100-mm plate, allowed to transfect for 5 h, then overlaid with 10 ml of culture media (Dulbecco's modified Eagle's medium, 10% fetal bovine serum, 1% antimycotics, and 1% nonessential Amino Acids; Mediatech, Inc., Herndon, VA) and cultured overnight. The cells were then plated at 100,000 cells per well for CXCR4 transfectants and 50,000 cells per well for CXCR7 transfectants into 48-well plates coated with poly-d-lysine (BD Biosciences, Franklin Lakes, NJ) and cultured overnight for binding studies. Cells were plated 2 × 106 cells/100-mm plate for parallel analysis by flow cytometry. Eighteen hours after plating, competition binding studies were performed using 45 to 160 pM 125I-CXCL12 (SDF-1α; GE Healthcare) and increasing doses of competitors. Based on the KD from preliminary saturation analyses (data not shown), these tracer concentrations would be expected to occupy ∼1.4 to 4.9% of the receptor population. In brief, cells were washed twice with 500 μl of ice-cold binding buffer (0.2% de-lipidized bovine serum albumin; BD Biosciences), 50 mM HEPES, 1 mM CaCl2, and 5 mM MgCl2) and then allowed to incubate for 1 h at 4°C. Buffer was replaced with 200 μl of binding buffer containing protease inhibitor cocktail (10 mM phenylmethylsulfonyl fluoride, 5 μg/ml aprotinin, 4 μg/ml leupeptin, and 10 nM phosphoramidon), 125I-CXCL12, and increasing doses of AMD3100 or CXCL12 (SDF-1β). Binding occurred at 4°C, for 4 h with gentle shaking. Tracer was removed with three washes of ice-cold wash buffer (0.5M NaCl, 50 mM HEPES, 1 mM CaCl2, and 5 mM MgCl2). To adapt the conditions of binding assays to those used for BRET, additional radioligand binding experiments were conducted as above, but at 37°C, and in BRET buffer (PBS with 0.1% bovine serum albumin), after 5-min preincubation with 10 μM phenylarsine oxide (Sigma) to prevent endocytosis, followed by washing. Cells were lysed with 150 μl of 2% nonidet P-40, collected, and binding was measured using a Wallace Wizard 1470 gamma-counter (PerkinElmer Life and Analytical Sciences, Waltham, MA).
Arrestin Recruitment. β-Arrestin recruitment was measured by BRET essentially as described previously (Hamdan et al., 2005). HEK293T cells were cotransfected in six-well plates with 1 μg of receptor-eYFP construct with 0.05 μg of arrestin2-Rluc (a generous gift of Michel Bouvier) completed to 2 μg with empty vector per well. After culture and transfer into 96-well plates as above, the cells were stimulated with indicated ligands. β-Arrestin recruitment to CXCR7 was rapid and was measured after 5 min, whereas β-arrestin recruitment to CXCR4 followed a slower time course and was therefore measured after 30 min. BRET measurements were performed as described above.
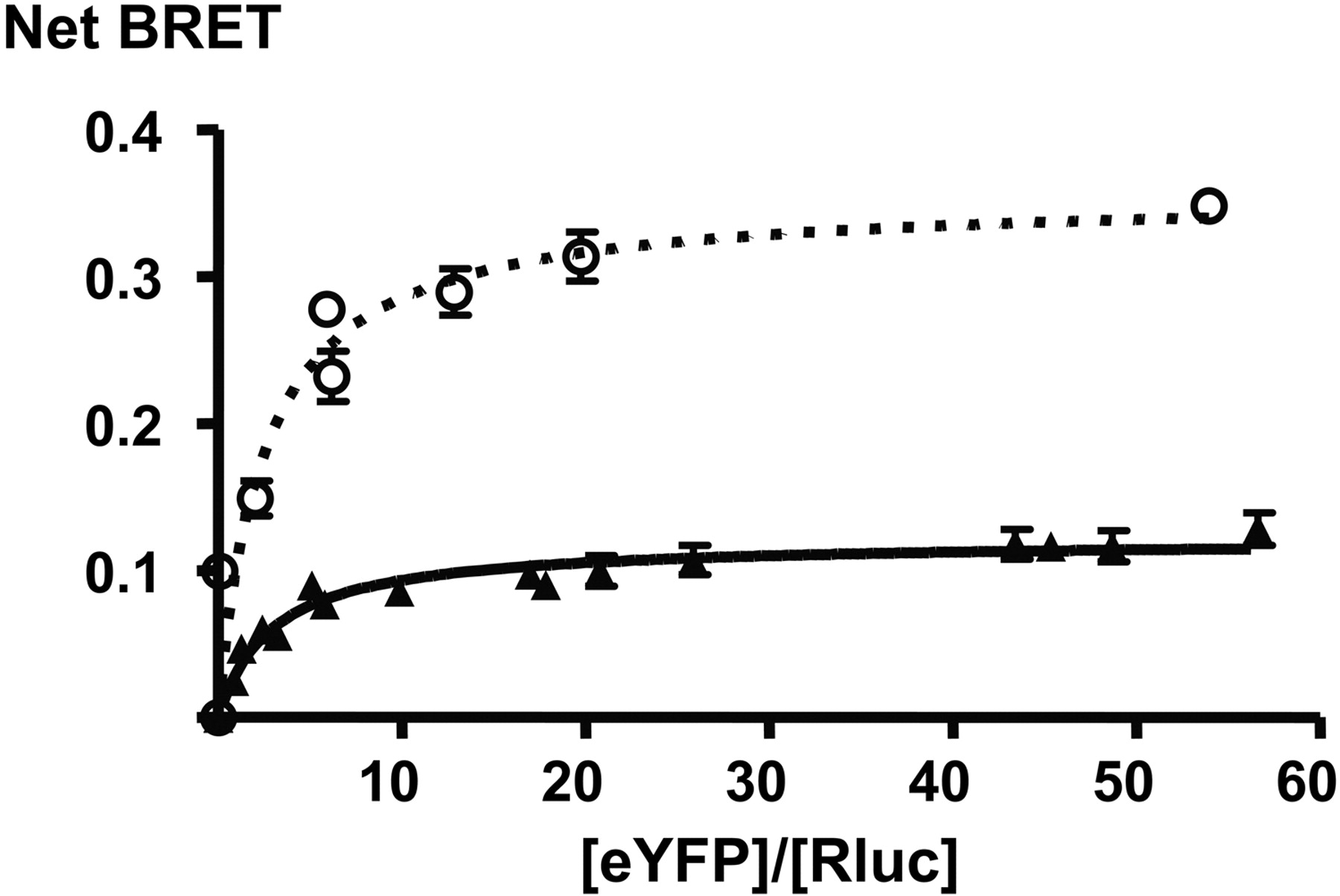
[acceptor]/[donor] titrations of CXCR4 (○) and CXCR7 homodimers (▴), demonstrating constitutive homodimerization of both receptors. Figure 1 shows representative curves for each receptor (The fitted curve parameters for each curve of this representative experiment are BRETmax 0.36 ± 0.02 and 0.12 ± 0.004 for CXCR4 and CXCR7, respectively, and BRET50 2.47 ± 0.64 and 2.95 ± 0.41 for CXCR4 and CXCR7, respectively). The errors are S.E.M. of the parameter estimate. The mean values obtained in repeated experiments are shown in Table 1.
Flow Cytometric Analysis. Receptor cell surface expression for both receptors was confirmed by flow cytometry. The following antibodies were used for staining: primary antibody, anti-FLAG M2 MAb (Sigma Corp., St. Louis. MO), followed by a secondary donkey antimouse (Fab′)2 (R)-phycoerythrin (Jackson ImmunoResearch Laboratories Inc., West Grove, PA). Cells were washed three times in ice-cold PBS resuspended and incubated on ice with goat serum for 1 min, after which they were stained with primary and secondary antibody for 30 min at 4°C. After a final wash, the cells were resuspended in 0.5% paraformaldehyde, incubated at 4°C for 10 min, followed by analysis by EPICS ALTRA Flow Cytometer (Beckman Coulter, Fullerton, CA).
Data Analysis. Experimental data were treated using Prism software v 4 (GraphPad Software, San Diego, CA). For BRET [acceptor]/[donor] titrations, the data were fitted using the one-site binding equation for the determination of BRET50 and BRETmax. For BRET CXCL12 dose-response experiments, all data sets were analyzed separately. Normalization was performed using the formula (net BRET - Bottom CXCL12 only curve)/(Top CXCL12 only curve - Bottom CXCL12 only curve) × 100; the top and bottom of the CXCL12-only net BRET curve was derived from an initial curve fit of the data obtained in the absence of AMD3100. The normalized CXCL12 only curve was constrained between 0 and 100, whereas the curve in the presence of AMD3100 was not constrained. Statistical significance was assessed using paired t test and data set comparison performed by assessing curve top, bottom, and logEC50 in simultaneous curve fittings. Binding data were analyzed by simultaneous comparison of one-site versus two-site competition binding models, with statistical significance for the optimal model determined by the extra sum-of-squares F test. Because our previous saturation isotherms for 125I-CXCL12 demonstrated that the radioligand bound to a single binding site in the heterologous system (data not shown), the binding model used was one in which the radioligand did bind to one site, but the unlabeled ligand was tested for discrimination of affinity between two sites. β-Arrestin recruitment data were analyzed using repeated-measures analysis of variance and Dunnett post test.
Results
CXCR7 Forms Constitutive Homodimers. To test whether CXCR7 forms constitutive homodimers that may serve as BRET conformational sensors, we constructed the CXCR7-RLuc and CXCR7-YFP fusion proteins. Coexpression of BRET partners at different [acceptor]/[donor] ratios yields BRET curves that, if specific, reach saturation at maximal BRET (BRETmax) (Mercier et al., 2002; Bouvier et al., 2007). As shown in Fig. 1, [acceptor]/[donor] titration of the CXCR7 BRET constructs results in saturation of the BRET signal, as does the previously reported constitutive CXCR4 homodimer, whereas titration of CXCR7-RLuc with V1Ra-YFP (vasopressin receptor V1Ra) as a control did not (data not shown) (Babcock et al., 2003; Percherancier et al., 2005). The CXCR7 BRET titration curve has a BRET50 that is in the same range as that of CXCR4, albeit slightly higher (Table 1). BRET50 has been described to characterize the propensity of receptors to form dimers (Mercier et al., 2002). The data shown in Fig. 1 therefore suggest a similar predisposition of the two receptors to form constitutive homodimers. The BRETmax values are different for the two receptors. Different maximal BRET of constitutive dimers does not represent their number or affinity but rather reflect differences of distance and/or orientation between the C-terminal fluorophore fusions in the CXCR4 and CXCR7 homodimers (Bouvier et al., 2007).
Fitted curve parameters of homodimer [acceptor]/[donor] titration
Summary of the curve parameters of homodimer [acceptor]/[donor] titrations, of three to six repeated experiments as shown in Fig. 1. The BRET50 values indicate the propensity of acceptor and donor to interact; they are in the same range for CXCR4 and CXCR7, suggesting that they form constitutive homodimers with similar propensity. BRETmax is a reproducible value intrinsic to each different receptor homodimer; the difference of BRETmax in CXCR4 and CXCR7 reflects solely the different distances/orientations of acceptor and donor in the CXCR4 and CXCR7 homodimers.
The CXCR7 Homodimer BRET Is Modulated by CXCL12 and AMD3100. BRET within a constitutive dimer depends on the distance between the fluorophores, which is a function of receptor dimer conformation. Ligand-induced changes of receptor conformation can therefore be picked up as changes in BRET. Ligand-induced BRET changes are also additional evidence that the BRET signal results from a specific dimer and is not due to random collision of the BRET partners, which would be expected to be unaffected by ligand binding. Cells expressing the respective BRET partners at BRETmax [acceptor]/[donor] ratios were incubated with single doses of different CXCR4 and CXCR7 ligands (100 nM for CXCL12 and 10 μM for AMD3100) (Fig. 2, A and B). CXCL12, the cognate ligand of both CXCR4 and CXCR7, increases BRET of both receptor dimers, as previously reported for CXCR4 (Percherancier et al., 2005). AMD3100 effects on the CXCR4 dimer are not well detected by BRET, as reported previously (Percherancier et al., 2005). It is noteworthy that 10 μM AMD3100 resulted in small but significant changes in BRET of the CXCR7 dimer, but not on nonspecific bystander BRET between CXCR7-RLuc and V1Ra-YFP (not shown), suggesting that AMD3100 interacts with CXCR7 and promotes a conformational change within the constitutive homodimer. Dose-response experiments of AMD3100 on the CXCR7 homodimer yielded sigmoid curves with a low maximum of 13 ± 3.7% BRET increase compared with the CXCR7 dimer without ligand, and an apparent EC50 of 46 μM (logEC50, -4.36 ± 0.08; n = 4), although this was difficult to assess as a result of the low amplitude of the signal (Fig. 2C).
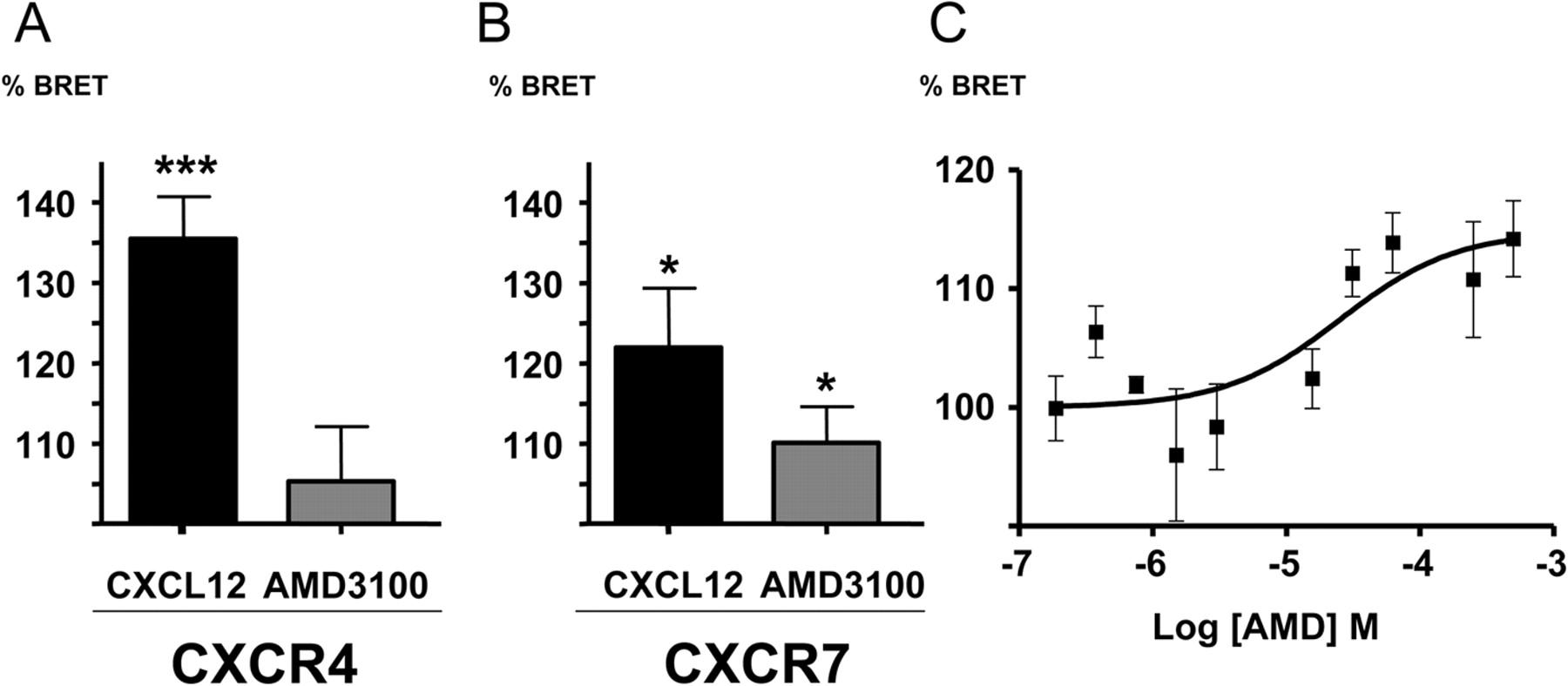
Changes in the BRET signal of CXCR4 (A) or CXCR7 (B) homodimers induced by AMD3100 and CXCL12. The changes are expressed in percentage of the basal BRET signal of the respective homodimer (in the absence of ligands). [acceptor]/[donor] ratios representing BRETmax were employed. Bars represent mean and S.E., the data are derived from n = 11 experiments (except AMD3100 on CXCR4 n = 5); *, p < 0.05; ***, p < 0.001 versus basal BRET. Dose-response curves with AMD3100 on the CXCR7 dimer show a low maximal increase of the BRET signal of 13 ± 3.7% compared with the CXCR7 dimer without ligand, and an apparent EC50 of 46 μM (n = 4); a representative example is shown in C.

Dose-response curves of BRET changes induced in the CXCR4 and CXCR7 homodimers, expressed as a percentage of the maximal change obtained at saturating CXCL12 concentration in the absence of AMD3100. Data obtained in the absence (▪) or presence (○) of 10 μM AMD3100 from one representative experiment are shown (CXCR4 without AMD3100: EC50 = 1.01 × 10-7M, with AMD3100 EC50 = 4.97 × 10-7M, and CXCR7 without AMD3100: EC50 = 1.93 × 10-8M, with AMD3100 EC50 = 0.59 × 10-8 M. On the CXCR7 dimer, the maximal BRET change of the curve with AMD3100 is 124% of that with CXCL12 alone).
BRET Changes Induced by CXCL12 Are Modulated by AMD3100. We then performed dose-response experiments with CXCL12 on the respective BRET partners. Representative results of these experiments are shown in Fig. 3. Table 2 shows that the average EC50 of CXCL12-induced BRET changes in the CXCR4 dimer is 5.34 × 10-8 M. The average EC50 of CXCL12 on the CXCR7 dimer is similar, at 1.60 × 10-8 M. Using this system, we assessed a possible modulation of the effect of CXCL12 on the different receptors by a given dose of AMD3100. We chose to add 10 μM AMD3100 to the CXCL12 dose-response experiment, because this concentration has been used in several in vitro studies investigating the role of CXCR4 in cancer (Cabioglu et al., 2005; Pattarozzi et al., 2008). The dose-response curve of CXCL12-induced BRET changes on CXCR4 was shifted by almost 1 log to the right by the presence of AMD3100, resulting in an increased EC50 of 5.10 × 10-7 M. However, the effect of 10 μM AMD3100 on the CXCL12-induced BRET changes of the CXCR7 dimer was the inverse: the dose-response curve was left-shifted, and the EC50 of CXCL12 was lowered 2- to 3-fold to 5.67 × 10-9 M. This finding is best explained by an increase of the effect of CXCL12 on CXCR7 by AMD3100 by a positive allosteric mechanism. In addition, the maximal BRET change observed in the CXCR7 dimer in the presence of both CXCL12 and AMD3100 was significantly higher than that in the presence of CXCL12 alone (paired t test, p = 0.0097, Table 2). We interpret this difference as additional evidence for an allosteric mode of action of AMD3100 on CXCR7, the different maximal BRET changes reflecting distinct receptor conformations in the presence of both ligands versus CXCL12 alone.
Fitted curve parameters of BRET changes
Table 2 summarizes the analyses of CXCL12-induced BRET changes of CXCR4 (n = 2) and CXCR7 (n = 6). The difference of logEC50 in the presence and absence of AMD3100 of the CXCR7 curves is statistically significant. The maximal BRET changes induced by CXCL12, in the presence or absence of 10 μM AMD3100, are also given, referring to the maximal change induced by CXCL12 alone as 100%. The difference is significant for CXCR7. In addition, simultaneous curve fitting for each set of curves (±AMD3100) confirmed the statistical significance for the difference between the CXCR4 curves (P < 0.001) and for five of six CXCR7 curves (P < 0.05) with AMD3100 treatment. Values are presented as mean ± S.E.M.
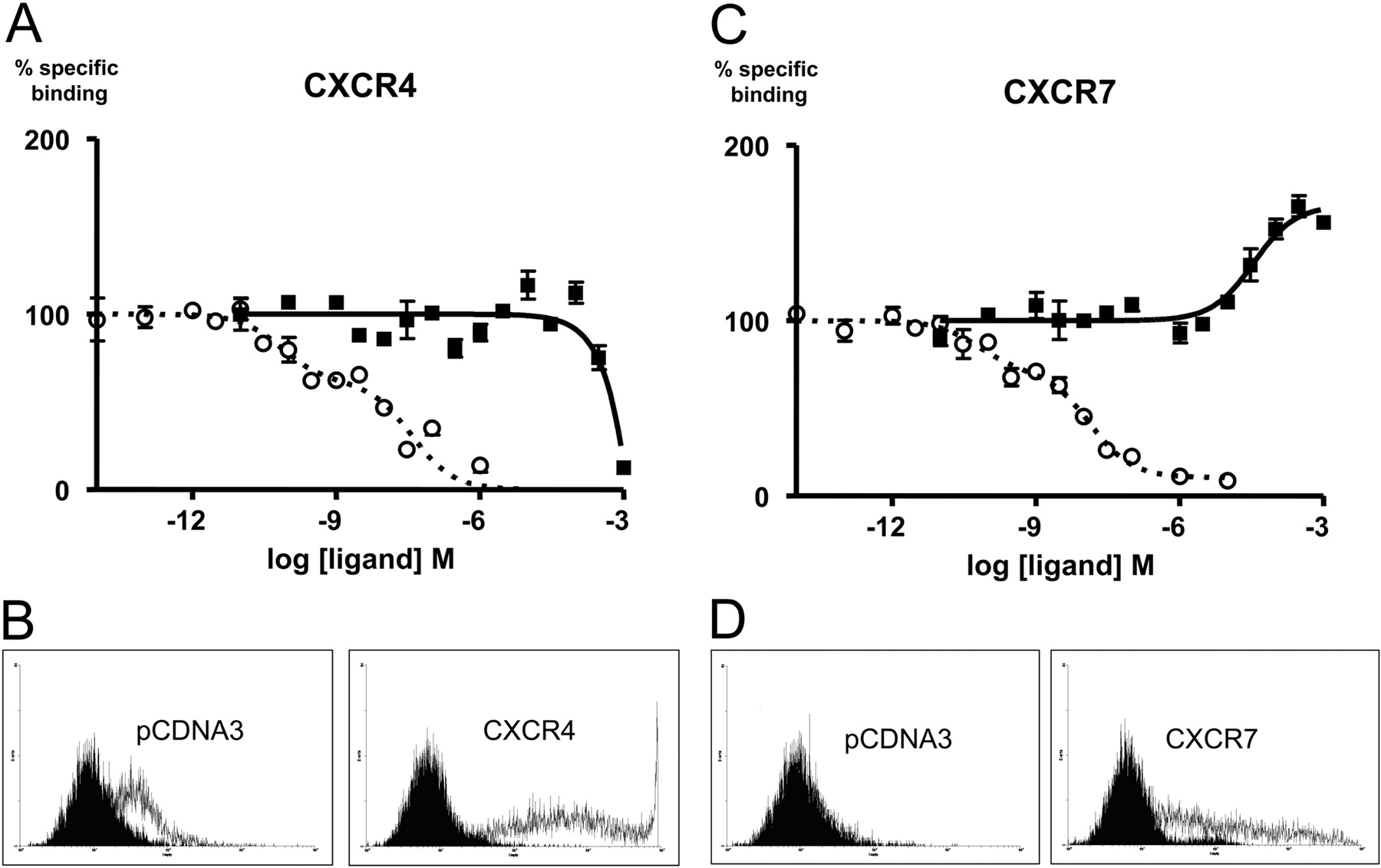
AMD3100 Increases 125I-CXCL12 Binding to CXCR7-Expressing Cells. To substantiate our observations, we performed radioligand binding assays using radiolabeled CXCL12 tracer on HEK293T cells transiently expressing either CXCR4 or CXCR7. Figure 4A indicates that AMD3100 does not compete for CXCL12 binding to CXCR4 until very high concentrations of AMD3100 (average Ki = 6.61 × 10-4 M, n = 6; see Table 3), suggesting that either AMD3100 is a low-affinity ligand for CXCR4 or the inability to efficiently compete in the radioligand binding assay with this radioligand is due to binding at a site that is somewhat distinct from that of [125I]CXCL12. It is unlikely, however, that the binding data in Fig. 4A represent low-affinity binding of AMD3100 to CXCR4, given the high potency with which AMD3100 antagonizes functional CXCR4 responses (see Discussion). In contrast, on CXCR7-expressing cells, increasing concentrations of AMD3100 lead to an increased binding of the radiolabeled chemokine to the cells, with an apparent average Ki of (3.15 × 10-5 M, n = 4) (Fig. 4B, Table 3). As a control, when binding assays were conducted under conditions similar to BRET assay conditions, similar results (Ki, 2.6 × 10-6 M, and maximal binding of 132 ± 0,1%, n = 2; data not shown) were obtained, ruling out that the observed increase in CXCL12 binding to CXCR7 was due solely to differences in temperature or salt content. The decreased EC50 of the effect at 37°C may suggest a temperature-dependent component in binding of AMD3100 to CXCR7. In any case, our observation is consistent with the conclusion that AMD3100 is a CXCR7 ligand that potentiates CXCL12 binding to CXCR7, thereby qualifying as a positive allosteric modulator of CXCL12 binding to the receptor. Taken together, the positive effect of AMD3100 on the AMD3100-CXCR7 interaction is in keeping between binding and homodimer BRET assays.
Summary of receptor binding parameters at CXCR4 and CXCR7
Binding was determined after over-expression of the individual receptors in HEK293T cells as described under Materials and Methods. The binding parameters were determined by fitting the data to a one- or two-site binding model as described under Materials and Methods, and the fitted curve parameters from the optimal fits of the data are summarized. Percentage inhibition in binding is defined relative to the magnitude of the specific 125I-CXCL12 signal in the absence of AMD3100 (100% indicating complete inhibition). The negative value for CXCR7 indicates an increase in CXCL12 binding relative to that observed in the absence of AMD3100 (P < 0.0001). Values are presented as mean ± S.E.M.
AMD3100 Induces β-Arrestin Recruitment to CXCR7 and Positively Modulates the Effect of CXCL12. We then investigated whether AMD3100 binding to CXCR7 was accompanied by functional consequences. Given that no G-protein signaling has been reported for this receptor, we studied recruitment of β-arrestin as a signaling readout, which can be G-protein independent (Wei et al., 2003). β-Arrestin is a scaffold protein that can be recruited to ligand-bound GPCRs independently of G-protein signaling and that is linked to downstream enzyme effectors. To quantitatively measure arrestin recruitment, we used a BRET-based system described previously (Hamdan et al., 2005). As shown in Figs. 5, A and C, we found that CXCL12 induces arrestin recruitment to CXCR7, thereby identifying arrestin recruitment as a previously unreported signaling pathway activated by CXCR7. We were surprised to find that AMD3100 alone also induced arrestin recruitment to CXCR7, showing a substantial effect at 10 μM, thereby qualifying as an agonist of this pathway. As a control, CXCL12 induced β-arrestin recruitment also to CXCR4, but this effect was blocked in the presence of 10 μM AMD3100 (Fig. 5B), suggesting that AMD3100 behaves as an antagonist of this pathway at CXCR4. To evaluate whether a positive allosteric effect of AMD3100 on CXCR7 would relate to this signaling readout, we performed a dose-response experiment with CXCL12 in the presence and absence of 10 μM AMD3100 (Fig. 5C). The resulting EC50 values indicate that AMD3100 approximately doubles the potency with which CXCL12 induces arrestin recruitment. This small, but reproducible effect is strongest at CXCL12 concentrations that are biologically relevant (1-10 nM).

Competition binding using 53 pM 125I-CXCL12 and increasing doses of AMD3100 or CXCL12 as the competitor. Percentage of control binding for each ligand was determined by normalizing the curves at each receptor to the percentage binding range of the CXCL12 curve(s) (Fig. 4, A and C). One representative experiment is shown. Experiments were repeated with similar results (n = 4-10), and the mean values obtained in repeated experiments are shown in Table 3. Cell surface expression for both CXCR4 and CXCR7 receptors were confirmed by flow cytometry (Fig. 4, B and D). The secondary antibody (α-mouse-phycoerythrin) was used as the negative control (black filled area). Positive expression was conferred using the primary (12G5 or αFLAG) + secondary antibodies (white filled area). A, HEK293T cells transfected with HA-Flag CXCR4 conferring dose-dependant competition of 125I-CXCL12 tracer with CXCL12 (○) or AMD3100 (▪). The CXCL12 competition of the representative experiment is best described by a two site model (p < 0.0001), where Ki-1 = 7.18 × 10-11 M and Ki-2 = 3.75 × 10-8 M, with 38.5% of the sites labeled at this tracer concentration being in the high affinity state for the uniodinated CXCL12 (0.65% of the total receptor population). The heterologous AMD3100 competition is best described by a one-site model with a Ki = 4.48 × 10-2 M. B, the 12G5 antibody was used to detect cell surface expression of CXCR4. C, HEK293T cells transfected with HA-Flag CXCR7 conferring dose-dependent competition of CXCL12 (○) and dose-dependent cooperative binding of AMD3100 (▪) in the presence of 125I-CXCL12. The CXCL12 competition is best described by a two-site model (p < 0.0001) where Ki-1 = 6.04 × 10-11 M and Ki-2 = 1.35 × 10-8 M with 32.06% of the sites labeled at this tracer concentration being in the high affinity state for the uniodinated CXCL12 (0.54% of the total receptor population). The AMD3100 data are best described by a one site model with a Ki = 3.45 × 10-5 M. D, the αFLAG (M2) antibody was used to detect cell surface expression of CXCR7.
Discussion
We report here that the small bicyclam AMD3100, a known negative modulator of the CXCR4/CXCL12 interaction (Gerlach et al., 2001; Percherancier et al., 2005; Fricker et al., 2006), is also a ligand for the alternative CXCL12 receptor CXCR7. We find that either CXCL12 or AMD3100 alone induce β-arrestin recruitment to CXCR7. In addition, and in contrast to the observed negative effects on CXCR4, we find positive modulation of the CXCL12-CXCR7 interaction in both BRET and radioligand binding assays, as well as a small, but consistent modulation of the CXCL12-induced recruitment of β-arrestin to CXCR7.
Taken together, these observations suggest that AMD3100 is an allosteric agonist at CXCR7. In itself, the increase of CXCL12 binding to CXCR7 by AMD3100 is incompatible with AMD3100 and CXCL12 binding to the same (orthosteric) site, demonstrating an allosteric binding mode (Fig. 4). In line with this conclusion, the maximal effect of CXCL12 observed on the CXCR7 homodimer conformation by BRET is increased by 40% by AMD3100, indicating increased efficacy (that is, an altered conformation of the receptor dimer in the simultaneous presence of both ligands on CXCR7) (Fig. 3). Finally, the small but reproducible increase in potency of CXCL12-induced β-arrestin recruitment to CXCR7 by AMD3100 is also consistent with allosteric binding to CXCR7 (Fig. 5C). The agonist property of AMD3100 on CXCR7 is highlighted by the recruitment of β-arrestin to the receptor by the compound alone (Fig. 5A). It thus appears that AMD3100 is behaving as an ago-allosteric modulator when bound in the context of the CXCR7 homodimer (Schwartz and Holst, 2006).
The existence of allosteric ligands for chemokine receptors is now well documented (Allegretti et al., 2008, and references therein). Operational models have been derived to relate the effects of increasing doses of an allosteric modulator on the dose-response curve of the orthosteric agonist, and these models include a series of independent parameters by which the allosteric ligand modulates the properties of the orthosteric ligand (Ehlert, 2005; Kenakin, 2005; Kenakin et al., 2006). These parameters include independent effects of the modulator on both the affinity and the efficacy of the orthosteric ligand, such that the effect for each modulator-ligand pair is unique at each receptor being studied, and for each signaling pathway that is being studied. In this manner, a particular ligand-modulator-receptor complex may be capable of stabilizing a distinct conformation, and the effect of this modulation is unique to any given receptor and signaling pathway being studied, a phenomenon recently referred to as “functional selectivity” (Urban et al., 2007). The operational models illustrate that the effect of the allosteric ligand on the signaling output is entirely dependent on the probe with which it is interacting [i.e., probe dependence (Kenakin, 2005)], and this has been underscored recently with the same small-molecule allosteric agonist acting through the CCR1 receptor as either a positive or negative modulator depending upon the endogenous chemokine (probe) tested (Jensen et al., 2008). In the case of AMD3100, we have demonstrated that this ligand behaves differently against the same probe (CXCL12) at two different receptors (CXCR4 and CXCR7).
Although our data are consistent with a positive ago-allosteric mechanism of AMD3100 at CXCR7, they are insufficient to clarify the nature of the negative interaction at CXCR4. In contrast to what was observed with CXCR7, we did not observe any appreciable effect of AMD3100 on 125I-CXCL12 binding to CXCR4 until high concentrations (Fig. 4A and Table 3), yet an effect of AMD3100 on the CXCL12-mediated BRET response through the CXCR4 homodimer was readily observed at 10 μM (Fig. 3, Table 2). The mechanism by which AMD3100 interferes with CXCL12 binding to CXCR4 and CXCR4 activity is complex and a matter of debate. No homologous binding competition studies using radiolabeled AMD3100 are published, and there is considerable discrepancy between the data reported for CXCL12 radioligand competition by AMD3100. It is noteworthy that AMD3100 has been referred to as an allosteric inhibitor of CXCR4 (Allegretti et al., 2008).
The absence of correlation between AMD3100 exhibiting antagonistic effects through CXCR4 and competition with radiolabeled CXCL12 binding has been noted previously (Fricker et al., 2006, as well as our own unpublished observations using HEK293T cells), and four major studies find 125I-CXCL12-displacement IC50 values largely above EC50 values of inhibition of CXCR4-mediated functions (Gupta et al., 2001; Zhang et al., 2002; Gerlach et al., 2003; Fricker et al., 2006). The reported IC50 values range from 74 and 106 nM when the receptor is expressed heterologously in COS-7 (Gerlach et al., 2001) or Chinese hamster ovary cells (Zhang et al., 2002) to 651 nM (Fricker et al., 2006) and 15 μM (Gupta et al., 2001) when the receptor is expressed endogenously in CCRF-CEM or HL-60 cells, respectively. Our own experiments resulted in an even higher IC50 with heterologous expression in the HEK293T cell background. The reason for this difference remains unknown and may lie in subtle differences in the experimental conditions. Alternatively, it is possible that the reported conformational heterogeneity of CXCR4 in different cell backgrounds may play a role (Baribaud et al., 2001). The discordance between the concentration at which competition for ligand binding and functional activity is observed at CXCR4 could indeed be explained by an allosteric mechanism, because the effects of allosteric modulators on ligand binding and functional readouts are independent (see above). However, ligand binding experiments are typically done at 4°C to prevent internalization, whereas the functional experiments or BRET experiments are typically done at 25-37°C to be more representative of physiological conditions, as alluded to elsewhere (Fricker et al., 2006), which might also contribute to the discrepancy between binding and functional assays. On the other hand, data mapping the binding site of AMD3100 on CXCR4 support a possible allosteric binding mode of AMD3100 to CXCR4 (Rosenkilde et al., 2004). Indeed, the binding site for AMD3100 on CXCR4 seems to be distinct from that of the orthosteric ligand CXCL12, as mutations that dramatically alter AMD3100 binding have little effect on CXCL12 binding (Gerlach et al., 2001). The picture that has emerged is that the binding site for the bicyclam is within the upper portion of the transmembrane domains, whereas the binding site for the orthosteric ligand CXCL12 involves mainly the extracellular loops (Labrosse et al., 1998; Brelot et al., 2000; Zhou et al., 2001). Because AMD3100 interacts with a distinct and overlapping binding site on CXCR4, it might be expected that it would behave as an allosteric ligand for its receptor in the presence of the orthosteric ligand CXCL12. Likewise, it may also be speculated that AMD3100 interacts at an allosteric site within the transmembrane domains of CXCR7, although this remains to be demonstrated.

Effects of CXCL12 and AMD3100 on the recruitment of β-arrestin. A, β-arrestin recruitment to CXCR7 in the presence of indicated doses CXCL12 (n = 4) or AMD3100 (n = 3). **, statistical significance (repeated measures analysis of variance, p ≤ 0.01). B, as a control, β-arrestin recruitment to CXCR4 induced by 100 nM CXCL12, and its inhibition by 10 μM AMD3100. C, dose-response curves of CXCL12 on arrestin recruitment to CXCR7, in the absence (○) or presence (▪) of 10 μM AMD3100. One experiment representative of four is shown. Arrestin recruitment was measured after 5 min (CXCR7) or 30 min (CXCR4) of incubation with ligands.
The finding that AMD3100 binds CXCR7 warrants reexamination of the selectivity of the compound. Previous reports, including our own, have described AMD3100 as being highly selective for CXCR4 (Labrosse et al., 1998; Hatse et al., 2002). However, CXCR7 was not included in these studies. The late discovery of CXCR7 to be a receptor for CXCL12 implies that the conclusions drawn from many data relating to biological effects must be reassessed, as they have previously been routinely ascribed to CXCR4, the only known CXCL12 receptor. Blockade of either CXCR4 or CXCR7 with selective inhibitors will be useful for such reassessment, and AMD3100 was a candidate of choice that is being used to demonstrate roles of CXCR4 (for example, see Cabioglu et al., 2005; Pattarozzi et al., 2008). Our findings call for special caution in the use of AMD3100 for the dissection of the respective roles of CXCR4 and CXCR7 as mediators of the biological effects of CXCL12.
Fitted curve parameters of β-arrestin recruitment to CXCR7
Summary of the analyses of CXCL12-induced recruitment of β-arrestin to CXCR7 (n = 4). The difference of logEC50 in presence and absence of AMD3100 of the curves is statistically significant. In addition, simultaneous curve fitting for each set of curves (±AMD3100) confirmed the statistical significance for the difference between the curves in three out of four experiments (P < 0.05). Values are presented as mean ± S.E.M.
Acknowledgments
We thank Geneviève Saint-Onge for expert technical assistance and Michel Bouvier and his laboratory for the gift of the V1Ra-YFP and RLuc-arrestin2 constructs.
Footnotes
-
This work was supported by the Canadian Institutes of Health Research [Grants HOP-79210 and CTP-79848], the “Fondation de l'Hôpital Sainte-Justine,” and the “Groupe de Recherche sur le Médicament”.
-
I.K. and Y.A.B. contributed equally to this work.
-
ABBREVIATIONS: GPCR, G-protein coupled receptor; BRET, bioluminescence resonance energy transfer; YFP, yellow fluorescent protein; RLuc, Renilla reniformis luciferase; V1Ra, vasopressin receptor 1a; CXCR, chemokine CXC motif receptor; CXCL, CXC chemokine ligand; HEK, human embryonic kidney; PBS, phosphate-buffered saline; HA, influenza virus hemagglutinin.
-
↵1 Current affiliation: CincyTechUSA, Cincinnati, Ohio.
- Received November 7, 2008.
- Accepted March 2, 2009.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}