


Abstract
Receptor antagonists can be classified as neutral antagonists or antagonists with inverse agonist activity based on their effectiveness to reduce the spontaneous agonist-independent activity of receptors. The goals of this study were to (1) demonstrate that A1-adenosine receptors (A1AdoRs) expressed at high density (4000–8000 fmol/mg of protein) in Chinese hamster ovary (CHO) cells cause constitutive activation of inhibitory G proteins and inhibition of adenylyl cyclase activity and (2) identify both neutral A1AdoR antagonists and antagonists with inverse agonist activity. The activity of A1AdoR agonists and antagonists was determined by assays of both specific binding of [35S]guanosine-5′-O-(3-thio)triphosphate ([35S]GTPγS) to membranes and cAMP content of intact cells in the presence of adenosine deaminase (2–5 units/ml). The A1AdoR agonist N6-cyclopentyladenosine (CPA) significantly increased binding of [35S]GTPγS by 241 ± 7% compared with control. The A1AdoR antagonists N-0861, N-0840, and WRC-0342 did not alter binding of [35S]GTPγS, whereas the antagonists 8-cyclopentyl-1,3-dipropylxanthine (CPX), CGS-15943, xanthine amine congener, and WRC-0571 significantly reduced binding of [35S]GTPγS by 28–53% from control, respectively. The effects of both the agonist N6-cyclopentyladenosine (CPA) and the antagonist CPX to alter binding of [35S]GTPγS were attenuated by 1 μm N-0861. CPA reduced cAMP content of forskolin-stimulated CHO:A1AdoR cells, and N-0861 and WRC-0342 did not alter cAMP content, but the antagonists CPX and WRC-0571 increased the cAMP content of CHO:A1AdoR cells. The effects of both CPX and WRC-0571 to increase cAMP content of forskolin-stimulated CHO:A1AdoR cells were attenuated by either N-0861 or WRC-0342. The results indicate that both N-0861 and WRC-0342 are neutral antagonists, whereas both CPX and WRC-0571 are antagonists with inverse agonist activity.
In recent years, it has become well established that many G protein-coupled receptors may exist in a spontaneously active form in the absence of agonist (Costa, 1992; Lefkowitz, 1993; Mewes, 1993;Chidiac, 1994; Milano, 1994; Kenakin, 1996). This agonist-independent spontaneous activity of receptors has been most readily observed in cell lines in which receptors are overexpressed or mutated (Adie and Milligan, 1994; Barker et al., 1994; Chidiac et al., 1994; Bond et al., 1995; Newman-Tancredi et al., 1997) and in reconstituted membrane systems with purified receptors and appropriate G proteins (Cerione et al., 1984;Freissmuth et al., 1991). In cells in which a constitutive response to agonist-independent receptor activity can be measured, antagonists that reduce the levels of activity and functional response are referred to as inverse agonists or antagonists with negative intrinsic activity, whereas antagonists that do not reduce activity are called neutral antagonists or antagonists with no intrinsic activity (Costa and Herz, 1989). For example, results of studies using NIH 3T3 fibroblast cells expressing constitutively active wild-type 5-hydroxytryptamine2C receptors revealed a wide spectrum of negative intrinsic activity for 5-hydroxytryptamine2C receptor antagonists, from full inverse agonists to neutral antagonists (Barker et al., 1994).
A1AdoRs are tightly coupled to G proteins (Stiles, 1985; Leung and Green, 1989), and activation of these receptors leads to inhibition of adenylyl cyclase activity (Cooperet al., 1980). A constitutive functional response caused by activity of A1AdoRs in the absence of an agonist has not yet been conclusively identified. However, evidence for constitutive inhibition by A1AdoRs of adenylyl cyclase activity was reported by Ma and Green (1992). These investigators found that the A1AdoR antagonists CPX and BW-A844U [3-(4-amino)phenethyl-1-propyl-8-cyclopentylxanthine] increased adenylyl cyclase activity in detergent-permeabilized embryonic chick ventricular myocytes in the absence of agonist. The action of CPX and BW-A844U was prevented by pertussis toxin treatment (Ma and Green, 1992). The authors postulated that “precoupled” A1AdoRs in cardiac myocytes exert a tonic inhibition of adenylyl cyclase and that CPX stimulates adenylyl cyclase activity by uncoupling the A1AdoR from its G protein.
The goals of this study were to demonstrate constitutive activity of A1AdoRs in intact cells and to identify both neutral antagonists and antagonists with inverse agonist activity at the A1AdoR. We used intact and broken cell preparations of CHO cells in which human A1AdoRs were cloned and expressed in high density (Kollias-Baker et al., 1997) as our experimental model. Using these CHO:A1AdoR cells, we demonstrate that activity of A1AdoRs in the absence of agonist causes activation of inhibitory G proteins and a tonic reduction of cAMP content. Evidence is also presented that certain A1AdoR antagonists (i.e., neutral antagonists) have no intrinsic activity in the absence of agonist, whereas others (i.e., inverse agonists or antagonists with inverse agonist activity) cause a response that is the inverse of that caused by agonist.
Experimental Procedures
Materials.
PD81,723 [(2-amino-4,5-dimethyl-3-thienyl)-[3-(trifluoromethyl)phenyl]methanone], the allosteric enhancer of agonist binding to the A1AdoR (Bruns and Fergus, 1990), and the A1AdoR antagonists N-0861, N-0840, WRC-0571 [C8-(N-methylisopropyl)-amino-N6(5′-endohydroxy)-endonorbornan-2-yl-9-methyladenine], and WRC-0342 [N6-(5′-endohydroxy)-endonorbornan-2-yl-9-methyladenine] (Martin et al., 1996) were gifts from Dr. Noel Cusack (Discovery Therapeutics, Richmond, VA). Adenosine deaminase, GTPγS, adenosine, and HEPES were purchased from Sigma Chemical (St. Louis, MO). Forskolin, 5′-N-ethylcarboxamidoadenosine, CGS-15943 [9-chloro-2-(2-furyl)[1,2,4]triazolo[1,5-c] quinazolin-5-amine], CPA, CPT, XAC, and CPX were purchased from Research Biochemicals (Natick, MA). Ham’s F-12 cell culture medium and fetal bovine serum were from GIBCO Life Technologies (Gaithersburg, MD). Antibiotic G-418, Falcon 150-mm culture plates, and Costar 12-well culture plates were from Fisher Scientific (Pittsburgh, PA). CHO cells were purchased from the American Type Culture Collection (Rockville, MD). [3H]CPX, carrier-free125I, and [35S]GTPγS were purchased from DuPont-New England Nuclear Research Products (Boston, MA). Penicillin/streptomycin antibiotic mixture was purchased from Mediatech (Washington, DC). The composition of HEPES-buffered Hanks’ solution was: 130 mm NaCl, 5.0 mm KCl, 1.5 mm CaCl2, 0.41 mmMgS04, 0.49 mmMgCl2, 0.5 mmNa2HPO4, 0.44 mm KH2PO4, 5.6 mm dextrose, and 5 mm HEPES (pH 7.4). Succinyl cAMP tyrosyl methyl ester (Sigma) was radiolabeled with125I in the presence of chloramine T as described by Steiner et al. (1972).
Cell culture.
CHO cells, referred to herein as CHO:Wild cells, were cultured as monolayers on plastic culture dishes in Ham’s F-12 medium supplemented with 10% fetal bovine serum, 100 units of penicillin G, and 100 μg of streptomycin in a humidified atmosphere of 5% CO2/95% air at 37°. The density of [3H]CPX binding sites in CHO:Wild cells was 26 ± 2 fmol/mg protein (four experiments). Cells were subcultured twice weekly after detachment using 1 mm EDTA in Ca2+/Mg2+-free HEPES-buffered Hanks’ solution. CHO cells stably expressing the recombinant human A1AdoR (CHO:A1AdoR cells) were prepared as described previously (Kollias-Baker et al., 1997) and cultured as for CHO:Wild cells, but with 0.5 mg/ml G-418 in the culture medium. Three different clones of CHO:A1AdoR cells were used for experiments, and all results were confirmed with cells from two or three clones. The density of A1AdoRs in these cells was 4000–8000 fmol/mg protein, as determined by assay of [3H]CPX specific binding.
Radioligand binding.
CHO cells grown onto 150-mm culture dishes were rinsed with HEPES-buffered Hanks’ solution and then removed with a cell scraper and homogenized in ice-cold 50 mm Tris·HCl, pH 7.4. Cell membranes were pelleted by centrifugation of the cell homogenate at 48,000 × gfor 15 min. The membrane pellet was washed twice by resuspension in fresh buffer and centrifugation. The final pellet was resuspended in a small volume of 50 mm Tris·HCl, pH 7.4, and stored in aliquots of 1 ml at −80° until used for assays.
To determine the density of A1AdoRs in CHO cell membranes, 100-μl aliquots of membranes (5 μg of protein) were incubated for 2 hr at 25° with 0.15–20 nm[3H]CPX and adenosine deaminase (2 units/ml) in 100 μl of 50 mm Tris·HCl, pH 7.4. Incubations were terminated by dilution with 4 ml of ice-cold 50 mmTris·HCl buffer and immediate collection of membranes onto glass-fiber filters (Schleicher & Schuell, Keene, NH) by vacuum filtration (Brandel, Gaithersburg, MD). Filters were washed quickly three times with ice-cold buffer to remove unbound radioligand. Filter discs containing trapped membranes and bound radioligand were placed in 4 ml of Scintiverse BD (Fisher), and the radioactivity was quantified using a liquid scintillation counter. To determine nonspecific binding of [3H]CPX, membranes were incubated as described above, and 10 μm CPT was added to the incubation buffer. Nonspecific binding was defined as [3H]CPX bound in the presence of 10 μm CPT. Specific binding of the radioligand to the A1AdoR was determined by subtracting nonspecific binding from total binding. Nonspecific binding was found to increase linearly with an increase of [3H]CPX concentration. Triplicate assays were done at each tested concentration of [3H]CPX.
To determine the affinities of antagonists of A1AdoRs for the human recombinant A1AdoR expressed in CHO cells, binding of 2 nm [3H]CPX in the presence of increasing concentrations of antagonist was measured. Aliquots of CHO cell membranes (100 μl; 5 μg of protein), [3H]CPX, antagonist (0.1 nm to 100 μm), and adenosine deaminase (2 units/ml) were incubated for 3 hr at 25° in 200 μl of 50 mm Tris·HCl buffer, pH 7.4. Assays were terminated as described above.
To determine the effects of agonist and antagonists on specific binding of [35S]GTPγS to G proteins in CHO cell membranes, membranes (5 μg of protein in 100 μl), agonist or antagonist, and 0.4 nm [35S]GTPγS were incubated for 45 min at 25° in 200 μl of 50 mmTris·HCl buffer, pH 7.4, containing 5 mmMgCl2, 1 mm EDTA, 1 mmdithiothreitol, 100 mm NaCl, 10 μm GDP, 0.5% bovine serum albumin, and 5 units/ml adenosine deaminase. Triplicate assays were done at each tested concentration of agonist or antagonist, in each experiment. Assays were terminated as described above, using 50 mm Tris·HCl containing 5 mmMgCl2 to wash the membranes. Binding of [35S]GTPγS in the presence of 10 μm GTPγS was considered to be nonspecific binding. Specific binding was determined by subtraction of nonspecific from total binding.
cAMP assay.
Culture medium was aspirated from nearly confluent cells grown in 12-well culture clusters as adherent monolayers, and warm (37°) HEPES-buffered Hanks’ solution was added to each well in the cluster. After 6 min, the Hanks’ solution was aspirated, and fresh warm Hanks’ solution containing appropriate drugs and adenosine deaminase (Type VIII, 2 units/ml), 0.3 μmforskolin, and 20 μm rolipram was added. Adenosine deaminase was present to degrade endogenous adenosine. Forskolin and rolipram were present to increase adenylyl cyclase activity and inhibit phosphodiesterase activity, respectively, and thereby amplify drug-induced changes in cell cAMP content. This incubation solution was aspirated at the end of 6 min of incubation and replaced immediately with 1 ml of ice-cold 50 mm HCl. cAMP contents of the acid extracts of cells were measured by radioimmunoassay. Cellular extract (100 μl), antibody to cAMP (a generous gift from Dr. Gary Brooker, Georgetown University), and 125I-labeled succinyl cAMP tyrosyl methyl ester (∼20,000 dpm) were mixed and incubated in glass tubes overnight at 2°. Hydroxyapatite (75 μl; 50:50 v/v in water) was then added to each sample, and the mixtures were incubated for 10–30 min at 2°. An assay was terminated by vacuum filtration of samples and collection of bound radioactivity on filter paper, using a Brandel cell harvester. The radioactivity of antibody-bound125I-labeled cAMP ester adsorbed to hydroxyapatite and trapped on filter paper was quantified by a gamma counter. cAMP content of sample extracts was estimated by comparison of sample results with results of parallel assays of standards of known cAMP content (0.06–8 pmol).
Protein determination.
Protein content of cell extracts and cell membrane preparations was determined according to the Bradford method using a kit purchased from BioRad (Richmond, CA). Albumin was used as a standard. Protein content of a nearly confluent monolayer of CHO cells was ∼125 μg/well of cells in a 12-well culture cluster.
Data analysis.
Values of experimental measurements are expressed either as mean ± standard error or as the negative logarithm of the mean ± standard error (for values of EC50 and Ki , which presumably are log normally distributed). Statistical analysis of differences among values in experiments with multiple-comparison groups was based on analysis of variance followed by Dunnett’s test (SigmaStat; Jandel, San Rafael, CA) unless otherwise indicated. Differences between group mean values were considered significant atp < 0.05. Results of radioligand binding assays were analyzed using the computer program KELL for Windows (Elsevier-Biosoft, Cambridge, UK).
Results
Effects of A1AdoR agonists and antagonists on G protein activation.
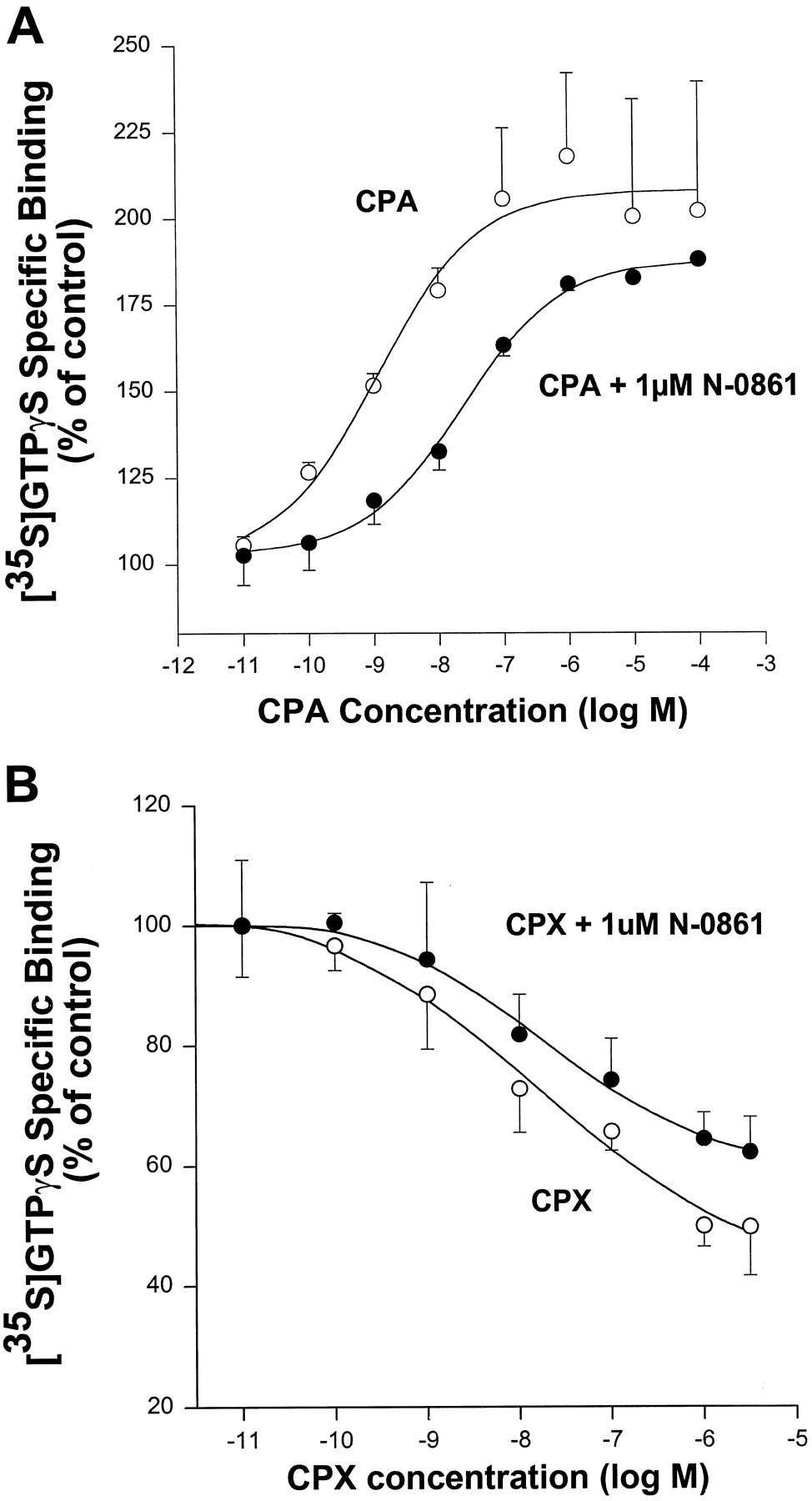
The activation of G proteins by G protein-coupled A1 adenosine receptors was assessed by quantification of binding of [35S]GTPγS to membranes prepared from CHO cells expressing these receptors (i.e., CHO:A1AdoR cells) and associated G proteins. Adenosine deaminase (5 units/ml) was added to the incubation medium to degrade endogenous adenosine. Results of assays of binding of [35S]GTPγS to membranes from CHO:A1AdoR cells are shown in Fig.1. Both the A1AdoR agonist CPA and the allosteric enhancer PD81,723 increased the binding of [35S]GTPγS, by 241 ± 7 (nine experiments) and 25 ± 3% (four experiments), respectively (p < 0.05 compared with control). Neither of the A1AdoR antagonists N-0861 (nine experiments), N-0840 (seven experiments), and WRC-0342 (four experiments) caused a significant change of [35S]GTPγS binding from control. In contrast, the selective and nonselective A1AdoR antagonists WRC-0571, CPX, CGS-15943, and XAC reduced the binding of [35S]GTPγS in a concentration-dependent manner (Fig. 1). For instance, a 1 μm concentration of WRC-0571, CPX, CGS-15943, or XAC significantly reduced the binding of [35S]GTPγS by 28 ± 3% (three experiments), 42 ± 1% (eight experiments), 53 ± 7% (seven experiments), or 47 ± 7% (four experiments), respectively (Fig.1). Thus, in the absence of an agonist of the A1AdoR, N-0861, N-0840, and WRC-0342 acted as neutral antagonists, whereas WRC-0571, CPX, CGS-15943, and XAC acted as inverse agonists in this assay. A neutral antagonist is expected to attenuate both the action of an agonist to increase [35S]GTPγS binding and the action of an antagonist with inverse agonist activity to decrease [35S]GTPγS binding. These expectations were confirmed. As shown in Fig. 2A, N-0861 (1 μm) caused a rightward shift of the CPA concentration-response relationship and a significant change in the value of pEC50 for CPA from 8.54 ± 0.23 (EC50 = 2.9 nm) to 7.74 ± 0.41 (EC50 = 18 nm) (five experiments). The action of CPX to reduce binding of [35S]GTPγS was also antagonized by 1 μm N-0861 (Fig. 2B). Thus, the data shown in Figs. 1 and2 suggest that neutral antagonists (N-0861, N-0840, and WRC-0342) of the A1AdoR and antagonists with inverse agonist activity (CPX, WRC-0571, XAC, and CGS-15943) were distinguished by analysis of their actions in [35S]GTPγS binding assays.

Effects of A1AdoR ligands on specific binding of [35S]GTPγS to membranes prepared from CHO cells expressing the recombinant human A1AdoR. Membranes were incubated for 45 min with indicated drug, 0.4 nm[35S]GTPγS, and adenosine deaminase (5 units/ml) as indicated in the text. Control specific binding of [35S]GTPγS was 3398 ± 466 dpm. Values of normalized responses to drugs are mean ± standard error of triplicate determinations in each of three to nine experiments. Specific binding of [35S]GTPγS in the presence of 1 μm of CPA, PD81,723, WRC-0571, CPX, XAC, or CGS-15943 was significantly different from control (p < 0.05, by one-way analysis of variance and Student-Newman-Keuls multiple-comparison procedure).

Attenuation by the neutral antagonist N-0861 (1 μm) of both the CPA-induced increase of [35S]GTPγS binding (A) and the CPX-induced decrease of [35S]GTPγS binding (B) to membranes prepared from CHO cells expressing the recombinant human A1AdoR. Control (absence of drug) specific binding of [35S]GTPγS was 2492 ± 275 dpm in A and 3939 ± 495 dpm in B. Values of normalized responses to drugs are mean ± standard error of triplicate determinations in each of five (A) or three (B) experiments. Experiments were done as described in legend to Fig. 1 and the text. Specific binding of [35S]GTPγS in the absence and presence of 1 μm N-0861 was significantly different for experiments presented in both A and B (p < 0.05, by two-way repeated measures analysis of variance for two factors: drug concentration and presence/absence of N-0861).
Effects of A1AdoR agonists/antagonists on cAMP content of intact cells.
To further demonstrate the distinctions among agonists, neutral antagonists, and antagonists with inverse agonist activity, experiments were done to study the effects of these classes of compounds on cAMP contents of intact CHO:A1AdoR and CHO:Wild cells. Two neutral antagonists (N-0861, WRC-0342) and two antagonists with inverse agonist activity (WRC-0571, CPX) were used in these experiments. The contents of cAMP of both CHO:Wild and CHO:A1AdoR cells in the absence of drugs were 7 ± 2 (six experiments) and 5 ± 2 pmol/mg protein, respectively. Additions of adenosine deaminase (2 units/ml) and CPX (0.1 μm) to the incubating medium of CHO:A1AdoR cells caused small (9–38%) increases of cAMP content. To increase cAMP content of cells and thereby amplify the effects of A1AdoR agonists and antagonists, forskolin, an activator of adenylyl cyclase, and rolipram, an inhibitor of cAMP phosphodiesterase, were used. In the presence of 0.3 μm forskolin and 20 μm rolipram, cAMP contents of CHO:Wild and CHO:A1AdoR cells increased to 462 ± 7 and 147 ± 13 pmol/mg protein (30 and 36 determinations, respectively, in three experiments). This result suggests that forskolin-stimulated adenylyl cyclase activity was lower in CHO:A1AdoR cells than in CHO:Wild cells. The addition of adenosine deaminase (2 units/ml) to the incubation medium caused a further increase of cAMP content of CHO:A1AdoR cells but not of CHO:Wild cells (Fig.3). In two separate experiments, it was determined that an increase of adenosine deaminase activity from 1 to 2, 5, and 10 units/ml did not significantly increase cAMP content of CHO:A1AdoR cells above that caused by 1 unit/ml adenosine deaminase (not shown). Results of assays of the action of adenosine (0.1 nm to 1 μm) to reduce cAMP content in the presence of 1 μm forskolin and 20 μm rolipram indicated that the concentration of endogenous adenosine in the incubation medium (i.e., adenosine that diffuses from CHO cells into the extracellular medium) in the absence of adenosine deaminase was ∼1–2 nm. This concentration of adenosine was thus apparently sufficient to cause a reduction of cAMP content of CHO:A1AdoR cells by 40% compared with cAMP content of CHO:A1AdoR cells incubated with adenosine deaminase (Fig. 3). The A1AdoR antagonist CPX (50 nm) caused an additional increase of cAMP content of CHO:A1AdoR cells by ∼2-fold in the presence of adenosine deaminase (2 units/ml), whereas it did not change cAMP content of CHO:Wild cells (Fig. 3). The data indicate that both endogenous adenosine and spontaneous activity of A1AdoRs in the absence of agonist act to inhibit adenylyl cyclase activity and reduce cAMP content of CHO:A1AdoR cells treated with forskolin.
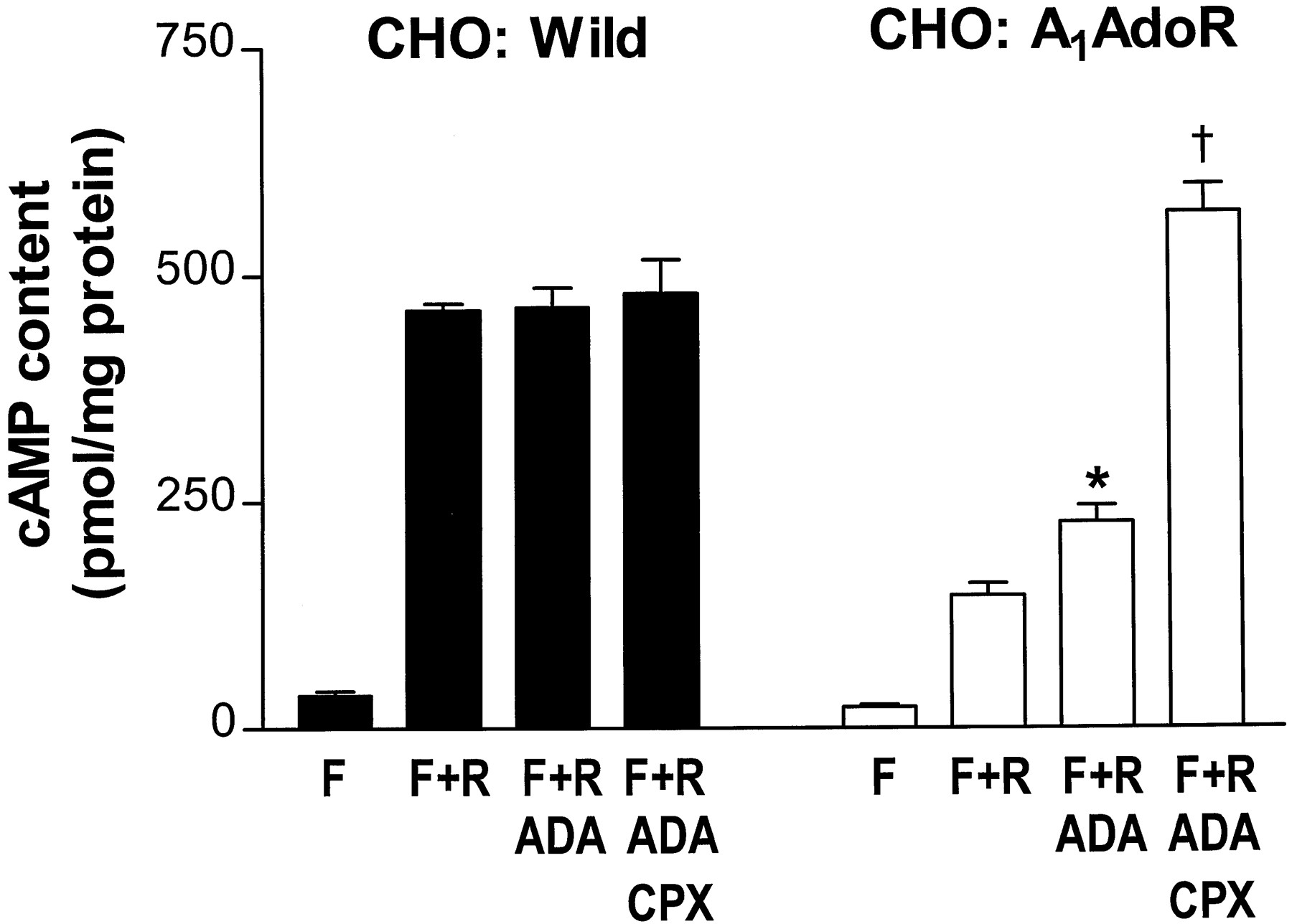
Endogenous adenosine-mediated and agonist-independent A1AdoR-mediated inhibition of forskolin-stimulated cAMP accumulation in CHO cells stably expressing the recombinant human A1AdoR (CHO:A1AdoR).CHO:Wild andCHO:A1AdoR cells were incubated with 0.3 μm forskolin (F), 20 μm rolipram (R), 2 units/ml adenosine deaminase (ADA), and CPX (50 nm for CHO:A1AdoR cells, 1 μm for CHO:Wild) in the combinations indicated. Bars, mean ± standard error for 6–36 independent determinations. ∗, Significantly different from F+R, p < 0.05. †, Significantly different from F+R+ADA, p < 0.05.
The A1AdoR agonist CPA reduced cAMP content of CHO:A1AdoR cells in the presence of 0.3 μm forskolin, 20 μm rolipram, and adenosine deaminase (2 units/ml). CPA decreased cAMP content from 324 ± 44 pmol/mg protein (six experiments) to 31 ± 3 pmol/mg protein (six experiments) in a concentration-dependent manner with an EC50 value of 0.1 nm. All four A1AdoR antagonists (WRC-0571, CPX, N-0861, WRC-0342) fully attenuated the action of 3 nm CPA to reduce cAMP accumulation in the presence of 0.3 μm forskolin (Fig.4). However, cAMP accumulation in cells exposed to either 1 μm WRC-0571 or 1 μm CPX was significantly greater than that in cells exposed to forskolin alone (Fig. 4). On the other hand, cAMP content of cells treated with either N-0861 or WRC-0342 was not significantly greater than that in cells treated with forskolin alone. These results (Fig. 4) are consistent with the results of [35S]GTPγS experiments and indicate that both CPX and WRC-0571 are antagonists with inverse agonist activity, whereas both N-0861 and WRC-0342 are neutral antagonists in intact CHO:A1AdoR cells and in membranes prepared from these cells.
Antagonism by inverse agonists (WRC-0571, CPX) and neutral antagonists (N-0861, WRC-0342) of the action of CPA on cAMP content of CHO:A1AdoR cells in the presence of forskolin. CHO:A1AdoR cells were incubated with 0.3 μmforskolin, 20 μm rolipram, and 2 units/ml of adenosine deaminase in the absence (control) and presence of 3 nm CPA alone or together with either 1 μm WRC-0571, 1 μm CPX, 50 μm N-0861, or 50 μm WRC-0342. Control cAMP content of cells incubated with forskolin, rolipram, and adenosine deaminase was 22.1 ± 0.6 pmol/well. Bars, mean ± standard error percent of control for 29–32 determinations in four experiments. ∗, Significantly different from control, p < 0.05.
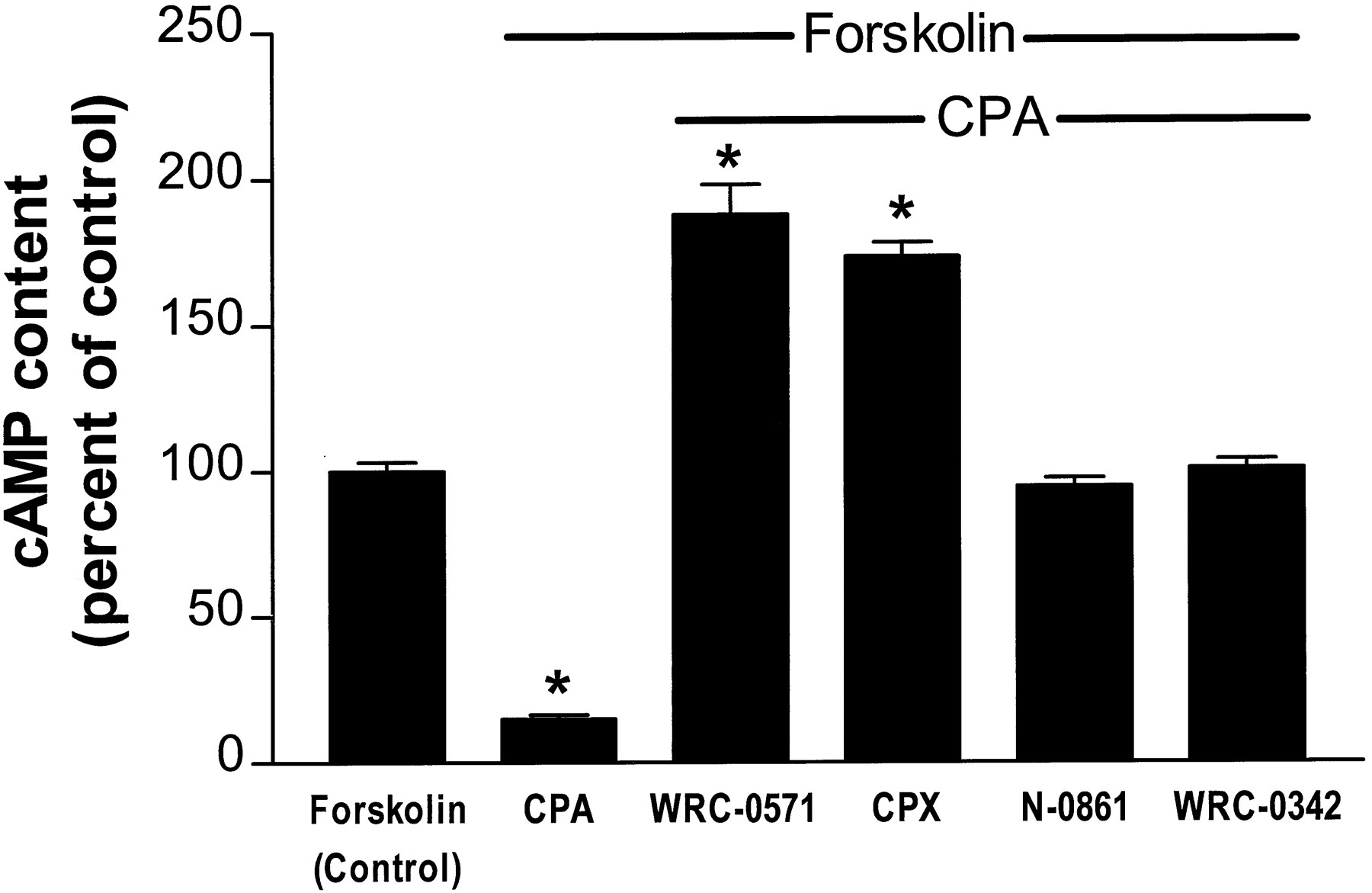
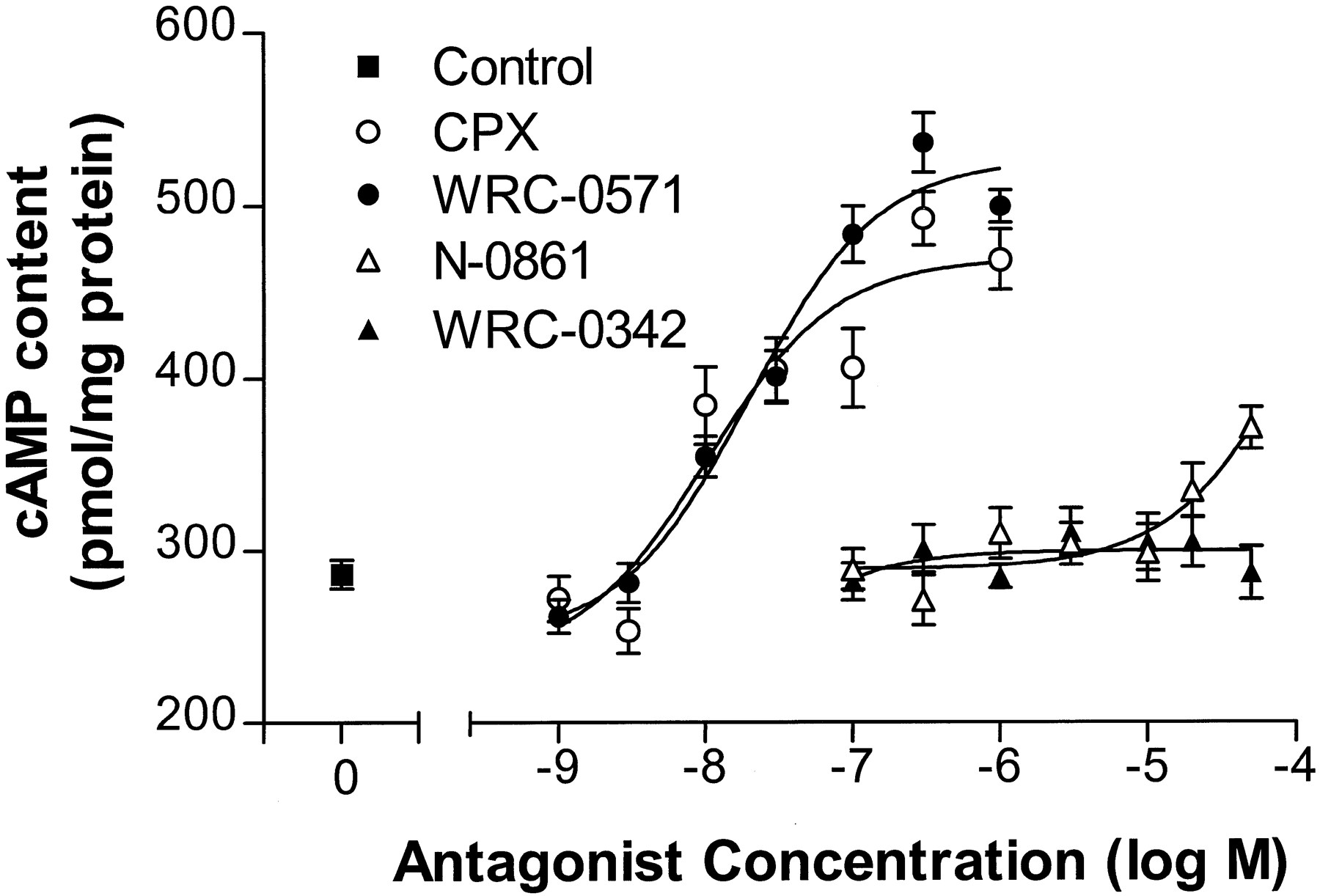
In the absence of an agonist (and in the presence of 2 units/ml of adenosine deaminase), CPX and WRC-0571 increased cAMP content of CHO:A1AdoR cells in a concentration-dependent manner, whereas WRC-0342 did not (Fig.5). N-0861 (50 μm) increased cAMP content of CHO:A1AdoR cells by 29% compared with control (p < 0.05). N-0861 (50 μm) also increased cAMP content of CHO:Wild cells by 27% (p < 0.05 compared with control), whereas 1 μm WRC-0571, 1 μm CPX, and 50 μm WRC-0342 (i.e., the maximal concentration of each antagonist used in intact cell experiments) did not significantly alter cAMP content of CHO:Wild cells. These data suggest that the action of N-0861 (50 μm) to increase cAMP content of CHO cells was unrelated to its antagonism of A1AdoRs. In contrast, the increase of cAMP content of CHO:A1AdoR cells caused by CPX and by WRC-0571 was associated with antagonism of A1AdoRs. The results of the cAMP experiments support those of the [35S]GTPγS experiments indicating a distinction between the neutral antagonists N-0861 and WRC-0342 and the antagonists with inverse agonist activity, WRC-0571 and CPX. The neutral antagonists inhibited only the response to agonist, whereas the antagonists with inverse agonist activity caused a response opposite that of the agonist in both [35S]GTPγS and cAMP assays.

Concentration-response relationships for stimulation by four A1AdoR antagonists of cAMP accumulation in CHO:A1AdoR cells. CHO:A1AdoR cells were incubated with 0.3 μm forskolin, 20 μmrolipram, and 2 units/ml of adenosine deaminase in the absence (control) or presence of either CPX (1 nm to 1 μm), WRC-0571 (1 nm to 1 μm), N-0861 (0.1–50 μm), or WRC-0342 (0.1–50 μm). Each point indicates mean ± standard error of 11–16 determinations in four experiments. The EC50 values for CPX- and WRC-0571-mediated increases of cAMP content were 10 and 19 nm, respectively.
The actions of both agonists and antagonists with inverse agonist activity to decrease and increase, respectively, cAMP content of CHO:A1AdoR cells are likely to be mediated by modulation of activity of inhibitory G proteins. To test this assumption, the effects of adenosine deaminase, CPA, CPX, N-0861, WRC-0571, and WRC-0342 on cAMP content of pertussis toxin-treated (50 ng/ml, 16 hr) CHO:A1AdoR cells were determined. Content of cAMP of pertussis toxin-treated CHO:A1AdoR cells incubated with 0.3 μm forskolin and 20 μm rolipram was significantly greater than that of untreated control CHO:A1AdoR cells (206 ± 27 versus 101 ± 15 pmol/mg protein, p < 0.05, three paired experiments). However, cAMP content of pertussis toxin-treated cells was not significantly different in the absence and presence of either 3 nm CPA, 2 units/ml adenosine deaminase, 50 nmand 1 μm CPX, 1 μm WRC-0571, 50 μm WRC-0341, or 50 μm N-0861. In contrast, 3 nm CPA reduced by 73%, whereas 2 units/ml adenosine deaminase and 50 nm CPX increased by 190% and 430%, respectively, the cAMP content of untreated control CHO:A1AdoR cells in these experiments. The results indicate that the actions of both agonists and antagonists of constitutively active A1AdoRs on cAMP content of CHO:A1AdoR cells are mediated by pertussis toxin-sensitive inhibitory G proteins.
To confirm that all seven compounds studied in [35S]GTPγS binding and cAMP assays were A1AdoR antagonists, competitive radioligand binding assays were used. Each of the seven compounds reduced the binding of [3H]CPX to membranes prepared from CHO:A1AdoR cells in a dose-dependent manner, with a Hill coefficient that was not significantly different from unity (the single exception was XAC, for which the Hill coefficient was 0.53 ± 0.07; three experiments). All compounds caused equal maximal reductions of [3H]CPX binding. Values ofpKi ± standard error for CPX, WRC-0571, XAC, CGS-15943, N-0861, N-0840, and WRC-0342 were 8.31 ± 0.14 (three experiments), 8.58 ± 0.10 (four experiments), 10.13 ± 0.26 (three experiments), 8.84 ± 0.06 (three experiments), 5.99 ± 0.06 (three experiments), 5.81 ± 0.07 (three experiments), and 5.98 ± 0.16 (four experiments), respectively.
Neutral antagonists are reported to attenuate actions of both agonists and antagonists with inverse agonist activity (Costa, 1989b). Thus, 1 μm N-0861 attenuated increases of [35S]GTPγS binding caused by either CPA or CPX (Fig. 2), and 50 μm N-0861 blocked the action of 3 nm CPA to reduce cAMP content of CHO:A1AdoRs (Fig. 4). To further demonstrate the antagonism by the neutral antagonists N-0861 and WRC-0342 of the inverse agonist responses caused by CPX and WRC-0571, assays of the effects of these compounds on cAMP content of intact cells were done. Both 50 nm CPX (Fig. 6,top) and 50 nm WRC-0571 (Fig. 6,bottom) caused significant increases of cAMP content of CHO:A1AdoR cells compared with control. Both 25 and 50 μm WRC-0342 and 25 μm N-0861 attenuated the actions of CPX and WRC-0571 to increase cAMP content of CHO:A1AdoR cells. These results thus confirm the results of the [35S]GTPγS binding experiments. The neutral antagonist WRC-0342 also antagonized the action of PD81,723, an allosteric enhancer of agonist binding to the A1AdoR. PD81,723 (5 μm) significantly reduced cAMP content of CHO:A1AdoR cells (but not of CHO:Wild cells) in the presence of 0.3 μm forskolin, 20 μm rolipram, and 2 units/ml of adenosine deaminase by 28%, from 34.2 ± 2.7 (seven experiments) to 24.6 ± 1.6 (seven experiments) pmol/well. In the presence of 25 μm WRC-0342, the action of 5 μm PD81,723 was attenuated by 93%, and cAMP content rose significantly, from 24.6 ± 1.6 to 33.5 ± 1.7 (four experiments) pmol/well.

Antagonism by WRC-0342 and N-0861 of actions of CPX and WRC-0571 to increase cAMP content of CHO:A1AdoR cells.Top, CHO:A1AdoR cells were incubated with 0.3 μm forskolin, 20 μm rolipram, and 2 units/ml of adenosine deaminase in the absence (control) and presence of 50 nm CPX alone or together with either 25 μm WRC-0342, 50 μm WRC-0342, or 25 μm N-0861. Bottom, cells were incubated as described for the top but in the absence (control) and presence of 50 nm WRC-0571 alone or together with either 25 μm WRC-0342, 50 μm WRC-0342, or 25 μm N-0861. Bars, mean ± standard error of 10–73 determinations in three to seven experiments. ∗, Significantly different from control, p < 0.05. ∗∗, Significantly different from either CPX (top) or WRC-0571 (bottom), p < 0.05.
Discussion
The two major findings of this study were that antagonists of A1AdoRs could cause responses (increased cAMP content of intact cells and decreased binding of [35S]GTPγS to membranes) in CHO cells expressing the transfected human A1AdoR and that two classes of A1AdoR antagonists could be distinguished by their effects on binding of [35S]GTPγS to CHO:A1AdoR membrane preparations and on cAMP content of intact CHO:A1AdoR cells. Based on these findings and other supportive evidence, we conclude that in the absence of agonist, the spontaneous activity of human A1AdoRs expressed at high density (4000–8000 fmol/mg of protein) in CHO cells causes constitutive inhibition of adenylyl cyclase and that N-0861, N-0842, and WRC-0342 are neutral antagonists of the A1AdoR, whereas CPX, WRC-0571, XAC, and CGS-15943 are antagonists with inverse agonist activity (inverse agonists).
A Two-State Equilibrium Model of Receptors Can Explain Our Results
The observation that a number of G protein-coupled receptors can be spontaneously active has led to a simple model in which receptors exist in an equilibrium between inactive (R) and active (R*) conformational states in the absence of agonist (Wreggett and DeLéan, 1984; Lefkowitz et al., 1993; Bond et al., 1995). Agonists bind preferentially to and stabilize the active state (R*) of the receptor and shift the equilibrium toward increased R*. In contrast, antagonists with inverse agonist activity bind preferentially to and stabilize the inactive state (R) of the receptor and shift the equilibrium toward increased R. On the other hand, neutral antagonists bind with equal affinity to both states of the receptor and hence do not alter the equilibrium between R and R*. The results of our experiments can be interpreted by use of this model. The agonist CPA and the allosteric enhancer PD81,723 both increased [35S]GTPγS binding to membranes prepared from CHO:A1AdoR cells (Fig. 1) and decreased cAMP content of intact CHO:A1AdoR cells (Fig. 4 and text), actions mediated by stabilization of the active R* state of the A1AdoR. It is noteworthy that PD81,723 was not found to have agonist-independent actions on either guinea pig isolated heart (Amoah-Apraku et al., 1993) or rat hippocampal brain slices (Janusz et al., 1991). We speculate that an agonist-independent action of PD81,723 may be indicative of the presence of constitutively active A1AdoRs. A1AdoR antagonists (Fig. 4), and pretreatment of cells with pertussis toxin blocked the action of CPA, the former by a competitive effect to reduce agonist binding and the latter by blocking G protein-mediated signal transduction. In addition, in the absence of agonist, CPX and WRC-0571 reduced the number of constitutively active (R*) receptors and thereby decreased the binding of [35S]GTPγS to G proteins in CHO:A1AdoR membranes and increased the cAMP content of intact CHO:A1AdoR cells. The neutral antagonists N-0861 and WRC-0342 bound equally well to both R and R* forms of the A1AdoR and consequently attenuated both the responses mediated by agonists and allosteric enhancer (Figs.2A and 4 and text) and those mediated by antagonists with inverse agonist activity (Figs. 2B and 6).
Consideration of Alternative Explanations of Our Data
Several alternative explanations of our data were considered, including (1) the postulated constitutive activity of A1AdoRs is caused by endogenous adenosine in the preparations and therefore all actions of antagonists are due to attenuation of the effects of this agonist; (2) the actions of A1AdoR antagonists to increase cAMP content of intact cells are caused by their inhibition of cAMP phosphodiesterase activity; (3) the putative neutral antagonists N-0861 and WRC-0342 are really inverse agonists but were used at concentrations too low to see their “true” effect. Each of these explanations of our data is discussed below and dismissed.
Endogenous adenosine.
Antagonism by A1AdoR antagonists of the action of endogenous adenosine in both membrane and whole-cell preparations may cause a decrease of [35S]GTPγS binding and an increase of cAMP content that could be falsely interpreted to be evidence of inverse agonism. Therefore, adenosine deaminase (5 units/ml in membrane preparations for [35S]GTPγS binding assay and 2 units/ml in whole-cell preparations) was present in all assays of A1AdoR agonist and antagonist activity. The addition of adenosine deaminase (0.1–10 units/ml) was found to increase cAMP content of CHO:A1AdoR cells (Results and Fig. 3), which suggests that adenosine in the bathing medium was acting on A1AdoRs to inhibit cellular adenylyl cyclase activity. The effect of adenosine deaminase was maximal at 1 unit/ml. In the presence of 2 units/ml of adenosine deaminase, both CPX (50 nm) and WRC-0571 further increased cAMP content of CHO:A1AdoRs (Fig. 3). These actions of CPX and WRC-0571 therefore are unlikely to be due to the antagonism of endogenous adenosine. Also, WRC-0571 and CPX increased the cAMP content of CHO:A1AdoR cells by 74% and 64% in the presence of adenosine deaminase, whereas N-0861 and WRC-0342 increased cAMP content of CHO:A1AdoR cells by only 29% and 0%, respectively (Fig. 5). In contrast, each antagonist caused a similar maximal reduction of [3H]CPX binding to A1AdoRs in CHO:A1AdoR membranes. Furthermore, N-0861 and WRC-0342 antagonized the increases of cAMP content of CHO:A1AdoR cells caused by either CPX or WRC-0571 (Fig. 6). Because all four A1AdoR antagonists are expected to fully and equally attenuate a response mediated by endogenous adenosine, our findings that the actions of CPX and WRC-0571 are clearly different from those of N-0861 and WRC-0342 are not consistent with a mechanism of action involving inhibition of the effect of endogenous adenosine.
Phosphodiesterase activity.
Adenosine receptor antagonists may act to inhibit phosphodiesterase activity and thereby increase cAMP content of cells. This action is a potential explanation of results of our assays of cAMP content of intact cells but not of results of assays of [35S]GTPγS binding to membrane preparations. For example, the phosphodiesterase inhibitor rolipram increased cAMP contents of both CHO:A1AdoR and CHO:Wild cells (Fig. 3) but did not alter binding of [35S]GTPγS. For assay of nonspecific actions (e.g., inhibition of phosphodiesterase activity) of A1AdoR antagonists on cAMP content, CHO:Wild cells were used in this study. Only N-0861 (50 μm) of the four A1AdoR antagonists tested caused an elevation of cAMP content of CHO:Wild cells in the presence of 0.3 μm forskolin. Assuming that N-0861 (50 μm) caused similar nonspecific increases of cAMP content in CHO:A1AdoR and CHO:Wild cells, the small increase in cAMP content of CHO:A1AdoR cells in the presence of 50 μm N-0861 (Fig. 5) also is unrelated to antagonism of A1AdoRs and is likely to be caused by inhibition of phosphodiesterase activity. The A1AdoR antagonists CPX, WRC-0571, and WRC-0342 did not appear to increase cAMP content of CHO:Wild cells at the concentrations tested and therefore are not likely to cause inhibition of phosphodiesterase activity in our experiments. Furthermore, the findings that treatment of CHO:A1AdoR cells with pertussis toxin abolished effects of both A1AdoR agonist and antagonists on cAMP content and that N-0861 and WRC-0342 attenuated increases of cAMP content caused by WRC-0571 and CPX strongly indicate that actions of A1AdoR antagonists in our experiments are not due to inhibition of phosphodiesterase activity but rather to antagonism of A1AdoRs.
Antagonist affinity for A1AdoRs.
The neutral A1AdoR antagonists N-0861, N-0840, and WRC-0342 at concentrations up to 0.1 mm did not reduce binding of [35S]GTPγS to membranes prepared from CHO:A1AdoR cells. In contrast, both antagonists competitively reduced specific binding of [3H]CPX to CHO:A1AdoR cell membranes with Ki values of ∼1 μm. These findings suggest that although the affinities of N-0861, N-0840, and WRC-0342 for the A1AdoR are low in comparison to those of CPX, WRC-0571, XAC, and CGS-15943, the concentrations of each antagonist used in functional assays of [35S]GTPγS binding and cAMP content were sufficiently high to cause occupancy of nearly all A1AdoRs. It therefore is not likely that N-0861, N-0840, or WRC-0342 at higher concentrations than those used in our experiments will act as inverse agonists. This conclusion is supported by the finding that actions of N-0861 and WRC-0342 were not additive with, but were antagonistic of, those caused by CPX and WRC-0571 (Figs. 2B and 6).
In conclusion, when human A1AdoRs are expressed in high density in CHO cells, constitutive activity of these receptors is manifested in the absence of agonist by the occurrence of responses to antagonists. Antagonists with inverse agonist activity, namely CPX and WRC-0571, caused both an increase in cAMP content of intact cells and a decrease of [35S]GTPγS binding to cell membranes. The neutral antagonists N-0861 and WRC-0342 antagonized both the actions of the agonist CPA and the inverse agonist actions of the antagonists CPX and WRC-0571. An assessment of the relevance of these findings is not possible at this time but rather awaits the demonstration of physiological and pathological actions mediated by constitutively active receptors in native tissues. Results of our study indicate potentially useful pharmacological probes and paradigms for investigation of constitutive activity of A1AdoRs in future studies.
Acknowledgments
We thank Peggy Ramsey for preparation of the manuscript; Jackie Ruble and Becky Hamilton for technical assistance with radioligand binding assays and cAMP assays, respectively; Dr. Donn Dennis for assistance with statistical analysis; and Dr. Cynthia Kollias-Baker (University of California School of Veterinary Medicine, Davis, CA) for preparation of clones of CHO cells expressing human A1 adenosine receptors.
Footnotes
-
Send reprint requests to: Luiz Belardinelli, M.D., Department of Medicine/Cardiology, University of Florida, P.O. Box 100277, Gainesville, FL 32610-0277. E-mail: ramsepd{at}medicine.ufl.edu
- Abbreviations:
- A1AdoR
- A1-adenosine receptor
- CPX
- 8-cyclopentyl-1,3-dipropylxanthine
- CHO
- Chinese hamster ovary
- N-0861
- (±)-N6-endonorbornan-2-yl-9-methyladenine
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- CPA
- N6-cyclopentyladenosine
- CPT
- 8-cyclopentyl-1,3-dimethylxanthine
- N-0840
- N6-cyclopentyl-9-methyladenine, XAC, xanthine amine congener
- GTPγS
- guanosine-5′-O-(3-thio)triphosphate
- Received October 23, 1997.
- Accepted January 30, 1998.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}