

Abstract
Astrocytes exhibit a prominent glycolytic activity, but whether such a metabolic profile is influenced by intercellular communication is unknown. Treatment of primary cultures of mouse cortical astrocytes with the nitric oxide (NO) donor DetaNONOate induced a time-dependent enhancement in the expression of genes encoding various glycolytic enzymes as well as transporters for glucose and lactate. Such an effect was shown to be dependent on the hypoxia-inducible factor HIF-1α, which is stabilized and translocated to the nucleus to exert its transcriptional regulation. NO action was dependent on both the PI3K/Akt/mTOR and MEK signaling pathways and required the activation of COX, but was independent of the soluble guanylate cyclase pathway. Furthermore, as a consequence of NO treatment, an enhanced lactate production and release by astrocytes was evidenced, which was prevented by downregulating HIF-1α. Several brain cell types represent possible sources of NO. It was found that endothelial cells, which express the endothelial NO synthase (eNOS) isoform, constitutively produced the largest amount of NO in culture. When astrocytes were cocultured with primary cultures of brain vascular endothelial cells, stabilization of HIF-1α and an enhancement in glucose transporter-1, hexokinase-2, and monocarboxylate transporter-4 expression as well as increased lactate production was found in astrocytes. This effect was inhibited by the NOS inhibitor l-NAME and was not seen when astrocytes were cocultured with primary cultures of cortical neurons. Our findings suggest that endothelial cell-derived NO participates to the maintenance of a high glycolytic activity in astrocytes mediated by astrocytic HIF-1α activation.
Introduction
In recent years, the concept that astrocytes, neurons and endothelial cells forming microvessels are organized into structural neurovascular units became fundamental for the understanding of cerebral blood flow regulation as well as blood–brain barrier (BBB) establishment and modulation (Iadecola, 2004). Astrocytes have been shown to be involved in the regulation and maintenance of physical, transport as well as metabolic characteristics of the BBB (Rubin et al., 1991; Hayashi et al., 1997; Haseloff et al., 2005). On the other hand, as partners in a bidirectional communication, endothelial cells release factors like the leukemia inhibitory factor (LIF) which are crucial for astrocyte differentiation (Mi et al., 2001). Furthermore, endothelial cells provide brain cells with glucose via a transcellular transport involving the glucose transporter isoform GLUT1 (Simpson et al., 2007). In this context, astrocytes have been proposed to provide an important link between endothelial cells and neurons. Coupling of neuronal activity with both vascular and metabolic responses are among the most important proposed functions of astrocytes in these neurovascular units (Zonta et al., 2003a; Giaume et al., 2010; Zhou et al., 2010). According to the astrocyte-neuron lactate shuttle model, astrocytes respond to neuronal activity by converting glucose into lactate that is subsequently taken up by neurons to satisfy their energy needs during activation (Pellerin and Magistretti, 1994). However until now, no explanation has been provided for the prominent glycolytic profile exhibited by astrocytes under these conditions. Considering the tight interactions between the different cell types forming neurovascular units, it could be possible that some intercellular signals determine specific metabolic characteristics in each cell type.
Nitric oxide (NO) has been identified as an important intercellular messenger in the CNS and can be produced by most brain cells which include astrocytes, neurons and endothelial cells (Garthwaite and Boulton, 1995). NO was shown to play a critical role in numerous brain functions such as the regulation of vascular tone (Akgören et al., 1996) as well as long-term potentiation (Hopper and Garthwaite, 2006). NO was also found to modulate energy metabolism. Thus, NO inhibits oxidative metabolism and enhances glycolysis in astrocytes but not in neurons (Almeida et al., 2001). More recently, it was shown that NO stimulates the expression of the monocarboxylate transporter MCT4 in astrocytes, an effect that was accompanied by a long-lasting enhancement in lactate production and release (Marcillac et al., 2011). Such an observation suggests that NO might coordinate the transcriptional activation of several genes related to glycolysis, although the specific transcription factor involved is unknown. Moreover, the cellular source of NO that could be responsible for modulating the metabolic profile of astrocytes has not been determined. Here we provide evidence that NO produced by brain endothelial cells may be a key factor to maintain elevated aerobic glycolysis capacity in astrocytes via HIF-1α activation.
Materials and Methods
If not indicated otherwise, chemicals and cell culture media were purchased from Sigma-Aldrich or Invitrogen.
Cell cultures and treatments
The immortalized mouse brain endothelial capillary cell line bEnd.3 was purchased from ATCC and cultured in DMEM supplemented with 10% fetal calf serum. Chinese hamster ovarian cells (CHO) were obtained from ATCC and cultured in HAM's F12 with 5% FCS. Murine primary cortical astrocyte-enriched, neuronal cultures or endothelial cells from brain capillaries were prepared from male C57BL/6J mice (The Jackson Laboratory).
Astrocytes.
Primary cortical astrocyte-enriched cultures were prepared from mice at postnatal day 1 as previously described (Sorg and Magistretti, 1992). Following isolation of cortices and removal of the meninges, dissociated cells were suspended in DMEM supplemented with 10% FCS and 1% 100 × Antibiotic-Antimycotic (v/v). Cells were seeded on 24-well imaging plates (zell-kontakt) with a gas-permeable bottom to avoid hypoxia in astrocytes. Experiments were performed after 21 (±2) d in culture at 37°C and 5% CO2. Medium was exchanged every 3–4 d.
Neurons.
Primary cortical neuronal cultures were obtained from mice at embryonic day 18 as described previously (Zerarka et al., 2001). In brief, cortices were isolated and neurons mechanically dissociated. Primary neurons were cultured in neurobasal medium supplemented with 1% B27 supplement and 0.25% l-glutamate. 2 × 105 or 3 × 105 cells were seeded per well on poly-l-ornithine-coated 24-well or 12-well plates, respectively. Experiments were performed after 7 d in culture at 37°C and 5% CO2.
Endothelial cells.
Endothelial cells from brain capillaries were isolated from 12-week-old mice (Song and Pachter, 2003). Briefly, brains were freed from meninges, homogenized and the cell suspension was centrifuged with 18% dextran solution to clear the suspension from myelin. The cell suspension was digested for 1 h 15 min at 37°C in DMEM containing 2% FCS, 0.1% collagenase/dispase (0.1 U/ml, 0.8 U/ml, respectively), 0.1% DNaseI, and 0.147 μg/ml tosyllysine chloromethyl ketone. Cells were washed with phosphate buffer saline (PBS) and resuspended in culture medium (DMEM, 20% FCS, 1% Antibiotic-Antimycotic (v/v), 1% l-glutamine (v/v), 1% endothelial cell growth supplement (ECGS), 50,000 U heparin) and 8 μg/ml puromycin. Medium was exchanged on Days 1 and 2 in culture. After Day 2, puromycin was omitted and medium was changed every 3 d. Cell suspension from one brain was seeded per well of a 12-well plate. Experiments were performed when cells reached confluence, usually after 1 week in culture at 37°C and 5% CO2.
Treatment.
Primary mouse cortical astrocytes were treated with 0.8 mm DetaNONOate (Enzo Life Sciences) and 20 μm of the phosphatidylinositol 3-kinase (PI3K) inhibitor LY294002 (2-[4-morpholinyl]-8-phenyl-1[4H]-benzopyran-4-one hydrochloride), 25 μm the mitogen-activated protein kinase kinase (MEK) inhibitor PD98059 (2-[2-amino-3-methoxyphenyl]-4H-1-benzopyran-4-one), 100 nm of the mTOR (mammalian target of rapamycin) blocker rapamycin, 20 μm of the guanylate cyclase inhibitor ODQ (1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one), 10 or 100 μm of nonselective inhibitor of cyclooxygenase (COX) indomethacin, or the selective COX-2 inhibitor celecoxib for 30 min before DetaNONOate treatment when indicated. Control experiments included the use of SulfoNONOate (does not release NO), degraded DetaNONOate and the NO scavenger CPTIO [2-(4-carboxyphenyl)-4,4,5,5-tetramethyl-imidadazoline-1-oxyl-3-oxide].
Coculture experiments.
For transwell coculture experiments, astrocytes were seeded in 12 well inserts (BD; 0.4 μm pore size) and cultured for 21 d. Inserts containing astrocytes were incubated for 12 h with 3 × 105 astrocytes, neurons, primary endothelial cells or bEnd.3 cells plated on 12 well plates (see Fig. 6C). During coculture 500 μm l-arginine was added to the corresponding medium as a substrate for nitric oxide synthase (NOS). For inhibition studies bEnd.3 cells or endothelial cells were preincubated for 30 min with 100 μm of the NOS inhibitor l-NAME (N-nitro-l-arginine methyl ester).
Fluorescence microscopy
Cells grown on Imaging Plates (zell-kontakt) were fixed with 4% ice-cold formaldehyde for 15 min, permeabilized with 0.3% Triton X-100 (v/v) in PBS and blocked for 1 h with 2% bovine serum albumin (BSA) (w/v) in PBST (0.1% Tween 20 (v/v). Primary antibodies were diluted in blocking buffer and incubated with cells for 1 h at room temperature. Subsequently, cells were washed and incubated with Alexa Fluor labeled secondary antibody in 2% BSA (w/v) in PBS for 1 h at room temperature. Nuclei were stained with 2 μg/ml DAPI. Cells were washed with PBS and microscopic analysis was performed immediately with the confocal laser-scanning microscope TCS SP5 (Leica). The following antibodies were used: rabbit anti-HIF-1α 1:500 (Santa Cruz Biotechnology), mouse anti-GFAP 1:1000 (Sigma), goat anti-rabbit Alexa 555 (Invitrogen) and goat anti-mouse Alexa 488 1:2000 (Invitrogen).
Transfection and siRNA knockdown
For transfection, Lipofectamine 2000 was used according to manufacturer's instructions. Transfection mixtures were prepared in a final volume of 100 μl/well OptiMEM medium containing 50 ng siRNA and 10 μg/ml of Lipofectamine 2000. Cells were incubated for 5 h at 37°C and 5% CO2. Subsequently, the transfection medium was replaced by complete medium without antibiotics and cells were incubated for 20 h at 37°C and 5% CO2. For the siRNA silencing Stealth/ siRNA duplex oligonucleotides specific for HIF-1α (5′-CAAGCAGCAGGAAUUGGAACAUUAU-3′) and control siRNA (5′-CAACGAGGAUAAGGUCAAUAGCUAU-3′) were purchased from Invitrogen.
Quantitative RT-PCR
For the quantification of mRNA, cells were washed with PBS, lysed and total RNA was extracted using the 6100 nucleic prepstation (Applied Biosystems) following the manufacturer's protocol. Total RNA (9 μl) was used for cDNA synthesis with the Cloned AMV First Strand Synthesis Kit (Invitrogen) according to the manufacturer's specifications. qRT-PCR was accomplished with Platinum SYBR Green qPCR Supermix (Invitrogen) and 2 μl cDNA in a volume of 25 μl on the ABI Prism 7000 Sequence Detection System (Applied Biosystems). Expression values were normalized to total RNA. For detection of specific cDNA, oligonucleotide primers were used as indicated (Table 1).
Sequences of primers used for RT-PCR of mouse RNA (orientation 5′ to 3′)
Western blot analysis
Cells were washed with PBS, lysed in 8 m Urea buffer (8 m Urea, 9.5 m Glycerin, 0.7 m SDS, 1 m DTT, 0.5 m Tris-HCl, pH 6.8, 22 mm Aprotinin, 2.2 mm Leupeptin, 1.46 mm Pepstatin), homogenized by centrifugation through Qiashredder tubes (Qiagen) and denatured for 5 min at 95°C in 4 × LDS Sample buffer (Invitrogen). Proteins were separated by 4–12% BIS-Tris gels (Invitrogen) and transferred onto PVDF membrane (Millipore). To detect nonphosphorylated proteins membranes were blocked for 30 min with 5% skim milk (w/v) in washing buffer (65 mm Na2HPO4 * 2 H2O, 25 mm NaH2PO4 * H2O, 100 mm NaCl, 0.1% Tween (v/v), pH7,5). For detection of phosphorylated proteins, a blocking buffer containing 1% BSA (w/v), 10 mm NaF, 2 mm Na3VO4 and 5 mm Na4P2O7 in TBST (10 mm Tris-HCl, pH 7.5, 150 mm NaCl and 0.1% Tween 20 (v/v)) was used. Membranes were incubated overnight in presence of the primary antibody diluted in appropriate blocking buffer at 4°C. The following antibodies were used: goat anti-β-actin 1:1000 (Santa Cruz Biotechnology), rabbit anti-HIF-1α 1:500 (Cayman Chemicals), rabbit anti-phospho-p70S6 kinase (Thr389) 1:500 (Cell Signaling Technology), rabbit anti-p70S6 kinase (Thr389) 1:1000 (Cell Signaling Technology), rabbit anti-Akt 1:1000 (Cell Signaling Technology) 1:1000, rabbit anti-phospho-Akt (Ser473) 1:1000 (Cell Signaling Technology). Afterward, the membranes were washed 4 times for 10 min with washing buffer and then incubated with either HRP-goat anti-rabbit IgG (DAKO) or HRP-rabbit anti-goat IgG (DAKO) 1:2000 for 60 min at room temperature. Blots were washed 4 times for 10 min with washing buffer and processed using the SuperSignal West Femto Maximum Sensitivity substrate (Pierce) in a 1:5 dilution. Chemiluminescence was detected and quantified using ImageJ software (NIH, Wayne Rasband, version 1.45e).
Measurement of NO
To determine total NO production in the supernatant of different cell types nitrite and nitrate were measured using the Nitrate/Nitrite fluorometric assay kit (Cayman Chemicals). Cells were seeded in equal cell numbers and the medium of different the cell types was replaced by fresh medium supplemented with 500 μm l-arginine. Samples were collected after 12 h of culture. The assay was performed as described by the manufacturer using 50 μl of medium. Fluorescence was measured with excitation wavelength of 360 nm and emission wavelength of 430 nm.
Lactate assay
To measure lactate release in the supernatant of astrocytes the medium was renewed and astrocytes treated as indicated. Samples were taken after 24 h. For lactate measurement after coculture astrocytes were cocultured with the indicated cells for 12 h. After separation of astrocytes from cocultured cells the medium was exchanged. Medium samples were taken after 6 h.
Medium (100 μl) was incubated for 5 min with 500 μl 0.3 m HClO4 to precipitate proteins from samples. After 5 min of centrifugation (13,000 rpm) 450 μl of clear supernatant were neutralized with 100 μl saturated KHCO3 solution on ice. Samples were centrifuged (13,000 rpm) and 150 μl of supernatant were incubated with 650 μl test buffer (250 nmol/L Tris-glutamate pH 8.75, 3.6 nmol/L NAD+) and 15 μl enzyme solution (400 U/ml lactate dehydrogenase (LDH) (Roche), 24 U/ml glutamate-pyruvate-transaminase (GPT) (Roche) in 0.1 m Tris buffer) for 30 min at room temperature. Absorbance was measured at 340 nm. Lactate release was normalized to protein content/well, which was measured by the method of Lowry et al. (1951).
Statistics
All data are presented as mean ± SD. All experiments were repeated at least three times. Statistical comparison between groups was done using the Student's t test or appropriate ANOVA followed by Bonferroni post-test.
Results
NO selectively enhances mRNA expression of glycolytic enzymes and glucose/lactate transporters in astrocytes
Application of the NO donor DetaNONOate (0.8 mm) on primary cultures of mouse cortical astrocytes led to a time-dependent increase in mRNA levels of genes coding for a variety of glycolysis-associated enzymes that include hexokinase-1 (HK1), hexokinase-2 (HK2), phosphofructokinase kinase L (PFKL), 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), aldolase A (ALDOA), phosphoglycerate kinase 1 (PGK1), enolase (ENO1), lactate dehydrogenase A (LDHA), and pyruvate dehydrogenase kinase 1 (PDK1) (Fig. 1A). Moreover, DetaNONOate enhanced the mRNA expression of glycolysis-related transporters such as the glucose transporter-1 (GLUT1) and the monocarboxylate transporter-4 (MCT4) (Fig. 1A) implying not only an increased glucose uptake into astrocytes but also enhanced glycolysis, lactate production and release (Fig. 1B). In contrast, DetaNONOate did not enhance the mRNA expression of other genes like hexokinase-2 (HK2), lactate dehydrogenase B (LDHB), and the monocarboxylate transporters MCT1 and MCT2 (Fig. 1A).

NO increases mRNA expression of selected glycolytic enzymes and glucose/lactate transporters in astrocytes. A, Primary cultures of murine cortical astrocytes were treated with 0.8 mm DetaNONOate for the indicated time periods (3, 12, 24 h) compared with untreated controls (C). B, glycolytic pathway and DetaNONOate regulated transporter/enzymes as indicated by arrows. Each data point represents the mean ± SEM of triplicate samples from one experiment. The experiment was repeated three times on different cell preparations with similar results. Glc, glucose; G6P, glucose 6-phosphate; F6P, fructose 6-phosphate; F1,6BP, fructose 1,6-bisphosphate; F2,6BP, fructose 2,6-bisphosphate; GADP, glyceraldehyde 3-phosphate; 1,3-BPG, 1,3-bisphosphoglycerate; 3PG, 3-phosphoglycerate; 2PG, 2-phosphoglycerate; PEP, phosphoenolpyruvate; Pyr, pyruvate; AcCoA, acetyl-CoA; Lac, lactate.
NO modulates glycolytic gene expression in astrocyte via HIF-1α stabilization
HIF-1α is a transcriptional regulator of hypoxia-induced glycolysis that is constitutively destabilized under normoxic conditions (Semenza et al., 1994). However, it has been shown that HIF-1α can be stabilized by NO in normoxia (Kasuno et al., 2004). Therefore, the putative involvement of HIF-1α in NO-regulated gene expression was investigated in cultures of astrocytes. Application of DetaNONOate on astrocytes led to a time- and concentration-dependent HIF-1α stabilization (Figs. 2A,B). HIF-1α protein was most efficiently stabilized after 6–12 h of DetaNONOate treatment. HIF-1α was already detected at a concentration of 0.4 mm DetaNONOate with a maximal effect between 0.8 and 1.2 mm. In contrast to the effect observed in astrocytes, NO-induced HIF-1α protein stabilization could not be detected in mouse primary cultures of cortical neurons (Fig. 2C). HIF-1α translocates to the nucleus after activation, where it regulates the transcription of various genes. Using immunocytochemistry HIF-1α was found not only to stabilize but also to be located within the nucleus of DetaNONOate-treated astrocytes (Fig. 2D). The NO scavenger CPTIO, at 250 μm, largely reduced the amount of HIF-1α protein stabilized by 12 h of DetaNONOate treatment (Fig. 3A) and abolished the induction of MCT4 mRNA (Fig. 3B). No effects on MCT4 mRNA expression were observed after 12 h of treatment with SulfoNONOate or degraded DetaNONOate (Fig. 3B).

NO stabilizes HIF-1α protein in astrocytes. A–C, HIF-1α protein levels were obtained by Western blot analysis of primary astrocytes treated with 0.8 mm DetaNONOate (DETA) for the indicated time periods (A), increasing concentrations of DetaNONOate for 12 h (B), or with 0.8 mm DetaNONOate for 12 h in comparison with DetaNONOate-treated primary cortical neurons (C). β-actin served as loading control. D, Immunocytochemical staining of HIF-1α in primary cultures of mouse cortical astrocytes treated with 0.8 mm DetaNONOate for 12 h compared with untreated controls (Ctr).

Inhibition of DETANONOate-induced HIF-1α stabilization and MCT4 mRNA expression by CPTIO. Primary astrocytes were pretreated for 30 min with 100 or 250 μm of the NO scavenger CPTIO or without scavenger before application of 0.8 mm DetaNONOate (DETA) for 12 h and Western blot analyses for HIF-1α and β-actin (A) as well as qRT-PCR for MCT4 expression (B) were performed. Control experiments included the treatment with SulfoNONOate (SULFO) and DetaNONOate that was degraded for 72 h in B. A representative Western blot for HIF-α and β-actin is shown from three independent experiments. MCT4 mRNA levels are shown as mean ± SEM of triplicate samples, repeated three times on different cell preparations with similar results.
To analyze the dependency of NO-induced glycolytic enzyme and glucose/lactate transporter expression on HIF-1α, HIF-1α was knocked down in cultured astrocytes using a HIF-1α-specific siRNA. HIF-1α mRNA levels were reduced by 90% using a specific HIF-1α siRNA compared with the application of an unspecific control siRNA (Fig. 4A). Treatment with HIF-1α-specific siRNA prevented the effect of DetaNONOate on HIF-1α protein stabilization while an unspecific control siRNA had no effect on NO-induced HIF-1α protein stabilization (Fig. 4B). Transfection of cultured astrocytes with the HIF-1α-specific siRNA before exposure with DetaNONOate greatly reduced or abolished the NO-induced increase of mRNA levels for the glycolytic enzymes HK2, PFKL, PFKFB3, PDK1, ALDOA, PGK1, ENO1, and LDHA as well as the glucose/lactate transporters GLUT1, GLUT3, and MCT4 (Fig. 4C). The expression levels of other genes like HK1 (Fig. 4C) and β-actin (data not shown) were not affected by HIF-1α siRNA with or without NO treatment.

HIF-1α siRNA knockdown prevents NO-induced gene expression. A, B, Primary astrocytes were transfected with HIF-1α-specific siRNA or unspecific scrambled siRNA (Scr) 24 h before DetaNONOate treatment and HIF-1α knockdown was confirmed by qRT-PCR (A) and Western blot analysis (B). C, mRNA expression levels of glycolytic enzymes and glucose/lactate transporters were obtained by qRT-PCR 12 h after application of 0.8 mm DetaNONOate (DETA) and compared with untreated controls (C). Each data point represents the mean ± SEM of triplicate samples from one experiment. The experiment was repeated three times on different cell preparations with similar results.
NO-dependent HIF-1α stabilization and transcriptional activation of glycolytic genes in astrocytes require the PI3K/Akt signaling pathway and COX activity, but not soluble guanylate cyclase signaling
Soluble guanylate cyclase (sGC) produces intracellular cGMP and is a main target of NO in many cell types. Therefore, to test the implication of this signaling cascade, astrocytes were treated with 100 μm of the sGC inhibitor ODQ before stimulation with DetaNONOate. ODQ was unable to neither prevent NO-induced HIF-1α stabilization (Fig. 5A) nor target gene expression, as exemplified by MCT4 gene expression (Fig. 5B). Concomitantly, the cGMP analog 8-Bromo-cGMP had no effect on MCT4 mRNA levels (Fig. 5B). DetaNONOate increased phosphorylation of Akt and the downstream transcriptional activator pS6K in astrocytes, an effect that was reduced by the PI3K inhibitor LY294002 as well as the mTOR inhibitor rapamycin (data not shown). Indeed, pretreatment of astrocytes with LY294002 and rapamycin, but also with the MEK-inhibitor PD98059 reduced NO-induced HIF-1α protein stabilization significantly indicating the involvement of this pathway. (Fig. 5C). In parallel, NO-induced MCT4 gene expression reflecting glycolytic genes sensitive to NO, was reduced in astrocytes pretreated with each of these inhibitors (Fig. 5D).

PI3K/Akt/mTor and MAPK kinase but not soluble guanylate cyclase pathways are involved in NO-mediated HIF-1α stabilization. A, B, Primary astrocytes were pretreated for 30 min with 100 μm selective guanylate cyclase inhibitor ODQ or without inhibitor before application of 0.8 mm DetaNONOate (DETA) or 8-Bromo-cGMP (8-Br-cGMP) for 12 h and Western blot analyses for HIF-1α and β-actin as loading control (A) as well as qRT-PCR for MCT4 expression (B) was performed. C, D, The effect of kinase inhibitors on DetaNONOate-dependent HIF-1α protein stabilization (C) and MCT4 mRNA expression (D) was analyzed in astrocytes pretreated with LY294002 (L), Rapamycin (R), PD98059 (P), or without inhibitor 30 min before addition of 0.8 mm DetaNONOate. E, F, The effect of COX inhibitors on DetaNONOate-dependent HIF-1α protein stabilization (E) and MCT4 mRNA expression (F) was analyzed in astrocytes pretreated with indomethacin (I) or celecoxib (C), or without inhibitor 30 min before addition of 0.8 mm DetaNONOate. Shown are representative Western blots for HIF-α and β-actin. HIF-1α was normalized to β-actin and quantification was performed on two to three independent experiments shown as mean ± SEM (*p < 0.05, **p < 0.01, and ***p < 0.001 compared with DetaNONOate alone). MCT4 mRNA levels are shown as mean ± SEM of triplicate samples, repeated three times on different cell preparations with similar results.
Prostaglandin E2 (PGE2) released by astrocytes plays a pivotal role in arteriole relaxation at the neurovascular unit (Zonta et al., 2003a, 2003b). As PGE2 was shown to induce HIF-1α and COX-2 inhibitors attenuated the PI3K/Akt-mediated effect of IL-1 on HIF-1α induction (Jung et al., 2003), we tested whether COX activation is involved in NO-dependent HIF-1α stabilization. In fact, NO-induced HIF-1α protein stabilization was reduced or abolished when astrocytes were pretreated with 100 μm of the nonselective COX inhibitor indomethacin or the selective COX-2 inhibitor celecoxib, respectively (Fig. 5E) and this effect was paralleled by largely reduced MCT4 mRNA expression (Fig. 5F).
Endothelial cells promote HIF-1α stabilization and glycolytic gene expression in astrocytes via NO production
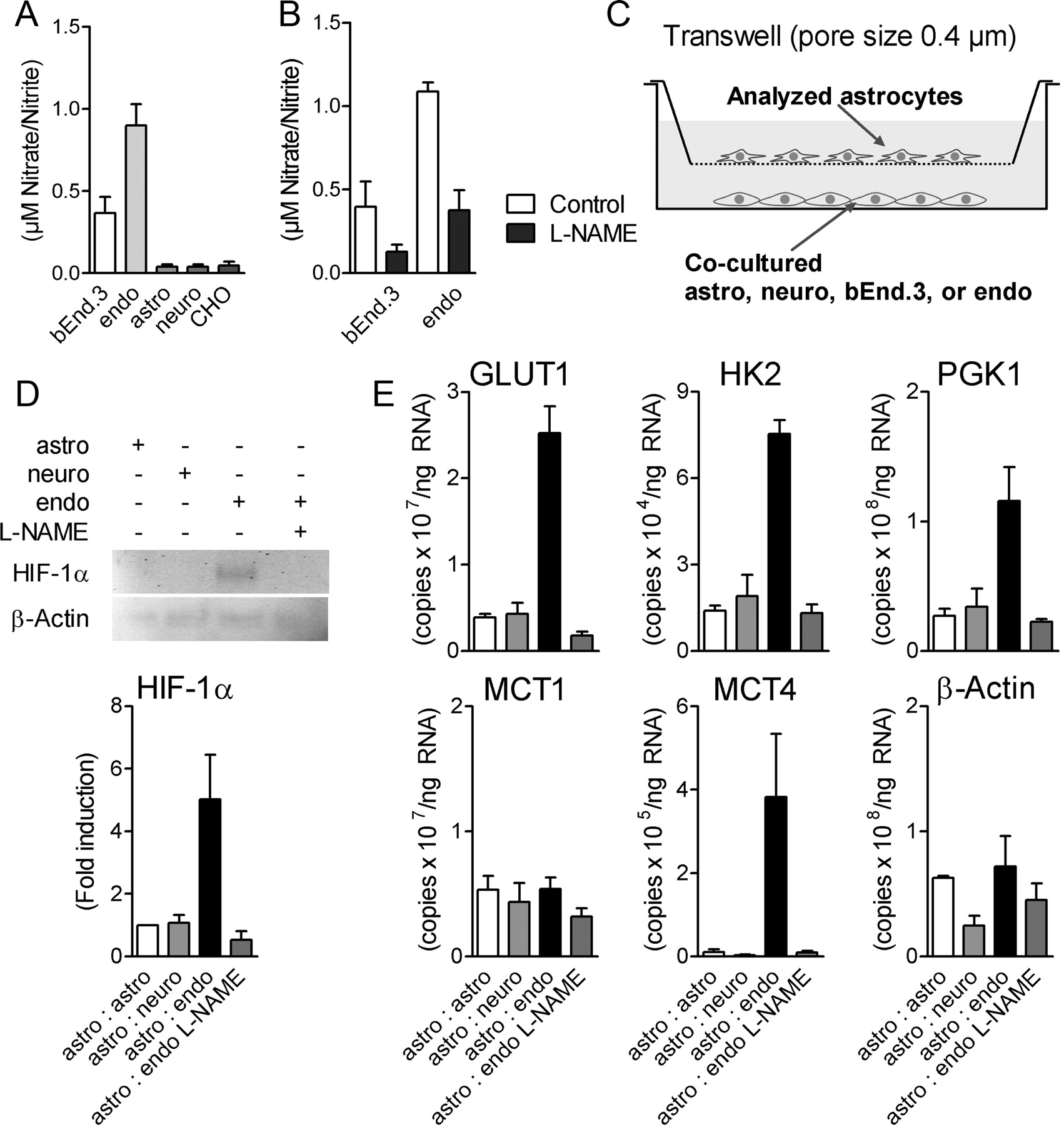
To identify the possible source of cell-derived NO, the production of NO by different brain cells in culture was evaluated by measuring the accumulation in their supernatant of two stable NO metabolites, nitrite and nitrate. bEnd.3 cells (a cell line derived from endothelial cells) and, to a larger extent, primary cultures of mouse endothelial cells derived from brain capillaries constitutively produced significant amounts of NO while primary cultures of mouse cortical astrocytes or neurons as well as CHO cells that served as negative control did not produce significant amounts of NO products (Fig. 6A). Treatment of bEnd.3 cells or endothelial cells with 100 μm of the NOS inhibitor l-NAME reduced their NO production (Fig. 6B).

HIF-1α and HIF-1α target gene expression is induced in astrocytes by coculture with endothelial cells. A, B, NO production in bEnd.3 cells, primary endothelial cells (endo), primary astrocytes (astro), primary neurons (neuro), and CHO cells after culturing for 12 h (A) and after additional treatment of bEnd.3 cells and primary endothelial cells with 100 μm l-NAME (B) as analyzed by nitrite/nitrate measurement. C–E, HIF-1α (D) and HIF-1α target gene mRNA levels (E) were analyzed in lysates from astrocytes (inset) cocultured for 12 h with indicated cells as shown in C by Western blot analysis or qRT-PCR. Primary endothelial cells were also pretreated with 100 μm l-NAME before coculture. Each data point represents means ± SEM from two to three independent experiments. MCT4 mRNA levels are shown as mean ± SEM of triplicate samples, repeated three times on different cell preparations with similar results.
The capacity of these different cell types to cause HIF-1α stabilization and enhance glycolytic gene expression in astrocytes was investigated in a coculture system. Primary cultures of cortical astrocytes were seeded in transwell inserts and cocultured cells were seeded on the bottom of the corresponding culture plates (Fig. 6C). Coculture of primary cultures of endothelial cells with astrocytes for 12 h led to HIF-1α stabilization in astrocytes (Fig. 6D). Furthermore, coculture with endothelial cells enhanced the expression of GLUT1 and MCT4 as well as glycolytic enzymes HK2 and PGK1 in astrocytes (Fig. 6E). These effects were abolished by pretreatment with l-NAME before coculture (Fig. 6D,E). No such effect was observed in astrocytes cocultured with neurons. Similar results were obtained when bEnd.3 cells were used for cocultures instead of primary cultures of endothelial cells (data not shown). In contrast to the mRNA levels of GLUT1/MCT4 and glycolytic enzymes, mRNA levels of MCT1 and β-actin in astrocytes were not changed by coculture with endothelial cells (Fig. 6E).
Endothelial cells induce lactate release from astrocytes via NO signaling
To evaluate the functional consequence of enhanced expression of glycolytic enzymes and glucose/lactate transporters, the effect of DetaNONOate on lactate production was measured in cultured astrocytes. After 24 h of treatment with DetaNONOate, a significant increase in lactate accumulation was observed (Fig. 7A). Transfection with HIF-1α-specific siRNA prevented the NO-induced increase in lactate (Fig. 7A). Finally, coculture of astrocytes with endothelial cells led to an increased lactate release in astrocytes compared with astrocytes cocultured with astrocytes or neurons (Fig. 7B). This effect was abolished by l-NAME.

NO and coculture with endothelial cells stimulate lactate release from astrocytes. A, Primary astrocytes were transfected with Hif-1α siRNA or scrambled (Src) control siRNA 24 h before the application of 0.8 mm DetaNONOate (DETA). After 24 h medium samples were taken and lactate was measured. B, Primary astrocytes were cocultured for 12 h with primary endothelial cells (endo), primary astrocytes (astro), or primary neurons (neuro) and lactate was measured in fresh medium after 6 h of incubation of separated astrocytes. (n = 6, ***p < 0.001 compared with untreated controls (C) or l-NAME-treated cells. Results were from two independent experiments.
Discussion
It has been well documented that astrocytes and neurons have a different metabolic profile, both at rest and upon activation (Pellerin et al., 2007; Pellerin and Magistretti, 2011). At rest, astrocytes have a higher glycolytic activity than neurons, an observation which is supported by results obtained with transcriptomic analysis on acutely isolated cells indicating a predominant expression of genes encoding glycolytic enzymes in astrocytes versus neurons (Rossner et al., 2006; Lovatt et al., 2007; Cahoy et al., 2008). In addition, recent results in vivo also suggest that astrocytes, in contrast to neurons, respond to neuronal activation by exhibiting a predominant glycolytic response (Chuquet et al., 2010). Whether such a distinctive feature depends only on a constitutive difference in expression of genes encoding glycolytic enzymes or whether it can be further reinforced by external signals was so far unknown. Our results provide support for the second possibility. Indeed, a selected group of genes encoding either glycolytic enzymes or transporters for glucose and lactate were found to be upregulated in cultured astrocytes by NO. In a previous report, NO was shown to induce the expression of the monocarboxylate transporter MCT4 in cultured astrocytes (Marcillac et al., 2011). The present data not only confirm this observation but further extend it to include this time a selective group of proteins belonging to the same pathway and identifies HIF-1α as the responsible transcription factor. Thus, it appears that the basal metabolic profile of astrocytes can be reinforced by an exogenous signal such as NO.
The transcription factor HIF-1 is a heterodimeric complex comprising the subunits HIF-1α and HIF-1β (Wang et al., 1995). HIF-1 controls transcription of genes involved in crucial aspects of cellular function like glucose and iron metabolism, energy homeostasis or vascularization. At physiological oxygen concentrations, two proline residues of HIF-1α (Pro-402 and Pro-564) are hydroxylated by prolyl hydroxylases (PHDs) (Schofield and Zhang, 1999). As a consequence, von-Hippel-Lindau protein binds to HIF-1α and ubiquitin-dependent proteasomal degradation is triggered (Kamura et al., 2000). Under hypoxic conditions PHDs are inactivated, which leads to HIF-1α stabilization. Upon stabilization, HIF-1α translocates to the nucleus where it dimerizes with HIF-1β and binds to hypoxia response elements (HRE) contained in the promoter of target genes such as enzymes and transporters involved in glycolysis (Semenza, 1999). In addition to hypoxia, some chemical compounds like deferoxamine and carbon monoxide have been described to stabilize HIF-1α under normoxic conditions (Wang and Semenza, 1993; Kasuno et al., 2004; Choi et al., 2010). NO has been shown also before to have an impact on HIF-1α stabilization under normoxic conditions (Sandau et al., 2001; Kasuno et al., 2004; Park et al., 2008) and destabilization under hypoxic conditions (Sogawa et al., 1998). Here, we detected HIF-1α stabilization under normoxic conditions in NO-treated astrocytes, but not in neurons. This is in accordance with earlier findings showing that NO induces glycolysis in astrocytes but not in neurons (Almeida et al., 2001). In primary astrocytes, the NO-induced enhancement of glycolysis was mediated by HIF-1α via the PI3K/AKT/mTOR pathway but independent of soluble guanylate cyclase as also shown in other cell types before (Sandau et al., 2000, 2001). Most interestingly, induction of HIF-1α via the PI3K/AKT/mTOR pathway had been shown previously to involve COX-2 activation in a lung cancer cell line (Jung et al., 2003). The NO-dependent stabilization of HIF-1α observed in the present study was also abolished by the COX-2 inhibitor celecoxib suggesting the involvement of prostaglandins which are also central astrocytic mediators of neurovascular coupling at the gliovascular unit (Zonta et al., 2003a, 2003b). However, the detailed mechanistic interactions between NO, the PI3K/AKT/mTOR pathway and COX activation for the regulation of HIF-1α stabilization in astrocytes remain to be clarified in future studies.
Our findings imply a specific regulation of glycolysis in astrocytes, as opposed to neurons, mediated at least in part by HIF-1α. In fact, it is not only NO-induced gene expression of glycolytic enzymes that is dependent on HIF-1α, but also lactate production and release. Interestingly, it was previously shown that NO enhances lactate production and release by astrocytes, but the mechanism invoked involved inhibition of respiration and activation of the glycolytic enzyme phosphofructokinase (Almeida et al., 2004). These two mechanisms are not mutually exclusive but probably represent temporally distinct levels of regulation. For example, respiration can be acutely reduced by NO via its inhibition of cytochrome c oxidase (Bolaños et al., 1994). In parallel, the activity of pyruvate dehydrogenase (and thus of the TCA cycle as well as of oxidative phosphorylation) is regulated by the enzyme pyruvate dehydrogenase kinase 1 (PDK1). NO treatment enhanced PDK1 expression in astrocytes (and not in neurons), providing another mean to reduce respiration while favoring glycolysis. In this regard, NO could represent a signal exerting either transient or long-lasting modifications in metabolic activity, depending of the requirements of neighboring cells and the type of activation.
In the CNS, NO is produced by three NO synthases that are expressed in neurons (nNOS), astrocytes (iNOS) and endothelial cells (eNOS) (Bredt et al., 1990; Förstermann et al., 1991). Taking into account the tight spatial structure in neurovascular units, we hypothesized that NO from one of those cell types could be a sufficient factor to stabilize HIF-1α in astrocytes, setting astrocytes in an enhanced glycolytic status. Endothelial cell-produced NO takes part in modulation of leukocyte-endothelial interaction, smooth muscle cell proliferation and regulation of the local vascular tone, i.e., vasodilation (Atochin and Huang, 2010). In the brain, 99% of the capillaries are enwrapped tightly by astrocytic end-feet and intense interactions have been shown in this gliovascular unit (Abbott et al., 2006). In our studies we could demonstrate that primary endothelial cells constitutively produce more NO then astrocytes or neurons. We could demonstrate that coculture of astrocytes with endothelial cells stabilizes HIF-1α in astrocytes and induces MCT4 mRNA expression. On the contrary, coculture with cortical neurons or astrocytes had no such effect, suggesting that endothelial cells represent the main source of NO. However, we cannot exclude that neurons or astrocytes with higher nNOS/iNOS activity or stimulated cells could also produce NO in amounts sufficient to stabilize HIF-1α in surrounding astrocytes.
It is well established that NO produced by endothelial cells induces vasodilatation of local capillaries allowing a certain local glucose flux. Based on our findings, we propose an extended model of the neurovascular unit that includes glucose metabolism and provides a molecular mechanism explaining production and release of lactate in astrocytes (Fig. 8). In this model, endothelial cell-derived NO, via its stabilization of HIF-1α and subsequent increase in expression of glycolytic enzymes and glucose/lactate transporter in nearby astrocytes, leads to enhanced glial glucose uptake via GLUT1, enhanced glycolysis and lactate production as well as release via MCT4. A fast lactate release from astrocytes to activated neurons, as was previously demonstrated to be triggered both by glutamate (Pellerin and Magistretti, 1994) and K+ (Bittner et al., 2011), should be facilitated if astrocytes are already in a preferred glycolytic status. The gliovascular unit would be able, by these mechanisms, to satisfy energy needs of most brain cell types.

Model of stimulated lactate production in astrocytes by nitric oxide (NO) released from endothelial cells. NO stabilizes HIF-1α in astrocytes leading to an increased expression of key glycolytic enzymes and glucose/lactate transporters. Glucose uptake from arterial supply is increased in astrocytes via GLUT1, lactate production is increased through enhanced glycolysis and export of lactate from astrocytes involves MCT4.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft (DFG) KFO-126 Grants ME 2741/2 (to J.R.M.) and JO 567/1-3 (to O.J.). We thank Christine Eichholz and Cornelia Magnussen for excellent technical assistance.
- Correspondence should be addressed to either of the following Dr. Olaf Jöhren, Institute of Experimental and Clinical Pharmacology and Toxicology, University of Lübeck, Ratzeburger Allee 160, D-23538 Lübeck, Germany, joehren{at}uni-luebeck.de, or Prof. Luc Pellerin, Département de Physiologie, 7 Rue du Bugnon, CH-1005 Lausanne, Switzerland. Luc.Pellerin{at}unil.ch





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}