


Abstract
Excretion of conjugated bile acids into bile is an essential function of the liver, and impairment of canalicular bile acid secretion leads to cholestatic liver injury. However, hepatic excretory function cannot be quantified in vivo because of the lack of suitable methods. Cholylsarcosine is an analog of the endogenous bile acid conjugate cholylglycine and exhibits characteristics in vivo that led us to hypothesize that the 11C-labeled form, that is, [N-methyl-11C]cholylsarcosine (11C-cholylsarcosine), would be a suitable PET tracer for quantification of hepatic excretory function. Methods: A method for radiosynthesis of 11C-cholylsarcosine was developed involving 11C-methylation of glycine followed by conjugation with cholic acid. Blood-to-liver uptake and liver-to-bile excretion were investigated in vivo by dynamic 11C-cholylsarcosine PET/CT of 2 anesthetized pigs. In pig 1, a second dynamic 11C-cholylsarcosine PET/CT examination was preceded by a high dose of the endogenous bile acid conjugate cholyltaurine to investigate possible inhibition of the transhepatocellular transport of 11C-cholylsarcosine. In pig 2, a second 11C-cholylsarcosine administration was given to determine the biodistribution of the tracer by means of 5 successive whole-body PET/CT recordings. Possible formation of 11C-metabolites was investigated by analysis of blood and bile samples from a third pig. Results: The radiochemical yield was 13% ± 3% (n = 7, decay-corrected) and up to 1.1 GBq of 11C-cholylsarcosine was produced with a radiochemical purity greater than 99%. PET/CT studies showed rapid blood-to-liver uptake and liver-to-bile excretion of 11C-cholylsarcosine, with radioactivity concentrations being more than 90 times higher in the bile ducts than in liver tissue. Cholyltaurine inhibited the transhepatocellular transport of 11C-cholylsarcosine, indicating that the tracer is transported by one or more of the same hepatic transporters as cholyltaurine. 11C-cholylsarcosine underwent an enterohepatic circulation and reappeared in liver tissue and bile ducts after approximately 70 min. There were no detectable 11C-metabolites in the plasma or bile samples, indicating that the novel conjugated bile acid 11C-cholylsarcosine was not metabolized in the liver or in the intestines. The effective absorbed dose of 11C-cholylsarcosine was 4.4 μSv/MBq. Conclusion: We have synthesized a novel conjugated bile acid analog, 11C-cholylsarcosine, and PET/CT studies on anesthetized pigs showed that the hepatic handling of tracer uptake from blood and excretion into the bile was comparable to that for the endogenous bile acid cholyltaurine. This tracer may be valuable for future studies of normal and pathologic hepatic excretory functions in humans.
- enterohepatic circulation
- bile salt export pump
- sodium–taurocholate cotransporting polypeptide
- liver function
- positron emission tomography
Biliary excretion of lipophilic compounds is a key liver function in which bile acids play an important role (1). Bile acids facilitate intestinal uptake of, for example, fats and lipid vitamins, and their hepatic secretion stimulates bile flow as well as serving to eliminate bilirubin, cholesterol, heavy metals, and drug metabolites. After intestinal reabsorption, bile acids enter enterohepatic circulation, with up to 15 circulations per bile acid per day. Both efficient hepatocellular uptake of bile acids from blood and biliary excretion are hence important processes. Intrahepatic cholestasis is a feature of a large variety of inherited and acquired conditions, and in these diseases disturbed bile flow and accumulation of bile acids in the hepatocytes is thought to be an important pathogenetic factor in liver injury (2–4). In this situation, a noninvasive method that assesses the transhepatocellular transport of bile acids would be of considerable interest given its potential to improve our understanding of normal physiology, pathophysiology, and the hepatic effects or side effects of new drugs (5,6). To date, such a method is not available; even dynamic planar 99mTc-mebrofenin scintigraphy does not provide the necessary quantitative information.
The high spatial and temporal resolution of contemporary PET suggests that PET/CT could be a suitable methodology to study the hepatic handling of bile acids, but so far no specific bile acid tracers have been developed. The ideal tracer should be biochemically similar to common bile acids, have a high first-pass hepatic extraction, and be excreted in high concentrations in bile. In addition, the transhepatocellular transport of the ideal tracer should be mediated by the same transporter proteins that facilitate transport of the common bile acid conjugate cholyltaurine, that is, the sodium–taurocholate cotransporting polypeptide from blood to hepatocytes and the bile salt export pump from hepatocyte to bile (7–9). Moreover, kinetic analysis is simpler if the tracer is not metabolized. Cholylsarcosine is an analog of the endogenous bile acid conjugate cholylglycine, derived from the bile acid cholic acid and the amino acid sarcosine (N-methylglycine) (10–12). Cholylsarcosine is nontoxic to humans and undergoes an enterohepatic circulation without hepatic or intestinal biotransformation in humans (10). We therefore hypothesized that the 11C-labeled form, that is, [N-methyl-11C]cholylsarcosine (11C-cholylsarcosine), would be a suitable PET tracer for quantitative PET/CT studies of bile acid excretion. Here, we present the radiosynthesis of 11C-cholylsarcosine and proof-of-concept PET/CT studies in anesthetized pigs. The study included dynamic PET/CT of the liver and analysis for 11C-metabolites of the tracer in blood and bile. Competition between 11C-cholylsarcosine and the endogenous bile acid conjugate cholyltaurine for the hepatic transporters was investigated. Biodistribution of 11C-cholylsarcosine was also determined.
MATERIALS AND METHODS
Chemicals
Cholic acid, cholyltaurine/taurocholate (used as its hydrated sodium salt), glycine methyl ester hydrochloride, 1,2,2,6,6-pentamethylpiperidine, diethyl cyanophosphonate, and dimethylsulfoxide (dry) were obtained from Sigma-Aldrich Ltd. and used as received. Acetonitrile (analytic grade) was obtained from VWR International Ltd. Water (sterile), ethanol (sterile), aqueous NaH2PO4 (70 mM; sterile), and aqueous NaOH (0.25 M; sterile) were prepared by the pharmacy at Aarhus University Hospital.
Radiochemistry
11C-cholylsarcosine was prepared by the 3-step radiosynthesis illustrated in Figure 1 and described in detail below.
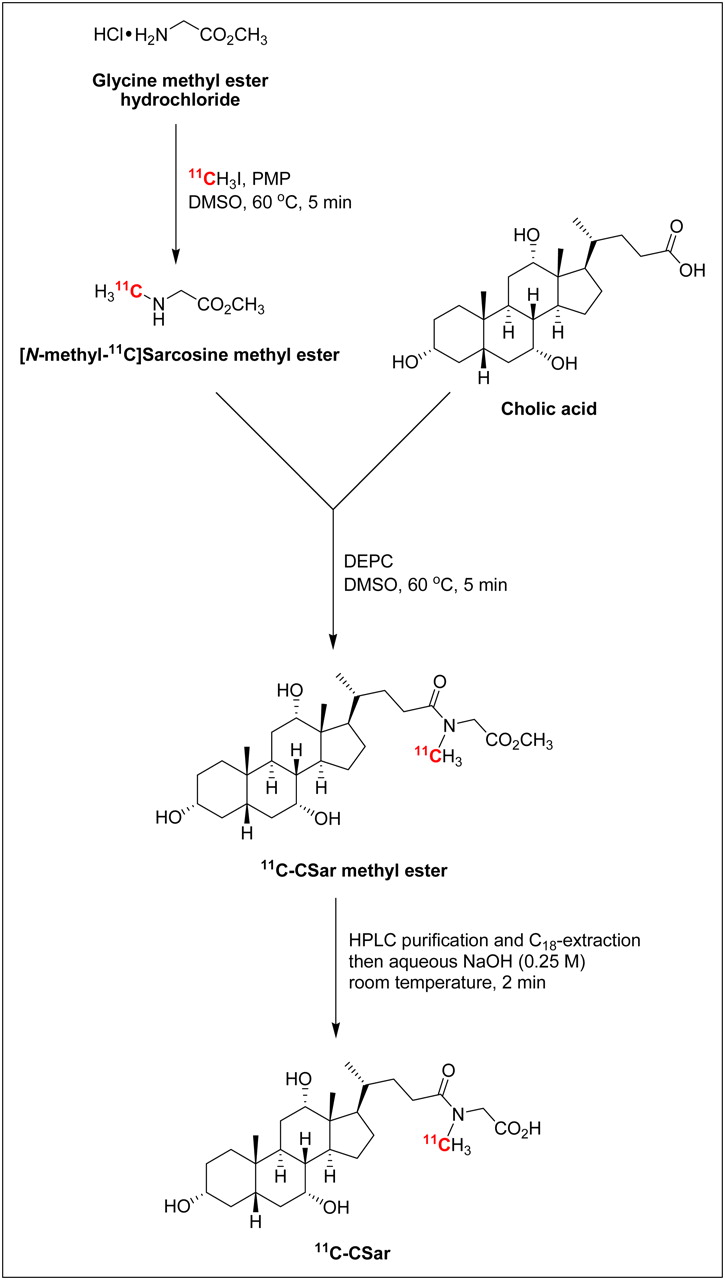
Three-step radiosynthesis of 11C-cholylsarcosine. CSar = cholylsarcosine; DEPC = diethyl cyanophosphonate; DMSO = dimethylsulfoxide; HPLC = high-performance liquid chromatography; PMP = 1,2,2,6,6-pentamethylpiperidine.
11C-methyl iodide (22–27 GBq, BioScan MeI-Plus) was delivered during 3 min to a mixture of glycine methyl ester hydrochloride (1 mg, 8 μmol) and 1,2,2,6,6-pentamethylpiperidine (5 μL, 28 μmol) in dry dimethylsulfoxide (300 μL) at room temperature. The sealed reaction vial was heated in an oil bath at 60°C. After 5 min, the vial was removed from the oil bath and solutions of cholic acid (12 mg, 29 μmol) in dry dimethylsulfoxide (250 μL) and diethyl cyanophosphonate (5 μL, 29 μmol) in dry dimethylsulfoxide (150 μL) were successively added. The reaction mixture was heated at 60°C for 5 min and then quenched with 40% ethanol in water (4 mL).
[N-methyl-11C]cholylsarcosine methyl ester was purified by preparative high-performance liquid chromatography (HPLC) followed by C18-cartridge extraction. Preparative HPLC was performed using a Waters-XTerra Prep RP18 OBD (5 μm, 19 × 100 mm) column with 40% acetonitrile in aqueous NaH2PO4 (70 mM) as eluent (isocratic, 10 mL/min). The fraction containing 11C-cholylsarcosine methyl ester (retention time, 8.0 min) was collected (from 7.7 to 8.4 min) in 70 mL of water and transferred to a Waters Sep-Pak Plus C18 cartridge (preconditioned with 10 mL of ethanol, followed by 10 mL of water), which was subsequently washed with water (10 mL) and eluted with ethanol (1.5 mL). Aqueous NaOH (2 mL, 0.25 M) was added to the ethanolic solution of 11C-cholylsarcosine methyl ester, and the mixture was kept at room temperature for 2 min to give 11C-cholylsarcosine. The basic solution was neutralized with aqueous NaH2PO4 (7 mL, 70 mM) and passed through a sterile filter into the final product vial. The radiochemical purity of the synthesized 11C-cholylsarcosine was determined by analytic HPLC using a Dionex Ultimate 3000 system (λ = 220 nm) connected to a GabiStar radiodetector (Nuclear Interface). The chromatographic column was a Phenomenex Luna 5μ C18(2) 100A (5 μm, 150 × 4.6 mm) with 30% acetonitrile in aqueous NaH2PO4 (70 mM) as eluent (isocratic, 2.5 mL/min). The chromatographic data were analyzed using Dionex Chromeleon software (version 6.80). The identity of 11C-cholylsarcosine (retention time, 3.4 min) was confirmed by coinjection of reference material (prepared as described in the Supplemental Data [available online at http://jnm.snmjournals.org]) and by electrospray ionization mass spectrometry (478.4 m/z [M-H], Bruker Daltonics HCT Plus ion trap mass spectrometer in negative ionization mode with a capillary voltage of +4.5 kV).
Optimization of reaction parameters, including solvent, concentrations, auxiliary base, temperature, and reaction time, are described in detail in the Supplemental Data.
In Vivo Studies
Three pigs (3-mo old female Danish Landrace and Yorkshire cross-breed; body weight, 35–40 kg) were kept fasting for 18 h with free access to water. The animals were premedicated with midazolam and s-ketamine; anesthetized with a mixture of midazolam, s-ketamine, and propofol, and mechanically ventilated as previously described (13). Catheters (Cordis) were inserted into the femoral vein and artery for intravenous administrations and blood sampling, respectively. The animals were placed on a thermostatically controlled heating blanket, keeping the rectal temperature at 38.5°C–39.5°C. By adjusting the mechanical respiration, arterial blood pCO2, pO2, and pH were kept between 5.3 and 7.2 kPa, 12 and 25 kPa, and 7.35 and 7.45, respectively. Blood glucose was 5.0–6.7 mM. After completion of the experiment, the animals were euthanized with an overdose of pentobarbital (100 mg/kg).
The studies were performed according to the Danish Animal Experimentation Act and the European convention for the protection of vertebrate animals used for experimental and other purposes (European Treaty Series no. 123).
For PET/CT studies, the pigs were placed on their back in a Siemens Biograph 64 Truepoint PET/CT camera with a 21.6-cm transaxial PET field of view. A CT scan (250 effective milliampere seconds with CARE Dose4D [Siemens], 120 kV, pitch of 1.0, and slice thickness of 2.0 mm) was obtained for definition of anatomic structures and attenuation correction of the PET recordings.
Pigs 1 and 2 underwent 90-min dynamic PET/CT recordings (list mode) with intravenous bolus administration of 400 MBq of 11C-cholylsarcosine. In pig 1, a second dynamic 11C-cholylsarcosine PET/CT scan was preceded by an intravenous 50-s infusion of 286 mg of cholyltaurine per kilogram of pig body weight to investigate possible inhibition of the transhepatocellular transport of 11C-cholylsarcosine. In pig 2, a second 11C-cholylsarcosine administration was given to determine the biodistribution of the tracer by means of whole-body PET/CT recordings. In both pigs, at least 6 half-lives of the 11C-isotope (half-life, 20.4 min) were allowed to pass between tracer administrations.
The decay-corrected dynamic PET data were reconstructed using the iterative TrueX algorithm (3 iterations, 21 subsets; Siemens) and postfiltered using a 3-mm gaussian filter yielding 3-dimensional images of 336 × 336 × 109 voxels. Time–activity curves were generated from volumes of interest drawn in liver tissue, intrahepatic bile ducts, and choledochus using fused PET/CT images.
During the dynamic PET recordings of pigs 1 and 2, blood samples (0.5 mL) were collected from the femoral artery at 12 × 5, 6 × 10, 6 × 30, 5 × 60, and 8 × 600 s. The radioactivity concentrations in the samples were measured in a well counter (Packard Biosciences) that was cross-calibrated to the PET camera. Additional blood samples were collected 2, 5, 10, 20, 40, 60, and 90 min after tracer administration for determination of 11C-metabolites in plasma.
Pig 3 did not undergo PET/CT but was used to collect bile samples 15, 30, and 90 min after administration of 11C-cholylsarcosine for analysis of possible 11C-metabolites. The plasma and bile samples were fractionated by HPLC (monitored by serial ultraviolet detection [λ = 220 nm] and radiodetection), and radioactivity concentrations were measured in the well counter. Two different HPLC conditions were used: Sphereclone SAX (Phenomenex, 250 × 4.6 mm) using 95% methanol and 5% acetic acid, pH 4, as eluent, or Sphereclone ODS(2) C-18 (Phenomenex, 250 × 4.6 mm) with a mixture of acetonitrile and aqueous 70 mM Na2HPO4 (60:40) as eluent.
The plasma free fraction of 11C-cholylsarcosine was determined in plasma samples collected before tracer administration in all 3 pigs; moreover, plasma was collected 10 s after the end of cholyltaurine infusion in pig 1, that is, before the 11C-cholylsarcosine administration. The plasma samples were mixed with aliquots of 11C-cholylsarcosine, pipetted into ultrafiltration units (Pall Nanosep centrifugal device; cutoff, 30,000 D; Sigma-Aldrich), and centrifuged at room temperature (10 min at 12,000 rpm). Radioactivity in plasma and ultrafiltrates was counted in the well counter. Filter retention of the tracer was determined using sodium phosphate buffer solution (35 mM; pH 7.2). The free fraction was calculated as the ratio of radioactivity concentration in the ultrafiltrate corrected for filter retention to the total radioactivity concentration in plasma (14).
For the biodistribution study, 11C-cholylsarcosine was administered intravenously in pig 2 followed by 5 whole-body PET scans with 1 min between scans and with a progressive increase in scan duration per bed position, that is, 1, 2, 3, 4 and 5 min, respectively. Organs with high accumulation of tracer relative to surrounding tissue were identified by visual inspection (liver, gallbladder, stomach, and small intestines) and were included as individual source organs. Virtually no radioactivity was detected in other organs, including the urinary bladder. The liver had a uniform radioactivity distribution, and accordingly the total liver radioactivity was estimated as the radioactivity concentration in a central volume of interest multiplied by the liver volume. Other source organs (gallbladder, stomach, small intestines, and kidneys) had a nonuniform radioactivity distribution, and for these organs the total activity was estimated using a large volume of interest encompassing all accumulated radioactivity. For each source organ, the time course of the non–decay-corrected total radioactivity was generated. Data were extrapolated from pig (40 kg) to human (74 kg) and recalculated into time courses of fractions of injected dose. Residence times were computed as the trapezoidal sum of the time course of fractions of injected dose assuming that the radioactivity decayed only by physical decay after the last scan. Residence time for the rest of the body was calculated as the total body residence time (without voiding) minus the sum of the residence times from the source organs. The residence times were entered into OLINDA/EXM 1.0 (15) to compute absorbed doses using the male reference phantom and to obtain effective dose values according to ICRP 60 (16).
RESULTS
Radiochemistry
The 3-step radiosynthesis provided 0.56–1.09 GBq of 11C-cholylsarcosine with a radiochemical yield of 13% ± 3% (mean ± SD; n = 7, decay-corrected) and with radiochemical purity greater than 99% within 40 min. The radiochemical yield of the intermediate [N-methyl-11C]cholylsarcosine methyl ester was approximately 20%, and the deprotection proceeded with full conversion to give 11C-cholylsarcosine exclusively. In its final formulation (0.5–1.0 μg of cholylsarcosine per milliliter in aqueous NaH2PO4 [approximately 47 mM] containing approximately 14% ethanol; pH 6–7), the tracer showed no alterations in chemical or radiochemical purity for up to 2.5 h after the end of the synthesis.
In Vivo Studies
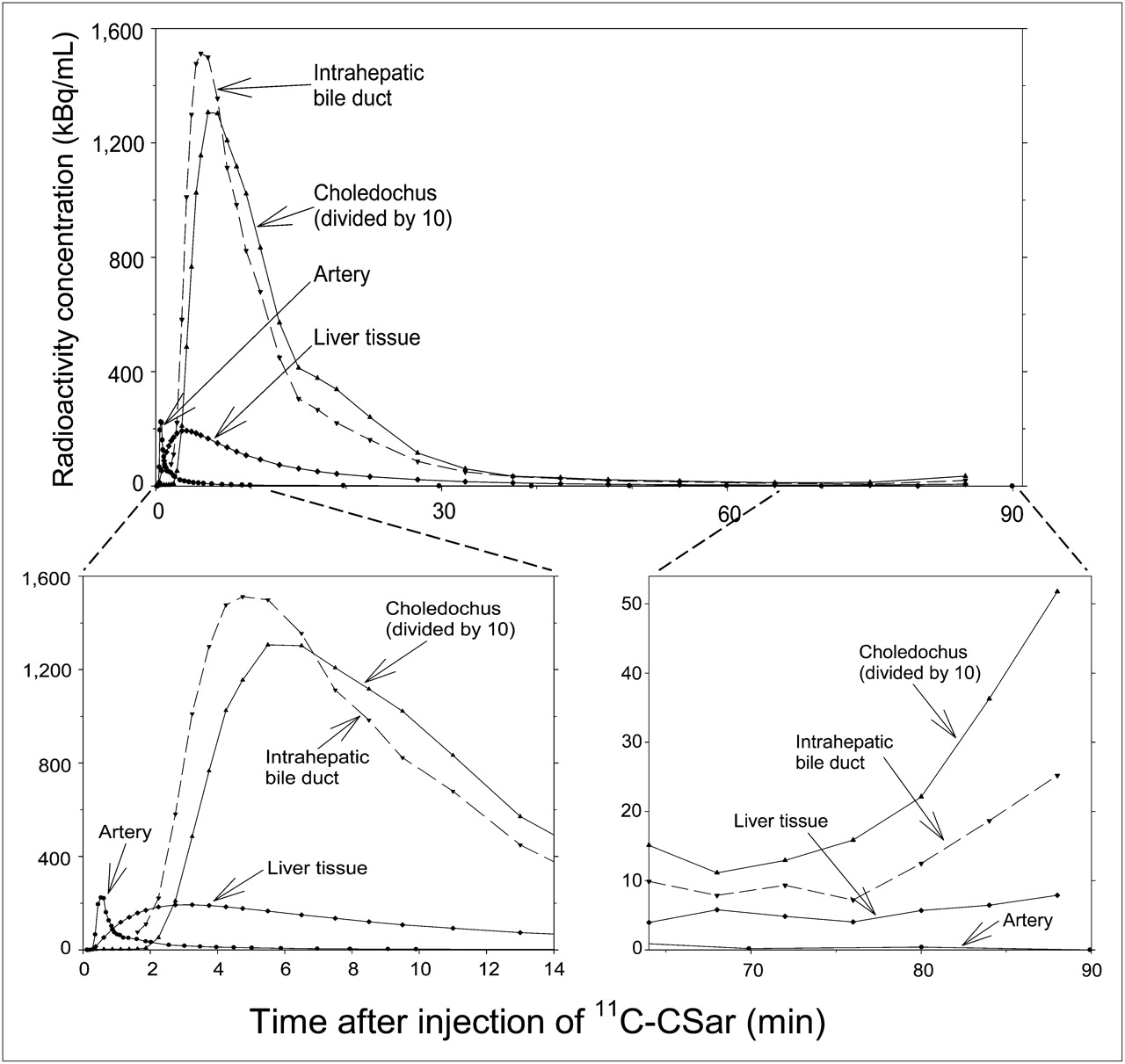
The dynamic PET/CT recordings in pigs 1 and 2 showed a rapid uptake of 11C-cholylsarcosine into liver tissue (Fig. 2A), with subsequent excretion into the intrahepatic bile ducts and the common hepatic bile duct (Fig. 2B). From there, most of the tracer was excreted via the choledochus into the duodenum, and the remainder of the tracer accumulated in the gallbladder (Fig. 2C). Eventually, the tracer was dispersed into the intestines or concentrated in the gallbladder (Fig. 2D). Figure 3 shows time–activity curves for these structures. The liver time–activity curve peaked within 3 min, with a radioactivity concentration comparable to the peak concentration of the arterial time–activity curve, and then decreased rapidly to low concentrations within 30–40 min. Radioactivity appeared in the intrahepatic bile ducts approximately 1 min after 11C-cholylsarcosine administration and in the choledochus 1 min later. The radioactivity concentration in the choledochus peaked 4–6 min after tracer administration, and the time–activity curves illustrate how 11C-cholylsarcosine was concentrated in the biliary tree (Fig. 3). As a result of enterohepatic circulation, radioactivity concentrations in the liver tissue and bile ducts increased again about 75 min after administration of the tracer. Analysis of plasma from pigs 1 and 2 and bile samples from pig 3 showed no 11C-metabolites up to 90 min after 11C-cholylsarcosine administration, verifying that 11C-cholylsarcosine does not undergo hepatic or intestinal metabolism. The free fraction of 11C-cholylsarcosine added to plasma samples collected before tracer administration was 18% ± 2% (n = 3), which is similar to that reported for endogenous cholyl-conjugates, that is, 20%–40% (3).
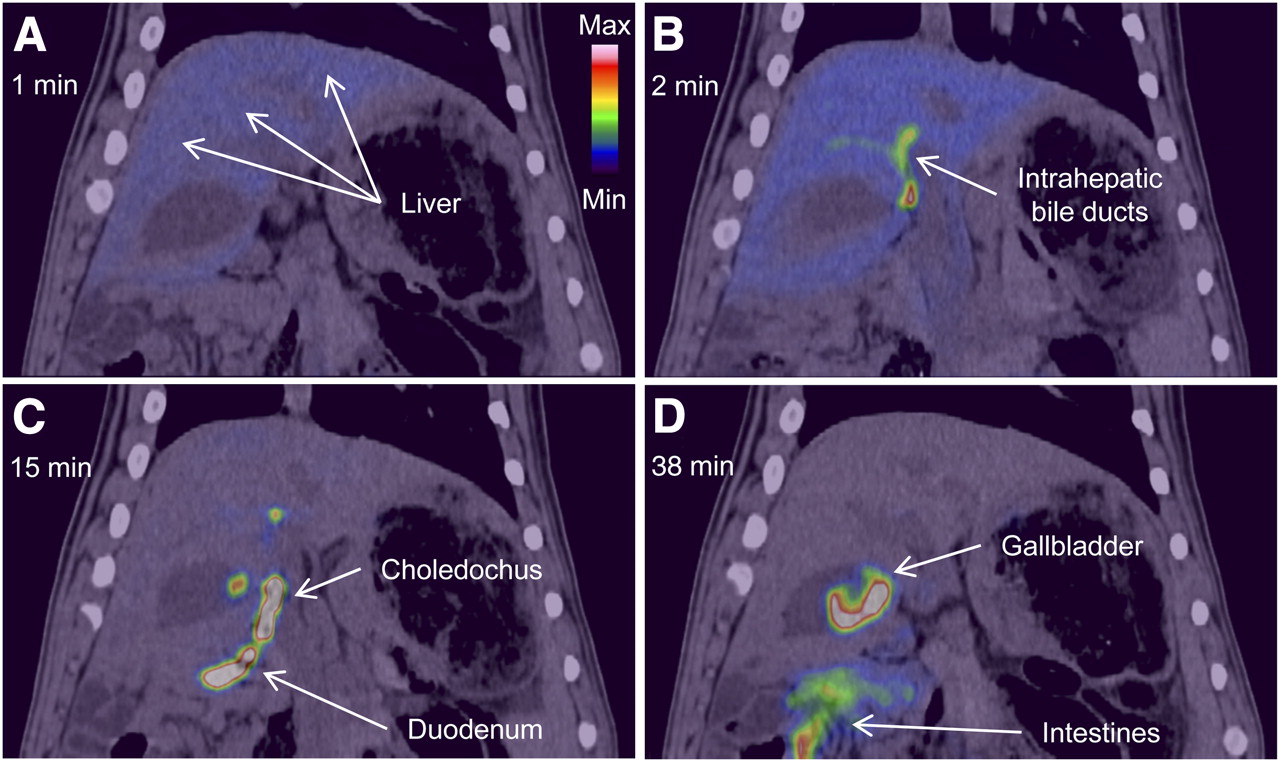
Coronal PET/CT images of time course of distribution of 11C-cholylsarcosine 1 min (A), 2 min (B), 15 min (C), and 38 min (D) after intravenous bolus administration of tracer (pig 2). Color scale is same for all images.
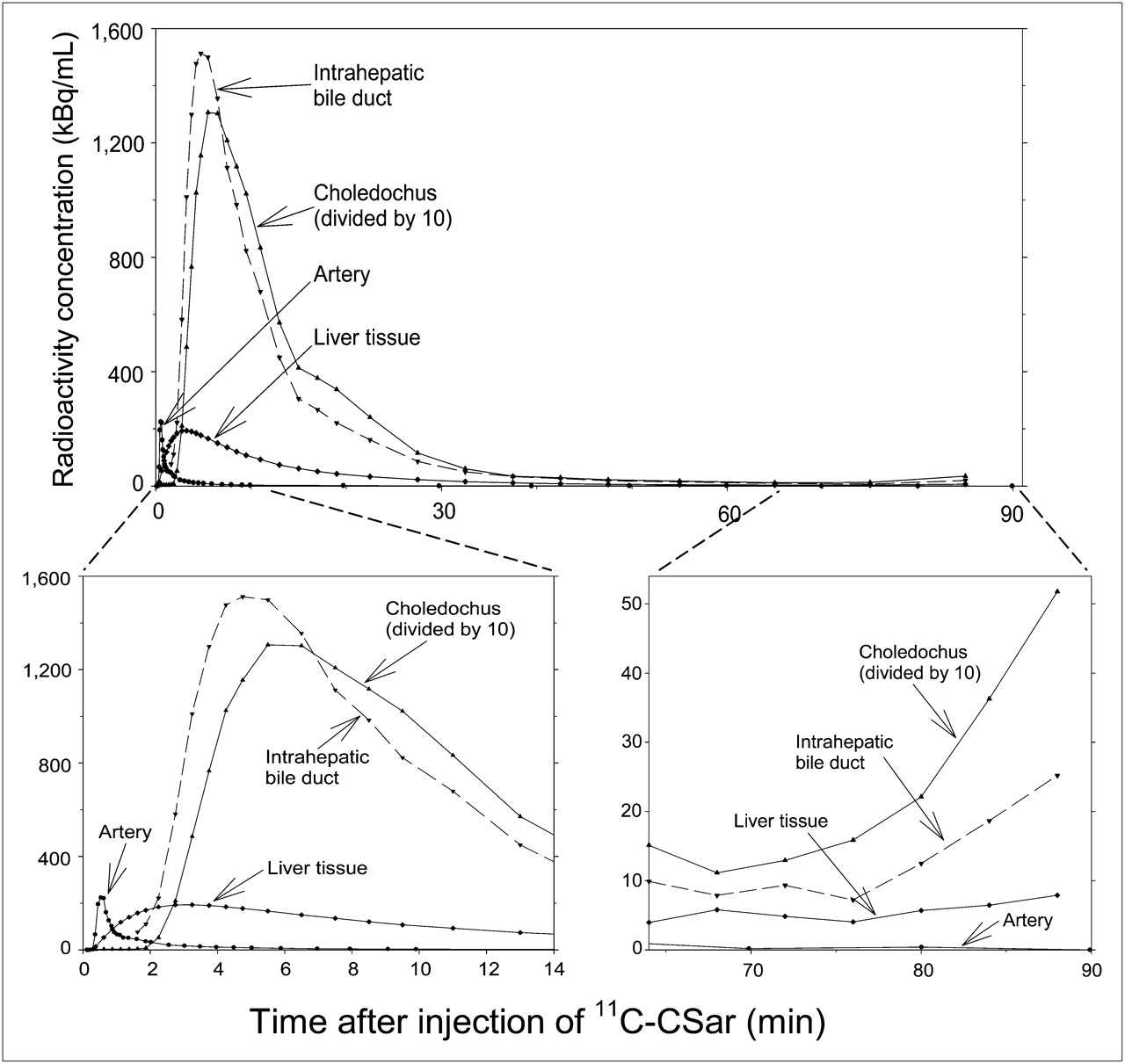
Time–activity curves in arterial blood (samples) and in liver tissue, intrahepatic bile ducts, and choledochus (dynamic PET/CT) after intravenous bolus administration of 11C-cholylsarcosine (pig 1). CSar = cholylsarcosine.
Pretreatment with cholyltaurine before the second scan of pig 1 markedly inhibited hepatic uptake and biliary excretion of 11C-cholylsarcosine as illustrated by the time–activity curves in Figure 4. Compared with the time–activity curves from the PET/CT studies without cholyltaurine, the liver tissue time–activity curves reached a maximum radioactivity concentration of only approximately 40% and the subsequent decrease with time was significantly slower. Moreover, the intrahepatic bile ducts were indistinguishable from surrounding liver tissue, and the time–activity curve in the choledochus increased at a much slower rate and reached a maximum value that was only 2% of that without pretreatment with cholyltaurine (Fig. 3 vs. Fig. 4). These findings are in accordance with the hypothesis that 11C-cholylsarcosine and cholyltaurine compete for one or more of the same transporters from blood to liver cells and from hepatocytes to bile capillaries. The free fraction of 11C-cholylsarcosine in plasma samples collected after administration of cholyltaurine was 100%, showing competition for protein binding between tracer and cholyltaurine.
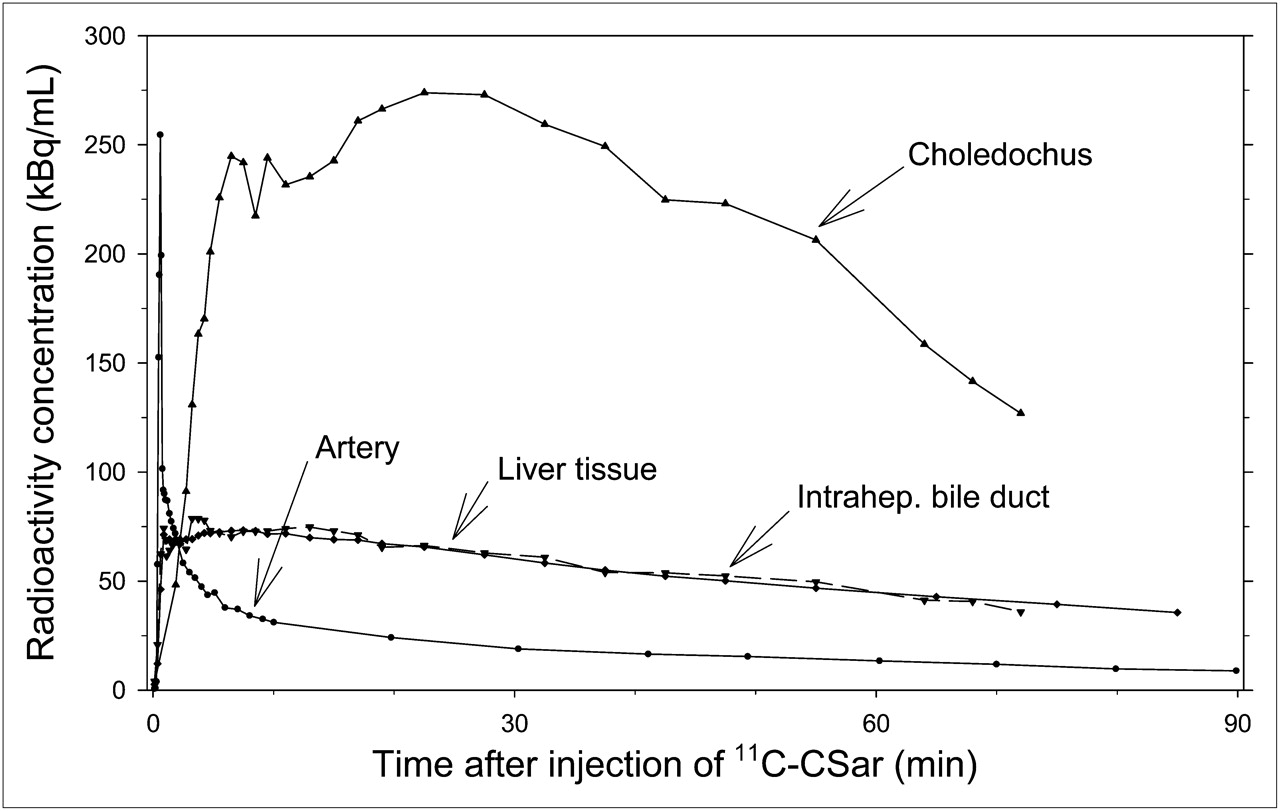
Time–activity curves in arterial blood (samples) and in liver tissue, intrahepatic bile ducts, and choledochus (dynamic PET/CT) after pretreatment with cholyltaurine and intravenous bolus administration of 11C-cholylsarcosine (pig 1). CSar = cholylsarcosine.
The estimated radiation doses were highest in the gallbladder wall, small intestines, liver, and upper large intestine wall (Table 1). The effective absorbed dose of 11C-cholylsarcosine was 4.4 μSv/MBq.
Absorbed Dose Estimates for 11C-Cholylsarcosine
DISCUSSION
We have developed a procedure for radiosynthesis of 11C-cholylsarcosine, which, to the best of our knowledge, is the first radiolabeled bile acid for PET/CT studies. In the initial phase of the development, we considered direct 11C-methylation of the amide nitrogen of cholylglycine protected at both the hydroxyl and the carboxylic acid groups. However, using this approach, several by-products that were impossible to separate from the tracer were formed. We therefore developed the present procedure involving 11C-methylation of glycine followed by coupling to cholic acid. The advantage of this procedure is that milder reaction conditions can be used and only the carboxylic acid group of glycine needs to be protected; protection of cholic acid is not necessary. As a consequence, the reaction proceeds with only minor amounts of by-products, and pure 11C-cholylsarcosine is easily obtained after deprotection of the carboxylic acid group. Even though 3 steps are involved in the radiosynthesis, the entire procedure can be completed within 40 min, that is, 2 half-lives of the 11C-isotope.
The in vivo studies revealed rapid transport of 11C-cholylsarcosine from blood through the liver into the bile ducts as unmetabolized tracer. The significant up-concentration of the tracer from liver tissue to bile supports our belief that 11C-cholylsarcosine behaves like an endogenous bile acid conjugate. Moreover, the inhibition of these steps by cholyltaurine pretreatment indicates that 11C-cholylsarcosine is transported by one or more of the same transporter proteins as cholyltaurine. This bile acid is transported from blood to hepatocytes predominantly via the sodium–taurocholate cotransporting polypeptide and from hepatocytes to bile capillaries via the bile salt export pump (7–9). Other transporters such as the organic anion transporting polypeptides and the multidrug resistance protein 2 may also be involved in the transport of 11C-cholylsarcosine. Further studies are necessary to elucidate the exact transhepatocellular transport of 11C-cholylsarcosine.
The binding of 11C-cholylsarcosine to plasma proteins (on average, 82%) is similar to that found for endogenous cholyl-conjugates (60%–80%) (3). This, together with the finding that cholyltaurine displaced 11C-cholylsarcosine from the plasma proteins, shows that 11C-cholylsarcosine is bound to the same plasma-binding proteins as cholyltaurine and further supports our view that 11C-cholylsarcosine is a bile acid tracer analog.
The present studies show that dynamic PET studies of hepatic removal kinetics in pigs can be performed up to approximately 40 min after tracer administration without the need to account for formation of 11C-metabolites or enterohepatic circulation of 11C-cholylsarcosine.
Our study suggests that 11C-cholylsarcosine may be not only a biochemical but also a physiologic cholyltaurine analog. If confirmed, the use of this PET tracer will allow noninvasive real-time analysis of central steps in hepatic bile acid transport, which is important in studies of normal physiology and pathophysiology. Thus, altered function of the sodium–taurocholate cotransporting polypeptide or the bile salt export pump has been hypothesized in inherited cholestasis as well as acquired liver disease such as drug-induced liver injury, acute hepatitis, primary biliary cirrhosis, posttransplantation liver dysfunction, and biliary obstruction (9,17,18). In addition, a large number of drugs use one or both transporters during their elimination pathway, with possible kinetic interactions between drug and bile acids. However, kinetic data to support such hypotheses in humans are almost completely missing since a suitable methodology has not been available. The 11C-cholylsarcosine tracer could prove a valuable new tool to study these questions.
The biodistribution study identified the liver, bile ducts, and gallbladder as target organs in agreement with the dynamic PET/CT findings. The high absorbed doses for these organs compared with other organs reflect the high organ specificity of 11C-cholylsarcosine. The present doses of 400 MBq of 11C-cholylsarcosine to 40-kg pigs gave high-contrast PET images, and it is likely that a dose of, for example, 100 MBq will be sufficient for obtaining similar data quality in human studies. With an effective dose for 11C-cholylsarcosine of 4.4 μSv/MBq, the radiation dose in humans may thus be no more than 0.4 mSv.
CONCLUSION
11C-cholylsarcosine was prepared from glycine by a simple 3-step radiosynthesis. This novel bile acid tracer undergoes rapid transhepatocellular transport in pigs resulting in high concentrations of unmetabolized 11C-cholylsarcosine in the bile. Competitive inhibition with cholyltaurine indicates that 11C-cholylsarcosine is transported via the same transporter proteins as this endogenous bile acid conjugate. We therefore believe that 11C-cholylsarcosine PET/CT may prove to be useful in humans for characterization and quantification of bile acid excretion during normal physiologic conditions and in patients with intrahepatic cholestatic liver diseases or drug-induced cholestasis.
DISCLOSURE STATEMENT
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank Prof. Karl Anker Jørgensen (Center for Catalysis, Department of Chemistry, Aarhus University, Aarhus, Denmark) for use of the nuclear magnetic resonance spectrometer and Associate Prof. Dirk Bender (PET Center, Aarhus University Hospital, Aarhus, Denmark) for valuable discussions during this project. This study was supported by the National Institute of Diabetes and Digestive and Kidney Diseases (R01-DK074419), the Danish Council for Independent Research (Medical Sciences, 09-073658), and the Novo Nordic Foundation (R179 A15289). No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Mar. 27, 2012.
- © 2012 by the Society of Nuclear Medicine, Inc.
REFERENCES
- Received for publication September 29, 2011.
- Accepted for publication December 27, 2011.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Hepatobiliary Secretion Kinetics of Conjugated Bile Acids Measured in Pigs by 11C-Cholylsarcosine PET
- Radiosynthesis of N-11C-Methyl-Taurine-Conjugated Bile Acids and Biodistribution Studies in Pigs by PET/CT
- Role of (Drug) Transporters in Imaging in Health and Disease
- Key discoveries in bile acid chemistry and biology and their clinical applications: history of the last eight decades