


Abstract
A variety of radiolabeled somatostatin analogs have been developed for targeting of somatostatin receptor (sst)–positive tumors. Bicyclic somatostatin-based radiopeptides have not been studied yet. Hypothesizing that the introduction of conformational constraints may lead to receptor subtype selectivity or may help to delineate structural features determining pansomatostatin potency, we developed and evaluated first examples of this new class of potential radiotracers for imaging or therapy of neuroendocrine tumors. Methods: The bicyclic peptides were synthesized by standard solid-phase peptide synthesis. DOTA was coupled to the resin-assembled peptide for labeling with 177Lu and 68Ga. Binding affinity and receptor subtype profile were determined using human ssts. Ca2+ flux, internalization, and efflux were studied in human embryonic kidney (HEK)-sst2 and HEK-sst3 cell lines. Biodistribution and PET/CT studies were performed in corresponding nude mice models. Results: Some of the new analogs showed high affinity for sst2 and sst3 and moderate affinity for sst1, sst4, and sst5, while exhibiting agonistic properties. The analog AM3, comprising an octreotide ring and a head-to-tail–coupled Arg-diaminobutyric acid(DOTA) cycle, showed the highest receptor affinity and agonist potency. 177Lu-AM3 showed high and receptor-mediated uptake in vivo in sst2 and sst3 tumors with low background. Kidneys were the only other tissue accumulating radioactivity that could be reduced by a preinjection of lysine. PET/CT studies of 68Ga-AM3 at 1 h after injection were characterized by clear localization of the tumor, visualization of the kidneys, and negligible background. Conclusion: The high rigidity of these new bicyclic somatostatin-based radiopeptides led to agonistic ligands with good affinity for all 5 ssts. The pharmacokinetic data of 177Lu/68Ga-AM3 make this peptide an excellent candidate as an imaging—and especially as a PET—radiotracer.
Radiolabeled regulatory peptide-based agents have become important radiopharmaceuticals for molecular imaging and targeted radionuclide therapy in nuclear oncology (1,2). Prototypic peptides are derivatives of somatostatin. Five somatostatin receptor subtypes (sst1–sst5) have been identified, all of which were found to be overexpressed on different tumors; the most important one is sst2 (2–4). Current clinical studies indicate that other ssts are also of relevance (5–7). Therefore, chelated pansomatostatin ligands have been developed for the potential targeting of a broader spectrum of tumors (8–10). 68Ga-DOTANOC, with affinity for sst2,3,5, has found its way into the clinic, showing properties superior to 111In-pentetreotide (OctreoScan; Covidien) (6,7). 68Ga is one of the most attractive β+-emitters because it can be produced from a generator system (11,12).
A neglected group of radiolabeled somatostatin-based peptides are bicyclic peptides. Such peptides were synthesized by the Merck group in the late 1970s, soon after the isolation of the natural hormone (13–15). The rational behind this work was severalfold, namely to understand the bioactive conformation (13) and pharmacophoric amino acid sequence of the natural peptide somatostatin-14 (SRIF-14) but also to increase the metabolic stability of the natural peptides (14) by increasing the rigidity. At that time, ssts were not known, and the peptides were studied functionally for their biologic activity.
Veber et al. (13–15) eliminated amino acids in SRIF-14 that were not required for biologic activity and introduced conformational constraints by head-to-tail coupling of a 16-atom ring with 7-aminoheptanoic acid (Aha) to the N- and C-terminally amino acid–deleted octreotide. The bicyclic peptide cyclo(Aha,cyclo(Cys-Phe-D-Trp-Lys-Thr-Cys)) showed higher potency than SRIF-14 and longer duration of biologic activity. In 2001, Falb et al. looked for selectivity toward receptor subtypes (16). They synthesized a backbone and disulfide-bridged bicyclic somatostatin analog. The inner cycle comprised the octreotide sequence, and backbone cyclization was through lactamization via the N and C terminus of the 2 cysteines. Falb et al. (16) found a high degree of sst2 selectivity, which they explained with the large hydrophobicity of the lactam ring Phe side chains. Intuitively, the introduction of conformational constraints should lead to subtype selectivity. This subtype selectivity was shown in a study by Rivier et al. examining short-chain somatostatin analogs at sst1–sst5 to identify sterically constrained sst1 selective scaffolds (17). They found a large affinity dependence on the ring size but also highly selective sst1 agonists if they used 4-(N-isopropyl)-aminomethylphenylalanine to replace lysine.
We were interested in the search for metabolically stable pansomatostatin analogs (9,10) and wanted to determine whether bicyclic peptides might form a class of radiopeptides with the required properties. In a first family of peptides, we followed partially the strategy of Veber et al. (13–15) and Falb et al. (16). We kept the octreotide 20-atom sequence as an inner cycle, and instead of using Aha we formed the second 16-atom ring with 2 amino acids, namely Arg and γ-aminobutyric acid (GABA). GABA could be exchanged with diaminobutyric acid (DAB) for chelator coupling. Arg was assumed to be important for a broad sst subtype profile; this was the conclusion of our studies presented earlier (10).
In this article, we describe the design, pharmacologic and receptor subtype profile, and agonist potency measurements of bicyclic somatostatin-based analogs. In vitro internalization, efflux, pharmacokinetic, and imaging studies in tumor xenografts bearing sst2- and sst3-expressing tumors of the 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA)-coupled bicyclic analogs labeled with 177Lu and 68Ga were performed. We were intrigued how the introduction of conformational constraints might influence receptor subtype profile and pharmacokinetics of the respective radiopeptides. In addition, we were interested in studying how increased metabolic stability might influence biodistribution, in particular background washout.
MATERIALS AND METHODS
General
All commercially obtained chemicals were of analytic grade. Rink-acid resin and 9-fluorenylmethoxycarbonyl amino acids were purchased from NovaBiochem AG and Bachem, respectively. 177LuCl3 (specific activity, 740 GBq/mg) was from PerkinElmer. The 68Ge/68Ga generator was obtained from Cyclotron Co. Ltd. The reversed-phase high-performance liquid chromatography (RP-HPLC) systems, γ-counter, and electrospray ionization mass spectrometer were the same as previously reported (5,8). The HPLC gradient was 0–25 min, 95%–45% A (A, 0.1% trifluoroacetic acid in water; B, acetonitrile); flow rate, 0.75 mL/min; and column, Nucleosil 120-C18 (Macherey-Nagel). All reagents used for the functional assays were purchased from common suppliers.
Synthesis of Bicyclic Analogs
The chemical structures of the investigated analogs are shown in Table 1. The synthesis of the lead peptide HR3005 is depicted in Figure 1. Four new bicyclic analogs based on the lead peptide, 1 replacing GABA with DAB (AM8) and 3 bearing DOTA (AM1, AM2, and AM3), were synthesized. Details of the synthesis will be published elsewhere. The final products were purified by semipreparative HPLC and characterized by electrospray ionization mass spectrometer and RP-HPLC.
Chemical Structures, Binding Affinity, and Agonistic Potency of Bicyclic Somatostatin-Based Analogs
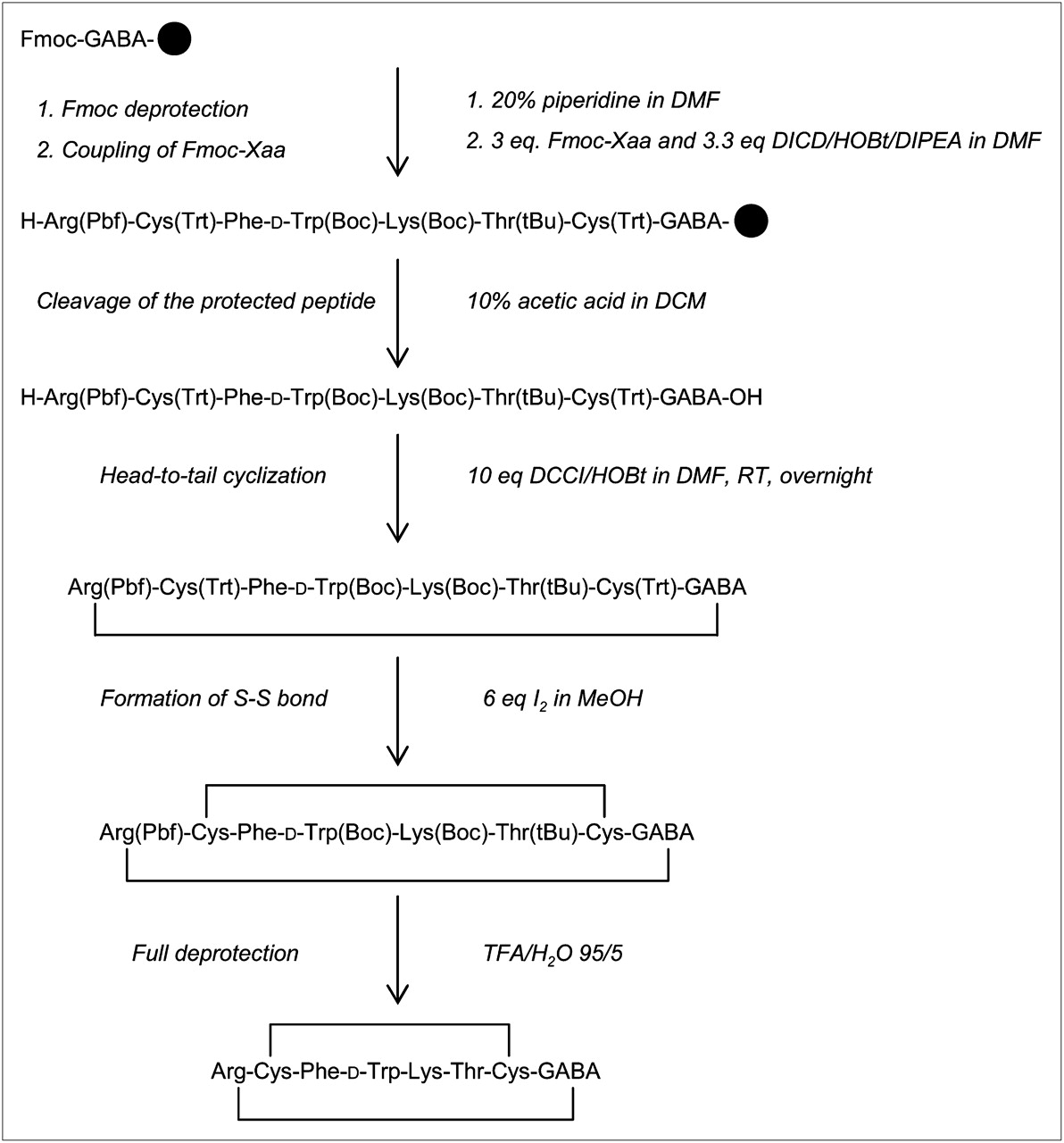
Synthesis of HR3005. Standard 9-fluorenylmethoxycarbonyl (Fmoc) solid-phase synthesis was applied to assemble linear peptide using side chain–protected amino acids (Xaa). Boc = tert-butoxycarbonyl; DCCI = dicyclohexyl-carbodiimide; DCM = dichloromethane; DICD = diisopropyl-carbodiimide; DIPEA = diisopropyl-ethyl-amine; DMF = dimethylformamide; HOBt = hydroxybenzotriazole; Pbf = 2,2,4,6,7-pentamethyl-dihydrobenzofuran-5-sulfonyl; RT = room temperature; tBu = tert-butyl; TFA = trifluoroacetic acid; Trt = trityl.
Receptor Autoradiography
Cell membrane pellets were prepared from human sst1-expressing Chinese hamster ovary cells; sst2-, sst3-, and sst4-expressing CCL39 cells; and sst5-expressing human embryonic kidney 293 (HEK293) cells and stored at −80°C. Receptor autoradiography was performed on 20-μm-thick membrane pellet sections and quantitated as previously described (18,19).
Immunofluorescence Microscopy
An immunofluorescence microscopy–based internalization assay for sst2,3 was performed with HEK293 cells stably expressing the human sst2 or sst3 using the sst2-specific primary antibody R2-88 (provided by Dr. Agnes Schonbrunn) or the sst3-specific primary antibody SS-850 (Gramsch Laboratories), respectively, as previously described (20).
Ca2+ Release Assay
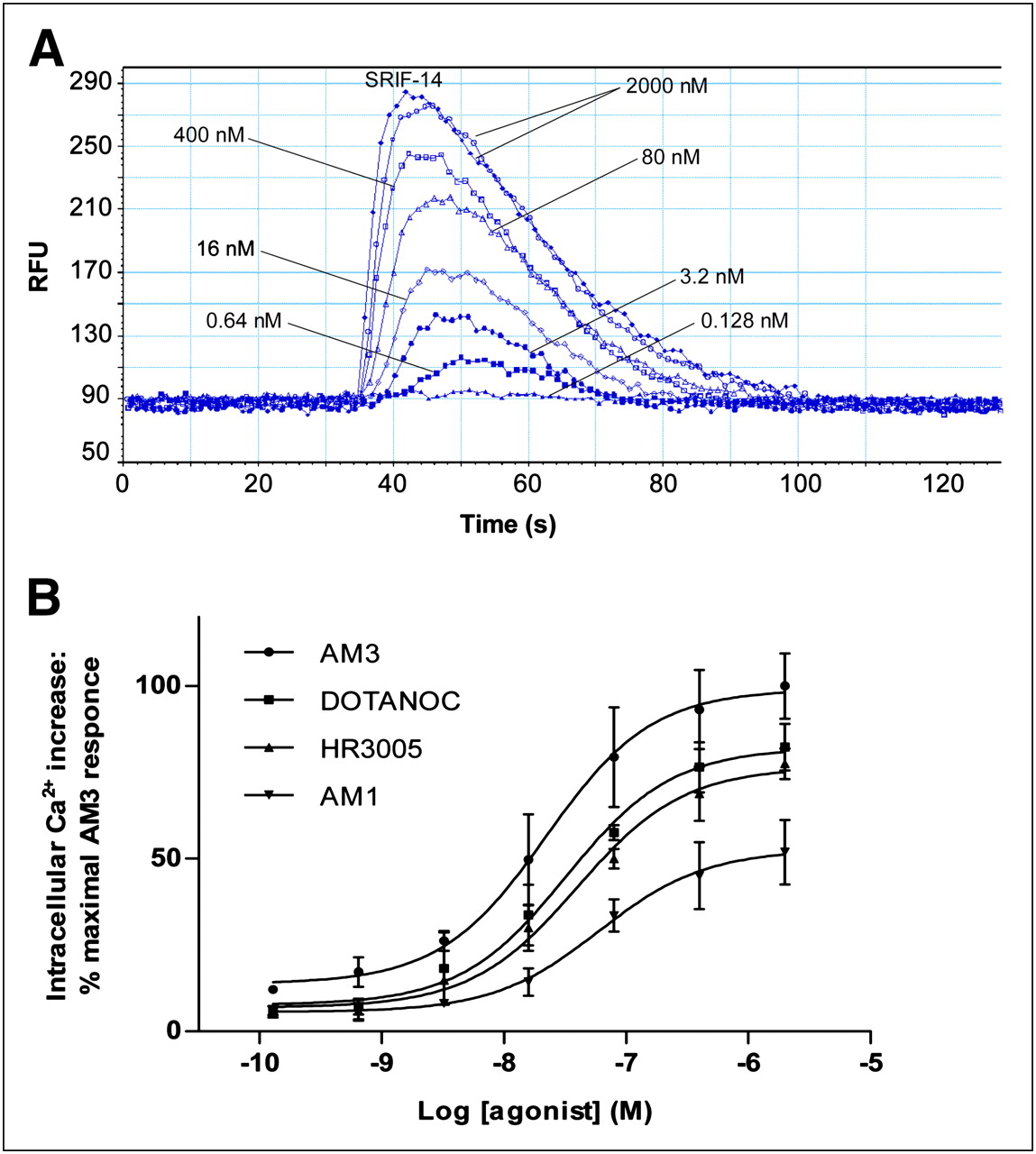
Intracellular Ca2+ release was measured in HEK-sst2,3,5 cells as described previously (19), using the Calcium 3 Assay Kit (Molecular Probes Inc.), according to the manufacturer's instructions. In brief, the cells were seeded (50,000 cells/well) in 96-well plates and cultured overnight at 37°C, 5% CO2. The day of the experiment, cells were washed with assay buffer containing 2.5 mM probenecid and then incubated with 100 μL of Ca2+ dye per well for 60 min. Seven different concentrations of HR3005, AM1, AM3, and DOTANOC (used as a reference analog) were dispensed into a reagent source 96-well plate. Cell- and peptide-containing plates were loaded into a FLEX station 3 Microplate Reader (Molecular Devices). Intracellular Ca2+ release was recorded for 130 s, monitoring fluorescence emission at 525 nm (with excitation wavelength at = 485 nm) in the presence of the analogs at concentrations ranging from 2,000 to 0.128 nM. Ionomycin and SRIF-14 were used as positive controls at a concentration of 2,000 nM. Baseline measurements were taken for dye-loaded, untreated cells. The data were analyzed by SoftMax Pro software (Molecular Devices).
Preparation of Radiotracers
177Lu-AM1 and 177Lu-AM3 were prepared after incubation of 10 μg of each analog with 37–111 MBq of 177LuCl3 at 95°C for 30 min in ammonium acetate buffer (0.4 M, pH 5.0). 68Ga-AM3 was prepared according to the method of Zhernosekov et al. (21), using a different cation exchange column (Strata-XC; Phenomenex) for the purification of the eluate. Quality control was performed by RP-HPLC. The radiotracer solutions were prepared by dilution with 0.9% NaCl (saline) containing 0.1% bovine serum albumin.
Enzymatic Stability in Human Blood and Serum
177Lu-AM3 was incubated with fresh human whole blood and serum (30 pmol/mL, 37°C). Blood samples were centrifuged at 1 and 24 h. The erythrocytes were washed with saline and measured for radioactivity, and plasma was removed, mixed with acetonitrile (2:3), and centrifuged. Serum samples at 1 h up to 7 d were mixed with ethanol (1:2) to precipitate proteins, followed by centrifugation. The supernatants from plasma and serum were analyzed by RP-HPLC to determine the relative amount of intact peptide and possible metabolites.
Radioligand Internalization and Cellular Retention Studies
The internalization rates of 177Lu-AM1 and 177Lu-AM3 and the efflux of 177Lu-AM3 were studied in HEK-sst2,3 cells. The cells were seeded in 6-well plates (0.8–1 million cells per well) and incubated at 37°C, 5% CO2, overnight. The radiotracer (2.5 pmol/well) was added to the medium, and the cells were incubated at 37°C. The internalization was stopped at preselected times (0.5, 1, 2, and 4 h) by removing the medium. The cells were treated as described previously (5,8,9). Nonspecific internalization was determined using a 1,000-fold excess of DOTANOC. Internalization was expressed as percentage of the applied radioactivity. In efflux studies, 177Lu-AM3 was allowed to internalize for 2 h at 37°C, followed by acid wash (5,8,9). Fresh medium was added, which was then collected at different times (15, 30, 60, 90, 120, and 240 min) for quantification. The recycled fraction was expressed as a percentage of the total internalized amount after 2 h of incubation at 37°C.
Animal Biodistribution Studies
Animals were kept and cared for according to Swiss regulations for animal treatment (approval no. 789). Five-week-old athymic female Swiss nude mice were implanted subcutaneously in the right front leg with 107 HEK-sst2, freshly suspended in sterile phosphate-buffered saline. In a dual-tumor model, the mice were also implanted with 107 HEK-sst3 in the other leg. The tumors were allowed to grow for 14–18 d (tumors weight, 100–150 mg).
The mice in the dual-tumor model were injected with 177Lu-AM3 (100 μL/10 pmol/0.15 MBq) into the tail vein. Nonspecific uptake was determined with a coinjection of a 2,000-fold excess of DOTANOC. The mice (groups of 4–7 animals) were sacrificed under anesthesia at 1, 4, and 24 h after injection. Additionally, 1 group was preinjected with a 20 mg/100 μL concentration of lysine in phosphate-buffered saline 10 min before the injection of the radioligand, to study kidney blocking; the mice were sacrificed at 4 h after injection.
68Ga-AM3 was evaluated in vivo in HEK-sst2 tumor–bearing mice injected with 68Ga-AM3 (100 μL/10 pmol/0.4 MBq) at 1 and 2 h after injection. Nonspecific uptake and kidney blocking studies were performed.
Organs of interest and blood were collected, rinsed of excess blood, blotted dry, weighed, and counted in a γ-counter. The results were expressed as percentage injected dose per gram of tissue (%ID/g). The total counts injected per animal were determined by extrapolation from counts of an aliquot taken from the injected solution as a standard.
PET/CT Studies
The mice bearing HEK-sst2 tumors were injected with 68Ga-AM3 (100 μL/10 pmol/0.4 MBq), including a group preinjected with lysine and a group injected with DOTANOC, as described in the “Animal Biodistribution Studies” section. One hour later, the mice were sacrificed; the bladder was emptied mechanically, and mice were scanned for 60 min using a routine combined PET/CT scanner (Discovery STE; GE Healthcare). A scout scan (180°, 10 mA, 120 kV) was acquired to establish a protocol for all other scans. CT scans were acquired with minimum slice distance (16 × 0.625 mm; pitch, 1.375:1) and the highest possible tube current for these settings (320 mA, 120 keV). PET emission events were collected in 3-dimensional scanning mode (septa out) over 60 min. Images were corrected for decay of 68Ga and random events and reconstructed using the manufacturer's 3-dimensional ordered-subset expectation maximization algorithm to 47 slices (display field of view, 6.4 cm; matrix, 128 × 128; resulting pixel size, 0.5 mm), once for each mouse separately in the center of the reconstruction cylinder.
RESULTS
Synthesis and Radiolabeling
The chemical structures of the investigated analogs are shown in Table 1. The purity of each analog, determined by RP-HPLC, was 95% or greater. The overall yields were approximately 30%. Labeling yields for 177Lu and 68Ga were greater than 97%. The specific activities were 17 GBq/μmol for 177Lu-AM1/AM3 and 35 GBq/μmol for 68Ga-AM3.
Binding Affinity Profiles and Structure–Activity Relationship
The binding affinity profiles of the bicyclic somatostatin analogs are listed in Table 1. The lead peptide HR3005 showed high affinity (comparable to SRIF-28) for sst2 and sst3 and moderate affinity for sst5, sst1, and sst4, conferring almost pansomatostatin potency to this peptide. The replacement of GABA by DAB (AM8) did not change the affinity profile, whereas DOTA-coupling (AM1) led to a 2-fold loss of affinity to sst1 and sst4, a 4-fold loss to sst2, a 5-fold loss to sst3, and a 3-fold loss to sst5. We reasoned that some affinity increase may be regained by replacing Phe3 (octreotide notation) with Tyr3 (AM2), at least in regard to sst2 affinity. This affinity increase was found in octreotide derivatives but not with these bicyclic peptides, resulting in a further loss of all receptor subtype affinities. By introducing Tyr as a spacer between the DOTA and the peptide (AM3), the affinity to sst2 and sst3 increased significantly, by a factor of 4–5. Slight improvements were also found for sst4 and sst5.
Immunofluorescence-Based Internalization Assay
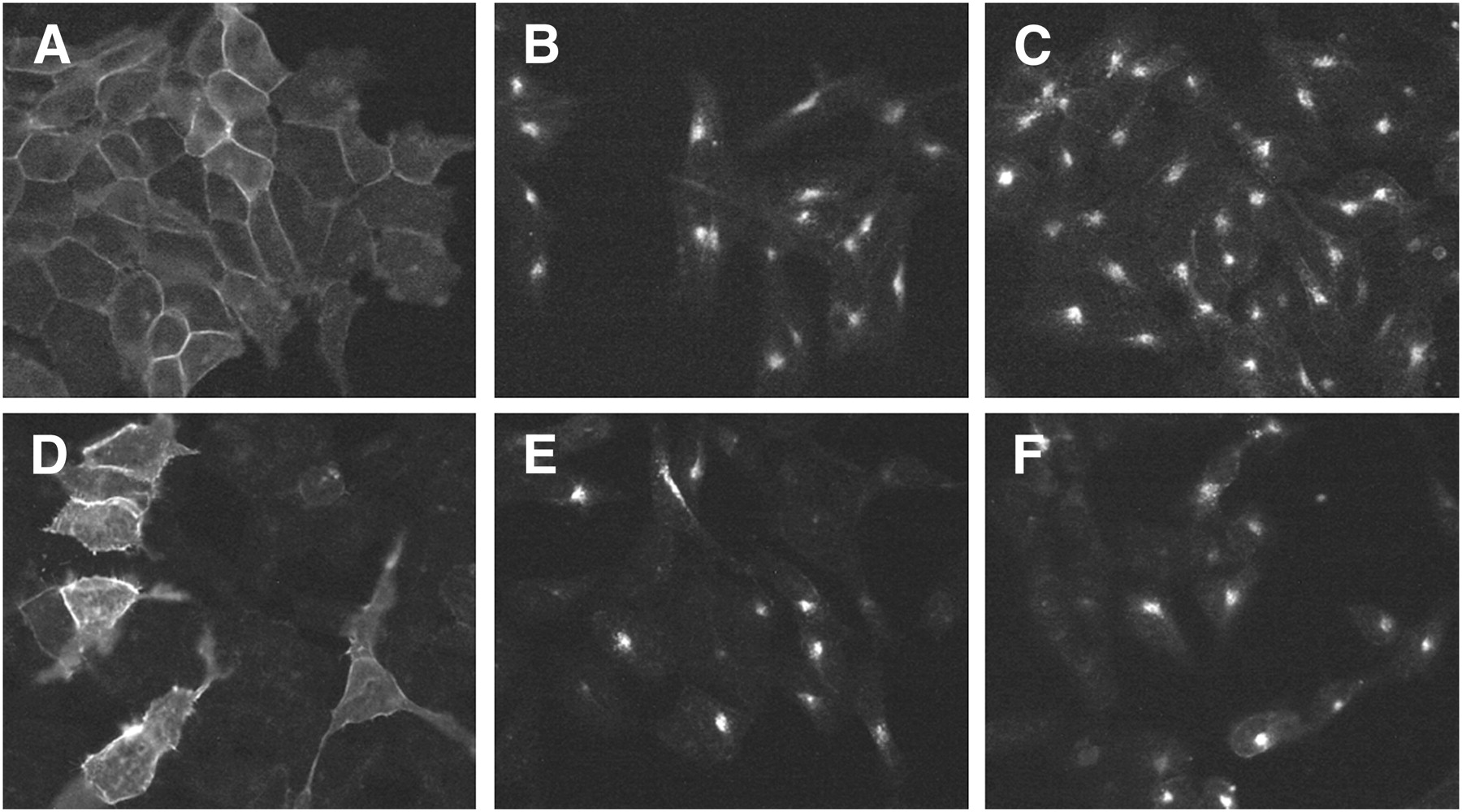
An immunofluorescence-based internalization assay using HEK-sst2 and HEK-sst3 was used to determine whether the analogs were able to stimulate receptor internalization and compared their effects with the analog of SRIF-28. Figure 2 illustrates the results of AM3, along with SRIF-28. AM3 applied to the cells for 30 min elicits a pronounced relocation of sst2 (Fig. 2C) and sst3 (Fig. 2F) from the plasma membrane to the cytoplasm, which is detectable by the strong, dotlike perinuclear staining using the R2-88 antibody for sst2 and the SS-850 antibody for sst3. SRIF-28 as a control showed the same stimulating effect (Figs. 2B and 2E).

sst2 and sst3 internalization assay showing agonistic properties of AM3. HEK-sst2 (A–C) and HEK-sst3 (D–F) cells were treated either with vehicle (A and D, negative control) or with 1 μM AM3 (C and F). As positive control, cells treated with 1 μM SRIF-28 are shown in B and E. After incubation with the peptides, cells were processed for immunocytochemistry as described in “Materials and Methods” section. AM3 exhibits agonistic activity for sst2 and sst3 internalization.
Ca2+ Release Studies
The bicyclic analogs were tested for their ability to stimulate intracellular Ca2+, a signaling pathway regulated by native somatostatin. On sst3,5, no signal was found with any of the tested analogs, except ionomycin. On sst2, a large concentration-dependent increase in intracellular Ca2+ (which corresponds to the fluorescence intensity) was revealed after stimulation with all analogs (Fig. 3). All peptides proved to be agonists, because they stimulated Ca2+ release and displayed high potencies in some cases similar to those of SRIF-14. AM3 yielded a 2-fold higher signal than AM1, HR3005 showed a response similar to that of DOTANOC, and addition of the buffer solution showed no response. The concentration-response curves, analyzed by nonlinear regression curve fitting, are shown in Figure 3B. The agonist-elicited response is quantified in terms of the half maximal effective concentration (EC50) values (nM) derived from this fit, calculated as 21.5 ± 3.5, 32.2 ± 8.5, 39.6 ± 12.7, and 60.0 ± 9.9 for AM3, DOTANOC, HR3005, and AM1, respectively.

Effect of somatostatin analogs on intracellular Ca2+ corresponding to fluorescence intensity. (A) Time course response of intracellular Ca2+ in HEK-sst2 cells loaded with Ca2+ dye and activated with indicated concentrations of AM3 and 2,000 nM SRIF-14. Data show representative experiment, typical of 3 experiments performed for each analog. (B) Concentration-response curves of intracellular Ca2+ in HEK-sst2 cells for all analogs. Results are expressed as percentage of maximal AM3 response (=100%) and represent mean ± SEM of all experiments (n = 3). RFU = relative fluorescence unit.
Enzymatic Stability in Human Blood and Serum
177Lu-AM3 was extremely stable in human blood and serum at 37°C. The erythrocytes isolated from whole blood contained less than 0.1% of the corresponding activity measured for the same blood volume. No metabolites were observed by HPLC analysis in plasma and serum after 24 h and 7 d, respectively, for which the only peak in the radiochromatograph observed was the one corresponding to 177Lu-AM3.
Radioligand Internalization and Cellular Retention Studies
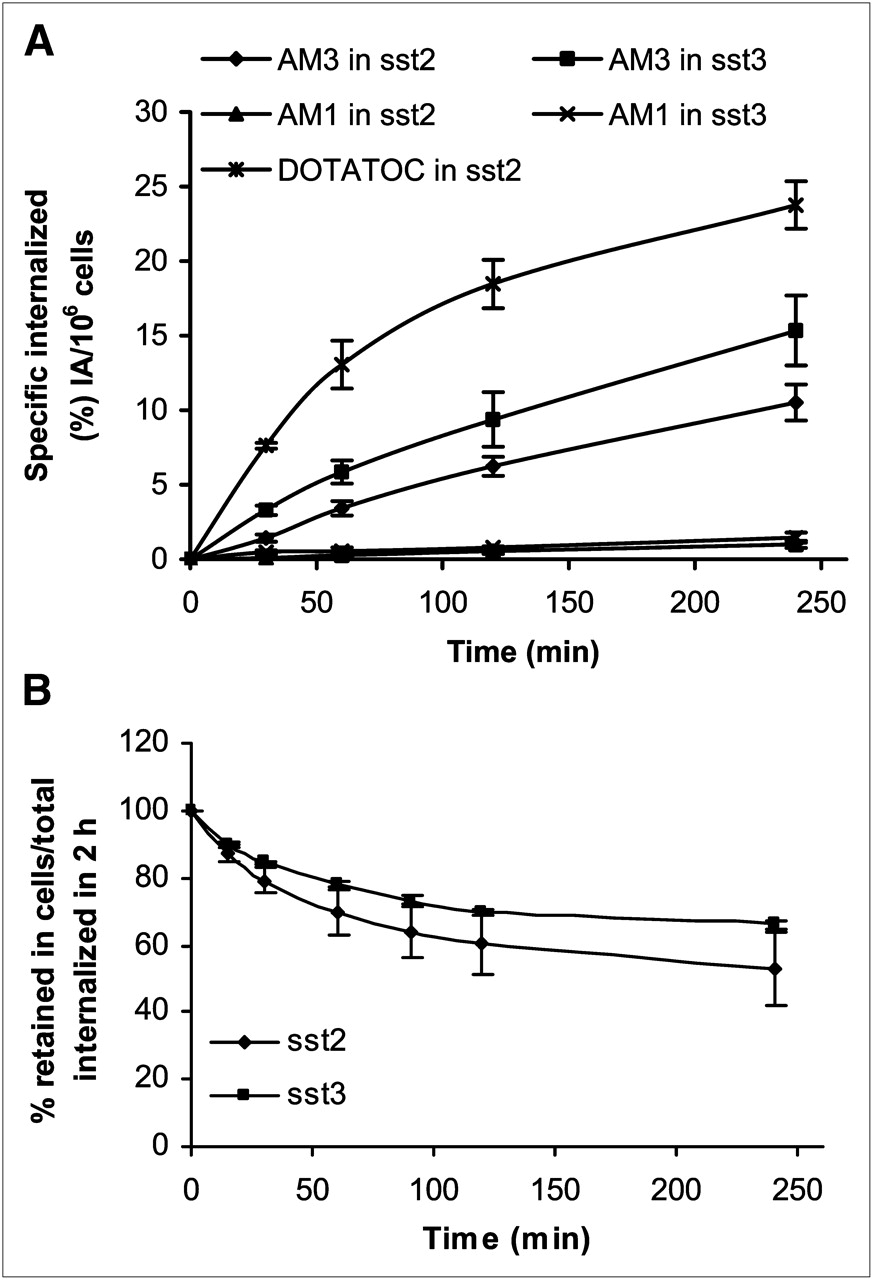
Figure 4A displays the internalization kinetics of 177Lu-AM1 and 177Lu-AM3. In HEK-sst2, 10.5 ± 1.2% of totally added 177Lu-AM3 was internalized at 4 h and in HEK-sst3 15.3 ± 2.3%. The corresponding values of 177Lu-AM1 were less than 3%. 177Lu-AM3 showed a lower internalization rate than the reference molecule 177Lu-DOTATOC (23.8 ± 1.6% in HEK-sst2). Nonspecific internalization was less than 1% (data not shown), demonstrating the receptor-mediated internalization.
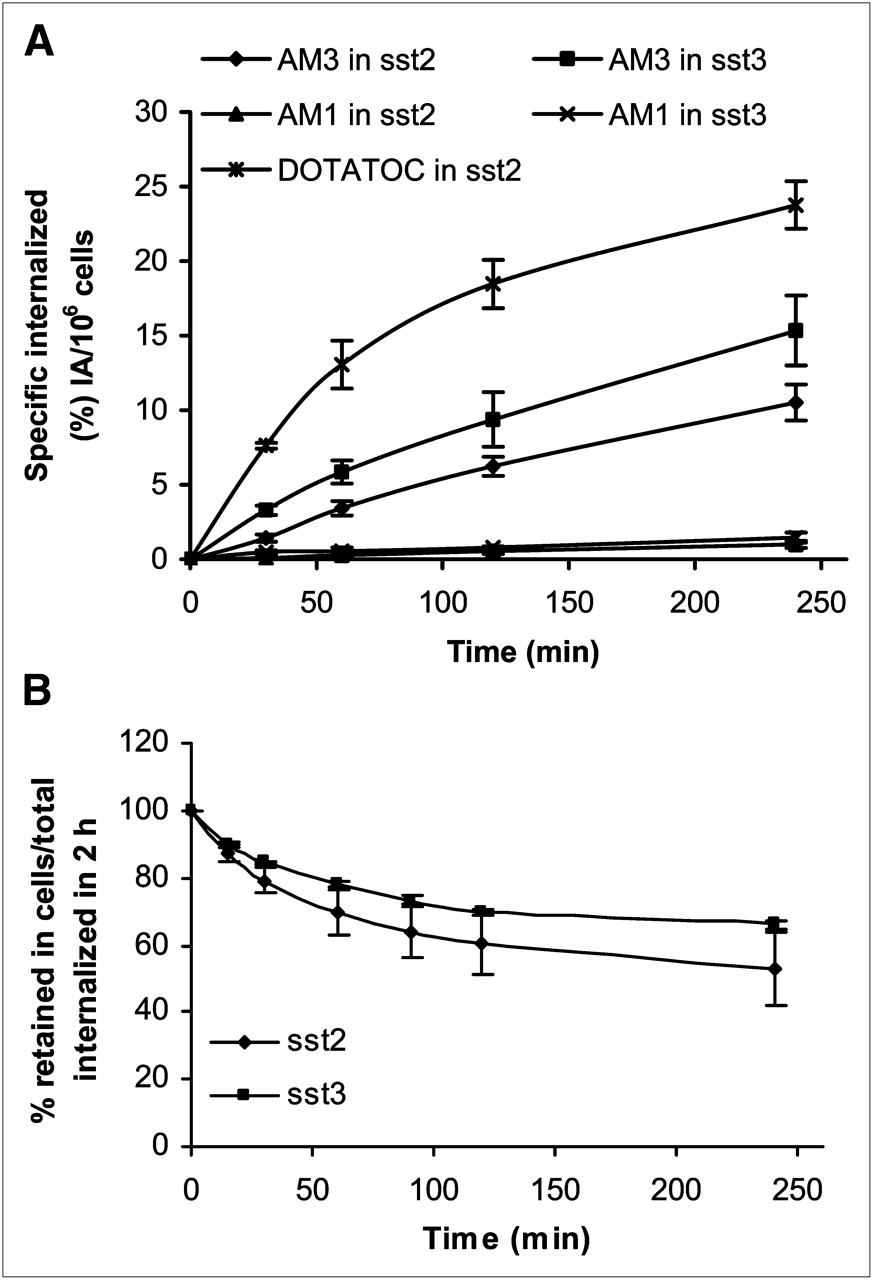
(A) Internalization rate of 177Lu-AM1 and 177Lu-AM3 into HEK-sst2 and -sst3 cells, within 4 h at 37°C, expressed as specific internalization, with bars representing ±SD. 177Lu-DOTATOC was used as reference molecule. (B) Cellular radioactivity retention (efflux) of 177Lu-AM3 in HEK-sst2 and -sst3 cells over time, expressed as percentage remaining in cells from total amount internalized (100%) at 2 h after incubation at 37°C, with bars representing ±SD. Data results are from 2 or 3 independent experiments, with triplicates in each experiment.
Cellular retention of 177Lu-AM3 in both HEK-sst2 and -sst3 cell lines is illustrated in Figure 4B. In both cases, 177Lu-AM3 seems to reach a plateau after 4 h. The efflux of 177Lu-AM3 was found to be somewhat faster in HEK-sst2 than in HEK-sst3. Within 4 h, 45%–50% of 177Lu-AM3 was released from HEK-sst2, whereas only about 35% was released from HEK-sst3.
Biodistribution Studies
The biodistribution profile of 177Lu-AM3 evaluated in nude mice bearing concomitantly sst2 and sst3 tumors showed fast blood clearance (only 0.01 %ID/g was left at 4 h after injection), fast nontarget clearance, and high receptor-mediated uptake in the tumors; the kidneys were the only other tissue accumulating radioactivity. The results are summarized in Tables 2 and 3. At 1 h after injection, both tumors accumulated the highest %ID/g, with the sst3 tumor exhibiting slightly higher uptake than the sst2 tumor. Four hours after injection, about 60% of the initially accumulated radioactivity remained in both tumors, and less than 30% was tumor-bound 24 h later. Blocking experiments with DOTANOC (high sst2,3,5 affinity) confirmed the specific and receptor-mediated uptake. The kidney uptake was not affected by the blocking agents, indicating that this uptake is not receptor-mediated. The tumor-to-kidney ratio was greater than 1, but the slower washout from the kidneys resulted in decreasing tumor-to-kidneys ratios with time (sst2/sst3-to-kidney, 1.3/1.6 at 1 h after injection and 0.8/1.2 at 24 h after injection). In the application of radiopeptides for targeted radionuclide therapy, the kidneys are dose-limiting (22). In an attempt to improve the tumor-to-kidney ratio by reducing the concentration of the radioactivity in this tissue, we preinjected lysine, which caused an approximately 50% reduction with no significant impact on the tumor uptake.
Biodistribution Results for 177Lu-AM3 in Nude Mice Bearing HEK-sst2 and HEK-sst3 Tumors
Tumor–to–Normal-Tissue Ratios for 177Lu-AM3 in Nude Mice Bearing HEK-sst2 and HEK-sst3 Tumors
The biodistribution profile of 68Ga-AM3 at early times (Fig. 5) is similar to that of 177Lu-AM3. The values were on the same level for the sst2 tumor uptake (11.92 ± 1.22 %ID/g for 68Ga and 10.37 ± 0.76 %ID/g for 177Lu), 1 h after injection, and for the tumor-to-kidney ratio (1.2 for 68Ga and 1.3 for 177Lu). In some organs of interest, such as the liver and blood, slightly higher values were observed with 68Ga than with 177Lu, but the tumor-to-tissue ratios were also high for 68Ga-AM3. A reduction of kidney uptake by 50% without any influence on the tumor uptake by preinjection of lysine was also confirmed, resulting in a significant improvement of tumor-to-kidney ratio from 1.2 to 2.1. Blocking experiments using DOTANOC proved the specific and receptor-mediated tumor uptake.

Biodistribution and blocking studies (tumor blocking [TB] with 2,000-fold excess of DOTANOC; kidney blocking [KB] with preinjection of 20 mg/100 μL concentration of lysine) of 68Ga-AM3 in sst2 tumor–bearing mice at 1 and 2 h after injection. Data are expressed as %ID/g ± SD (n = 4–7).
PET/CT Images Studies
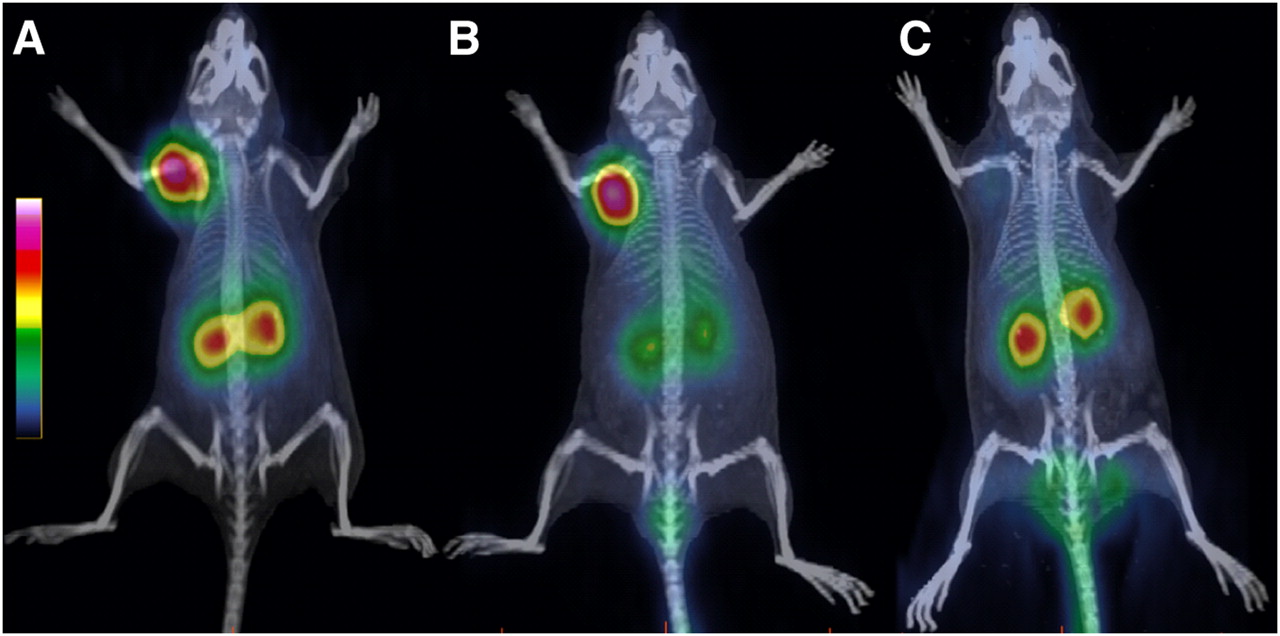
68Ga-AM3 was evaluated in nude mice bearing sst2 tumors at 1 h after injection using PET/CT (Fig. 6). The images showed clearly the high tumor and kidney uptake but negligible background. Preinjection of lysine resulted in a significant reduction of the kidney uptake. The specific uptake of 68Ga-AM3 was also confirmed using DOTANOC as a blocking agent, with which only the kidneys were visualized. The imaging studies confirmed the biodistribution data.
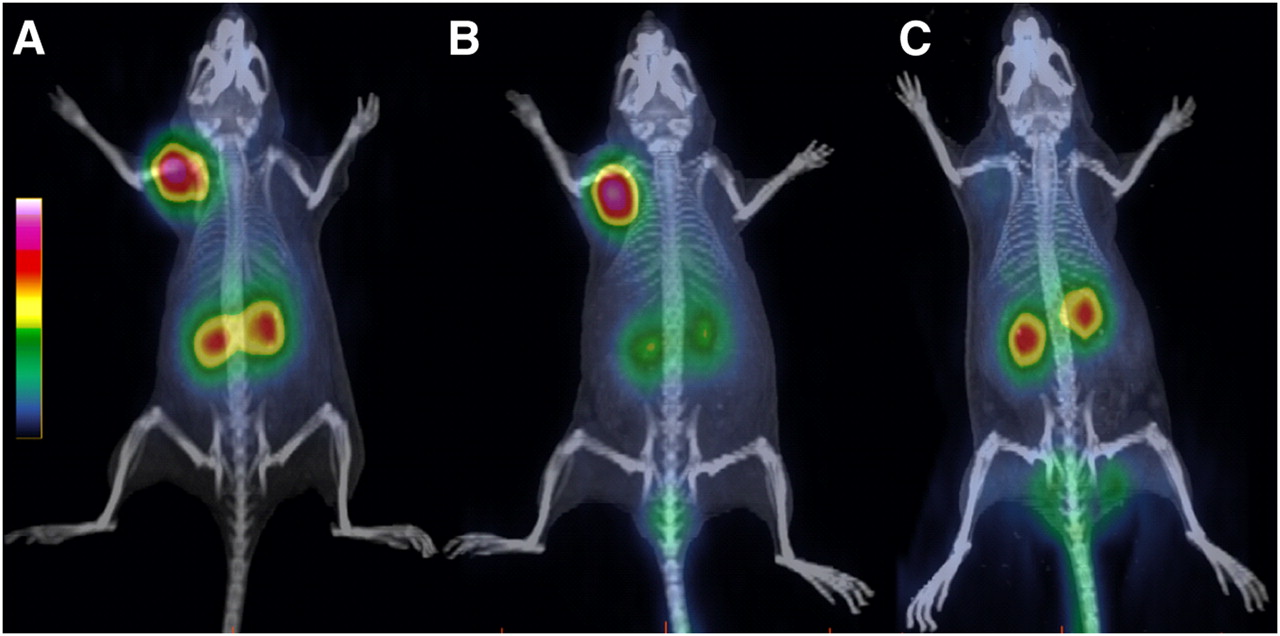
PET/CT images of sst2 tumor–bearing nude mice at 1 h after injection of 68Ga-AM3 (A) show clearly high uptake in tumor, accumulation of radioactivity in kidneys, and negligible background. (B) Preinjection of lysine reduces kidney uptake significantly and increases tumor-to-kidney ratio. (C) Coinjection of DOTANOC in excess demonstrates specific receptor-mediated uptake of 68Ga-AM3.
DISCUSSION
Radiolabeled analogs of somatostatin are being increasingly used to diagnose and treat patients with sst-positive tumors. A variety of octapeptides have been synthesized over the years to improve receptor affinity, receptor subtype profile, pharmacokinetics, and agonist and antagonist properties. Surprisingly, bicyclic somatostatin-based radiopeptides were overlooked. They were studied as cold peptides by Veber et al. (13–15), mainly to increase the metabolic stability but also to better understand the structural parameters necessary for functional activity. Later, Rivier et al. and the Peptor group along with Falb et al. used this strategy to introduce conformational constraints for sst subtype selectivity (16,17).
In a first family of peptides, we partially followed the approach of Veber et al. by keeping the 20-atom sequence of the N- and C-terminally amino acid–deleted octreotide as an inner cycle and by adding a 16-atom ring composed of the 2 amino acids Arg and GABA head-to-tail. This lead peptide showed high affinity for sst2 and sst3 comparable to SRIF-28 and also moderate affinity for sst1,4,5, indicating that the introduced conformational constraints did not result in subtype selectivity; on the contrary, the sst binding profile is broader than the profile of most octapeptides, such as DOTATOC or DOTATATE (18). GABA was exchanged with DAB (AM8), which affords an amino group for coupling of a chelator. Coupling of DOTA resulted in a distinct loss of affinity to all receptor subtypes, an effect that was also seen in octreotide-derived octapeptides but not to the same extent. We assumed that some of the loss to sst2 can be recovered by replacing Phe3 for Tyr3 (AM1 vs. AM2), as found in octreotide derivatives, but in this family of peptides the reverse effect was observed. The affinity loss with the introduction of the chelator was almost regained when introducing Tyr as a spacer between DOTA and the peptide (AM3 vs. AM1), indicating that the introduction of spacer molecules may be a means to further improve receptor affinity and subtype profile of this family of peptides.
Both the immunofluorescence-based internalization assays for sst2,3 and the Ca2+ flux for sst2,3,5 showed that the peptides are agonists on sst2 and sst3. A side-by-side comparison of the DOTA-bearing analogs with the lead peptide showed the superiority of AM3 in Ca2+ mobilization.
The internalization kinetics into HEK-sst2 of the 177Lu-labeled analogs are time-dependent but relatively slow compared with those of octapeptides such as 111In/177Lu-DOTATOC, despite the higher affinity of AM3. Also AM1, having the same sst2 affinity as DOTATOC, shows low internalization. The internalization into HEK-sst3 is somewhat faster than that into HEK-sst2, as may be explained by the higher receptor density of HEK-sst3 (23). It seems possible that the increased rigidity of the bicyclic peptides is responsible for the lower internalization rates. We have seen earlier that carbocyclic octapeptides having pansomatostatin character internalize in some receptor subtypes but not in others (10). Furthermore, the increased rigidity of the present peptides may also be the reason for the high metabolic stability found for 177Lu-AM3 in human blood and serum.
The in vivo pharmacokinetic data of 177Lu-AM3 and 68Ga-AM3 reflect the internalization and efflux data with respect to somewhat low tumor uptake and fast washout—only a hypothesis because we do not know the internalization rate in vivo of these radiopeptides. Recently, we demonstrated that in vivo internalization may be a fast process, happening within minutes if potent agonists are being injected into AR4-2J tumor–bearing rats (24).
The pharmacokinetic data are promising in respect to the fast clearance from nontargeted organs, allowing for a high tumor-to-blood and tumor-to-muscle ratio already at 1 h, increasing to around 1,000 at 4 h for both tissues. The tumor-to-tissue ratios represent important parameters to evaluate the quality of imaging agents. In our case, all organs except the kidneys show low uptake and retention. The kidney uptake of radiometallated peptides is a serious concern; usually the tumor-to-kidney-ratios are less than 1. We found higher tumor-to-kidney ratios, which are due to the relatively low kidney uptake considering the 2-plus overall charge of the radiopeptide. We and others found that positive charges increase kidney uptake (10,25,26). Recently, we found a kidney uptake of 58.7 ± 11.0 %ID/g at 4 h—a value 10-fold higher than with the present study—in the same tumor model using a pansomatostatin DOTA-coupled peptide with the same amino acid composition (10). This example shows the difficulty in understanding the exact mechanism of kidney uptake and retention (27) and that charge distribution, and possibly the metabolic stability of the peptides, are decisive for kidney uptake. The kidney uptake could be reduced by 50% by the preinjection of lysine, which is in agreement with what is known from the usual octreotide-based monocyclic radiopeptides (28).
The pharmacokinetics of 177Lu-AM3 makes AM3 an excellent candidate to be developed into an imaging agent, although we do not know whether a similar rapid washout from sst2-positive organs, such as the pancreas, will also occur in humans. AM3 seems to be comparable or even more promising than analogs with similar broad sst binding profiles, such as DOTANOC (5,8). 111In-DOTANOC showed similar uptake in sst3 tumors (17.5 ± 4.3 and 6.5 ± 0.7 %ID/g at 0.5 and 4 h, respectively) in the same tumor model (29) but tumor–to–normal-tissue ratios are superior for 177Lu-AM3. Another potent sst2-selective agonist, 111In-diethylenetriaminepentaacetic acid-TATE (inhibitory concentration of 50% (IC50), 1.3 ± 0.2 nM), shows higher tumor uptake at 4 h (15.8 ± 3.9 %ID/g) (29). However, tumor–to–normal-tissue ratios, such as tumor-to-blood and tumor-to-muscles, are superior for 177Lu-AM3, whereas the tumor-to-kidney ratio is similar. The efficient background clearance, most probably due to the high stability of the bicyclic analog and the fact that there is an increased interest in the development of new PET tracers based on the generator-produced 68Ga, led us to label this peptide with 68Ga and to evaluate 68Ga-AM3 in the sst2 tumor model. 68Ga-AM3 showed pharmacokinetics similar to 177Lu-AM3, clear tumor delineation, and receptor-mediated uptake. Besides the tumor, only the kidneys were clearly visible on PET/CT images—kidney uptake could be blocked by injecting lysine. On the other hand, in the case of diagnostic radiotracers, compared with therapeutic radiopharmaceuticals, the kidney uptake is not such a serious drawback, especially when PET isotopes with a short half-life are used.
CONCLUSION
The high rigidity of the family of bicyclic somatostatin-based analogs studied here led to agonistic ligands with almost pansomatostatin properties. This family of radiopeptides appears to constitute another group of radiopeptides, showing the importance of a high internalization capacity for a high and long-lasting tumor uptake. This importance was shown recently in a study of a group of pansomatostatin radioligands (10). Interestingly, this finding does not correlate with the properties of low or noninternalizing antagonists. Why this is the case is not clear yet and needs further study. On the other hand, high-contrast imaging depends not only on high absolute target uptake but also on an efficient background clearance, clearly demonstrated with 68Ga-AM3. This family of radiopeptides may not be suitable for targeted radionuclide therapy with a long-lived radionuclide such as 177Lu but may be a good candidate for PET/CT studies of sst-expressing tumors.
Acknowledgments
We thank Dr. Stephan Schulz for the sst2,3,5-transfected HEK cells; Dr. Andreas Bauman, Sibylle Tschumi, and the staff of the Radiological Chemistry and Nuclear Medicine Departments, University Hospital Basel, for their assistance; Novartis Pharma for analytic assistance; and the COST D38 action. This work was supported by the Swiss National Science Foundation (no. 320000-118333).
- © 2010 by Society of Nuclear Medicine
REFERENCES
- Received for publication February 26, 2010.
- Accepted for publication August 5, 2010.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- International Union of Basic and Clinical Pharmacology. CV. Somatostatin Receptors: Structure, Function, Ligands, and New Nomenclature
- Optimizing Strategies for Immune Checkpoint Imaging with Immuno-PET in Preclinical Study
- Evaluation of 177Lu-DOTA-sst2 Antagonist Versus 177Lu-DOTA-sst2 Agonist Binding in Human Cancers In Vitro
- Novel 64Cu- and 68Ga-Labeled RGD Conjugates Show Improved PET Imaging of {alpha}{nu}{beta}3 Integrin Expression and Facile Radiosynthesis
- PET of Somatostatin Receptor-Positive Tumors Using 64Cu- and 68Ga-Somatostatin Antagonists: The Chelate Makes the Difference
- Somatostatin Receptors as Targets for Nuclear Medicine Imaging and Radionuclide Treatment