Article Text

Abstract
Congenital hyperinsulinism (HI) is a clinically and genetically heterogeneous entity. The clinical heterogeneity is manifested by severity ranging from extremely severe, life threatening disease to very mild clinical symptoms, which may even be difficult to identify. Furthermore, clinical responsiveness to medical and surgical management is extremely variable. Recent discoveries have begun to clarify the molecular aetiology of this disease and thus the mechanisms responsible for this clinical heterogeneity are becoming more clear. Mutations in 4 different genes have been identified in patients with this clinical syndrome. Most cases are caused by mutations in either of the 2 subunits of the β cell ATP sensitive K+ channel (KATP), whereas others are caused by mutations in the β cell enzymes glucokinase and glutamate dehydrogenase. However, for as many as 50% of the cases, no genetic aetiology has yet been determined. The study of the genetics of this disease has provided important new information about β cell physiology. Although the clinical ramifications of these findings are still limited, in some situations genetic studies might greatly aid in patient management.
- hypoglycaemia
- sulphonylurea receptor
- ATP sensitive potassium channel
- hyperinsulinism
Statistics from Altmetric.com
Congenital hyperinsulinism (HI) is a heterogeneous entity, the genetics of which have only recently begun to be elucidated. The incidence of HI in the general population ranges from one in 27 000 in the Irish population (P Thornton, unpublished observations) to one in 40 000 in Finland,1 and one in 50 000 in the Netherlands.2 In areas of high consanguinity, the incidence can be as high as one in 3000.1 ,3 The clinical heterogeneity includes a highly variable age of onset, severity, and responsiveness to medical treatment. The pathological findings are also variable, and two distinct histopathological forms have been described. Most cases show diffuse involvement of β cells throughout the pancreas (diffuse HI), whereas some have focal adenomatous hyperplasia (focal HI), in which a distinct region of the pancreas appears to be involved, the remainder of the pancreas being histologically and functionally normal. Genetic heterogeneity has also been described. Mutations in four different genes have been detected in patients with HI, and both autosomal recessive and dominant inheritance has been demonstrated. However, in many cases the genetic aetiology is still not known. In this review, we will attempt to provide an update for each of the known subclassifications of HI, while emphasising those areas where research is still needed.
Control of insulin secretion
To understand the pathophysiology of the various forms of HI, one must understand the mechanisms responsible for glucose homeostasis. Under normal conditions, the circulating glucose concentration is regulated, primarily by insulin, to within a very tight range. Unregulated release of insulin will result in hypoglycaemia with resultant neuroglycopenia and, if uncontrolled, irreversible brain damage.
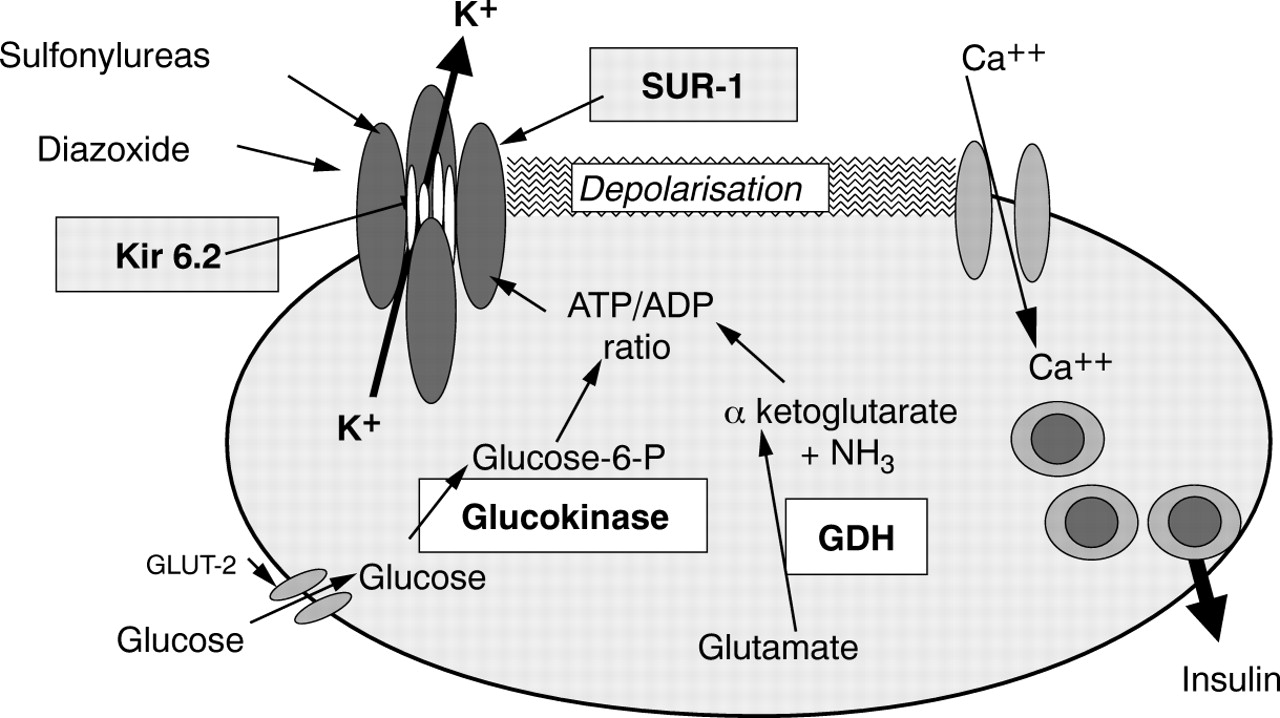
The major factors that control glucose stimulated insulin secretion are described in fig 1. Although this figure gives a highly simplified description of the regulation of insulin secretion and fails to consider multiple other pathways that modulate the response to glucose and to other stimuli, it is sufficient for the purpose of the discussion here.

The major pathways responsible for glucose regulation of insulin secretion are shown. Hyperinsulinism (HI) can be caused by mutations in the genes encoding the four proteins highlighted with boxes. The ATP sensitive potassium channel (KATP) is composed of four molecules of SUR1 and four of Kir 6.2, as shown schematically. Glucokinase is the rate limiting step in the metabolism of glucose and thus regulates changes in the intracellular ATP/ADP ratio in response to extracellular glucose concentrations. The mechanism by which activating mutations in the glutamate dehydrogenase (GDH) gene cause unregulated insulin secretion has not yet been confirmed experimentally.
The normal, resting β cell membrane is maintained in a hyperpolarised state by the Na+–K+ ATPase pump and open, ATP sensitive potassium channels (KATP). These channels sense the metabolic state of the cell. When plasma glucose increases, it enters the β cell through a specific membrane-bound glucose transporter—GLUT-2. It is then phosphorylated by the enzyme glucokinase and metabolised, resulting ultimately in the phosphorylation of ADP to ATP, thereby increasing the ATP/ADP ratio. This causes the KATP channels to close, which results in depolarisation of the cell membrane, opening of voltage dependent Ca2+ channels, and a rise in the free intracellular Ca2+ concentration. This, in turn, activates the insulin secretory mechanisms. Glucokinase is the rate limiting step in the metabolism of glucose and is thus a key step in the regulation of the secretion of insulin. The ATP/ADP ratio can be increased by metabolism of substrates other than glucose, such as amino acids. Activating mutations of glutamate dehydrogenase (GDH) cause unregulated insulin secretion, possibly by increasing the flux of substrates into the citric acid cycle, thus increasing the ATP/ADP ratio.
The KATP channel can be regulated by drugs, the most common of which are the antidiabetic sulfonylureas (tolbutamide, glibenclamide, glipizide, and others), which cause closure of the channel, membrane depolarisation, and insulin secretion. Diazoxide has the opposite effect, increasing the channel's mean open probability, thus inhibiting insulin secretion, and is commonly used in states of unregulated insulin secretion, particularly insulinomas and some cases of HI. A natural ligand for the KATP channel has been described recently (endosulphine), but its physiological role in the regulation of insulin secretion is still unknown.4 ,5
Mutations in several different genes might be predicted to cause a similar phenotype: inappropriate insulin secretion and hypoglycaemia. Mutations that decrease or destroy KATP channel activity will result in continuous depolarisation and unregulated insulin secretion. In most cases, insulin secretion will not respond to diazoxide or to tolbutamide because a functional channel is required for these drugs to exert their effect. However, some KATP mutations might cause lack of function in the natural state, yet retain the ability to respond to pharmacological intervention.
In contrast, mutations that increase nutrient metabolism and thus increase the ATP/ADP ratio will result in insulin secretion that is suppressible with diazoxide. As described below, mutations at both of these sites have been found in patients with HI. Theoretically, mutations in genes responsible for functions downstream from the KATP channel, such as the voltage gated Ca2+ channels or genes involved in the mobilisation of secretory granules, could also cause unregulated insulin secretion, although no such mutations have yet been identified.
Mutations in the β cell KATP channel
The first evidence that HI is a genetic disease came with the identification of rare families with affected siblings.6-14 More concrete evidence for autosomal recessive inheritance came from statistical analysis of relatively large groups of patients and their families.15 ,16 Linkage studies carried out in a subgroup of families selected specifically for evidence of recessive inheritance (at least two affected siblings with unaffected parents) showed linkage to chromosome 11p15.1, which was subsequently confirmed.17 ,18
Soon after the linkage was established, the β cell sulfonylurea receptor gene (SUR1) was cloned.19 The gene was located within the crucial region for the HI gene, as defined by linkage analysis, and mutations were identified in nine families with HI.20 Thus, the first HI associated gene was discovered. Soon thereafter, the gene encoding the inward rectifying K+channel (KIR6.2) was cloned and it was discovered that together with SUR1 the product of this gene formed the β cell KATPchannel.21 Dunne et al proved the association between SUR1 mutations, KATP channel function, and HI by demonstrating a lack of KATP channel function in β cells obtained at the time of pancreatectomy from a patient known to be homozygous for an SUR1 mutation.22 The KIR6.2 gene is located adjacent to the SUR1 gene on chromosome 11p and, based on the linkage data, it too is a candidate gene for HI associated mutations.23
To date, three KIR6.2 mutations24-26 and more than 40 SUR1 mutations have been reported.1 ,20 ,26-30 SUR1 mutations are spread throughout the coding region of the gene (fig 2), although there is an apparent clustering of mutations in the second nucleotide binding domain. In vitro studies of how these mutations affect channel function are providing important new information about how this protein regulates channel function.31-33 Some mutations completely eliminate channel activity, whereas others alter channel density or channel activity in response to intracellular nucleotide concentrations. A lack of response to increased MgADP concentrations appears to be a defect common to several different mutations.31 ,32 All reported SUR1 and KIR6.2 mutations appear to be recessive, because heterozygous parents and siblings are clinically normal. However, because the functioning KATP channel is a hetero-octomer composed of four molecules of SUR1 and four molecules of KIR6.2,34 and because a dominant negative KIR6.2 mutation has been engineered into the mouse homologue of this gene,35 it is possible that dominant mutations occur in humans.

Schematic of the SUR1 gene showing 39 exons (rectangles) and introns (lines connecting the exons). Neither is drawn to scale. The two nucleotide binding domains are shaded in grey. Mutations published to date are shown. Missense and nonsense mutations are shown above the gene, whereas intronic mutations, insertions, and deletions are depicted below the gene. Superscripts indicate references in which the specific mutations are described: 1, Thomas and colleagues20; 2, Nichols and colleagues31; 3, Thomas and colleagues27; 4, Nestorowicz and colleagues28; 5, Aquilar-Bryan and Bryan26; 6, Nestorowicz and colleagues29; 7, Otonkoski and colleagues1; and De Lonlay-Debeney et al.30
Most of the mutations are extremely rare, and most were found to be unique to specific families. However, some exceptions exist. In the Ashkenazi Jewish population, at least 88% of the disease associated chromosomes carry one of two specific mutations.28 More recently, a founder mutation (V187D) was found in Finnish patients with HI. Eighteen of 42 disease associated chromosomes from Finnish patients carried this mutation.1 In contrast, out of a group of 16 Irish patients, mutations have been identified in only three probands, and in each case there is a different mutation (P Thornton and B Glaser, unpublished data, 1999).
Interestingly, in over half of the patients screened for mutations in the entire SUR1 and KIR6.2 coding regions, no mutation was identified. To date, no systematic search of the promoter regions of these genes has been reported, although such studies are currently in progress.
Mutations in the glutamate dehydrogenase gene
Recently, patients were described with HI and apparently asymptomatic hyperammonaemia. No known defects of ammonia metabolism were identified.36 ,37 Increased glutamate dehydrogenase (GDH) activity was found in peripheral lymphocytes from these patients, and genomic mutations in the regulatory domain of the enzyme were identified.38 To date, six mutations in this gene (GLUD-1) have been described in patients with the hyperinsulinism hyperammonaemia syndrome. All are located in the allosteric regulatory domain of the enzyme, and all are dominant mutations (fig 3). Some families with multigeneration dominant inheritance have been identified, whereas the other mutations were identified as de novo mutations in affected probands. One particular mutation (S445L) appears to represent a mutation hot spot because it has been found to occur de novo in three independent cases. GDH catalyses the reversible reaction converting glutamate to α-ketoglutarate, a substrate for the citric acid cycle in the β cell (fig 1). The precise mechanism by which these mutations cause the hyperinsulinism hyperammonaemia syndrome has not been confirmed; however, the known function of this enzyme has led to a plausible hypothesis, although this has yet to be tested experimentally.38 Increased enzyme activity might result in increased flux of amino acid substrates through α-ketoglutarate into the citric acid cycle, resulting in increased conversion of ADP to ATP, closure of the KATP channel, membrane depolarisation, and finally insulin secretion. This increased secretion will occur without any correlation with glucose concentrations, but is triggered by high protein diets, which increase the amino acid substrate availability and therefore increase insulin secretion. Indeed, these patients exhibit a pronounced worsening of their hypoglycaemia after high protein intake and have prolonged, severe hypoglycaemia after an oral leucine load. Thus, many of these patients would have been described in the early HI literature as having leucine sensitive hypoglycaemia. In the liver, increased activity of this enzyme also results in increased glutamate to α-ketoglutarate conversion, thus increasing ammonia production and decreasing glutamate concentrations. Glutamate is an essential substrate for the formation of N-acetyl glutamate (NAG), an allosteric stimulator of carbamoyl phosphate synthetase. Decreased concentrations of NAG cause decreased carbamoyl phosphate synthetase activity and accumulation of ammonia by inhibition of the urea cycle. Treatment with diazoxide is effective in most patients, consistent with the hypothesis that the defect is proximal to the KATPchannel. Recently, however, two unrelated patients with hyperinsulinism hyperammonaemia syndrome were described who have mutations in the allosteric domain of GDH and are unresponsive to diazoxide treatment (P Delonlay et al, unpublished data). This suggests that the mechanism by which GDH mutations cause hyperinsulinism might be different from that presented above. The hyperinsulinism hyperammonaemia syndrome appears to be a rare cause of hyperinsulinaemic hypoglycaemia because only five of 170 probands with HI in our series were found to have mutations in this gene (B Glaser, unpublished data, 1998). Eleven other patients with hyperammonaemia are currently being investigated; three were resistant to diazoxide. So far, in five, no mutations have been found in the allosteric domain (C Junien, unpublished data, 1999).

{kind=link}
{kind=link}
{kind=link}
Schematic diagram of the glutamate dehydrogenase gene (GLUD1) showing the exons as rectangles and the introns as lines connecting the exons. The locations of the six published mutations in exons 11 and 12 are shown.38
Glucokinase mutations
A family with a unique form of HI with five affected individuals in three generations was described recently.39 Two patients had detailed evaluation of the control of insulin secretion that revealed qualitatively normal control, with good insulin responses to both intravenous and oral glucose load, and suppression of insulin secretion during insulin induced hypoglycaemia. However, fasting and postprandial glucose concentrations were low, below the threshold of symptomatic neuroglycopenia. The data suggested a defect in the glucose threshold at which insulin secretion is turned off. In normal individuals, insulin secretion is completely inhibited at about 4 mmol/litre glucose.40 In these patients, however, apparent inhibition of insulin secretion occurred at 2 mmol/litre, resulting in both fasting and reactive hypoglycaemia. Glucokinase is the rate limiting step in the metabolism of glucose, and acts as the cellular sensor of glucose concentrations. Indeed, mutations in glucokinase that decrease enzymatic activity cause one form of autosomal dominant type 2 diabetes (maturity onset diabetes of the young). Genomic DNA from the proband of this family was screened for mutations in the glucokinase gene (GK) and a missense mutation (V455M) in exon 10 was identified. Linkage analysis confirmed that the mutation co-segregated with clinical disease, and in vitro studies of the mutant enzyme revealed a considerably increased affinity for glucose. This defect readily explains the clinical phenotype in these patients. As expected, these patients respond well to diazoxide treatment. We did not find this mutation in any other proband with HI, and neither did we find any GK mutation in any of the other six families with apparent autosomal dominant inheritance. In a separate cohort of 30 infants with hyperinsulinism, systematic screening of the entire coding sequence failed to identify any GK mutations (C Bellané-Chantelot, unpublished data, 1999). Thus, we conclude that this form of HI is probably exceedingly rare.
Phenotype–genotype correlations
The clinical phenotypes associated with GDH and GK mutations are described above, and for these mutations, the phenotypes and genotypes appear to correlate quite well. In contrast, when trying to correlate the clinical phenotype with specific SUR1 mutations, the situation is quite different. All mutations identified to date are recessive, and most are rare. Therefore, most patients with HI are compound heterozygotes for HI mutations, or only a single mutation is identified. In either case, correlations between the clinical disease and the in vitro function of the mutant channel is difficult or impossible. However, some patients are homozygous for specific mutations, usually because the patients come from a genetically isolated population, or because of consanguinity.
Mutations “SUR1 949 del c” and “KIR6.2 T12X” result in termination codons (the former by means of a frameshift) early in the coding sequence of SUR1 and KIR6.2, respectively. Individuals homozygous for each of these mutations have been identified and all have clinically very severe disease that is unresponsive to diazoxide or somatostatin analogue. The latter drug has been used effectively in some patients with HI,41 and appears to work by inhibiting both KATP channel closure and insulin secretion at more distal sites in the insulin secretory cascade. Interestingly, these mutations do not appear to be associated with any defects in other organ systems, suggesting the neither of these two proteins are crucial for the normal function of other tissues or organs.25 ,29
Mutation “SUR1 4310 g→a” results in abnormal exon splicing and the complete removal of the second nucleotide binding domain of SUR1.20 β Cells isolated from a patient homozygous for this mutation failed to show any KATP channel activity.22 The resultant phenotype was, as expected, very severe disease, unresponsive to diazoxide treatment. Two other mutations have been described in homozygous patients. The delF1388 mutation is found in approximately 20% of the Ashkenazi Jewish HI associated chromosomes, but only two patients homozygous for the mutation have been described. As expected from the in vitro studies that show no channel activity if this mutation is present, both patients had very severe disease. Both were treated with somatostatin analogue along with frequent feeding and night time continuous intragastric feedings, and neither underwent surgery. The V187D mutation has been reported in 15 of 24 cases from central Finland. Six of the patients were homozygous and nine heterozygous for the mutation. Interestingly, all of these patients were equally severely affected. This mutation eliminates KATP channel function and, as expected, results in a very severe, drug unresponsive phenotype.1
The splice site mutation (3992 –9 g→a), found in approximately 70% of Ashkenazi Jewish HI associated chromosomes, is of particular interest. Most patients homozygous for this mutation have severe disease that responds poorly to treatment with diazoxide. However, we have identified two families with multiple siblings in which the proband was homozygous for this mutation and had severe HI, whereas three siblings, who were also homozygous for the same mutation, had very mild disease or were clinically unaffected (H Landau, personal communication, 1997). These are both Ashkenazi families in which this is a founder mutation.42 Therefore, the severe and the mild cases were all genetically identical at this locus, a finding that was confirmed by haplotype analysis. This observation suggests that some splice mutations might be variably expressed and that, with different genetic backgrounds, a different percentage of normal protein might be produced. A similar phenomenon has been found in patients with a splice site mutation in the gene encoding 17 hydroxylase.43 Most patients homozygous for this mutation have severe congenital adrenal hyperplasia; however, asymptomatic homozygous patients with apparent near normal enzyme function have been identified.
Occasionally, the genotype and phenotype can be correlated with electrophysiological properties of β cells obtained from pancreatectomy specimens. Although in some cases the correlation is excellent,1 ,22 in others clinical and in vitro findings do not correlate (P Thornton and M Dunne, unpublished data, 1999). This might be because of the difficulty in differentiating cases of focal disease (see below) from diffuse disease using the current techniques of β cell isolation.
HI as a result of focal lesions
Ever since HI was initially described, there has been controversy about the histopathological findings associated with the clinical syndrome. One of the major controversies related to the existence of a group of patients in whom only a part of the pancreas is affected—so called focal HI.11 ,44 Although careful histological examination did demonstrate focal HI in some cases, the pathophysiology of the entity was not known until very recently. de Lonlayet al reported loss of maternal chromosome 11p in the focal lesions of 10 probands with HI.45 A similar finding was previously reported in one adult patient with insulinoma.46 The chromosome loss apparently occurs during development of the pancreas, and affects only β cells in one specific region of the gland. The precise timing and location of the somatic event determines the location and extent of the affected region. The chromosome deletion contains at least two imprinted, maternally expressed genes (p57KIP2 and H19) that encode important cell cycle regulators, and an imprinted, paternally expressed gene encoding a growth stimulator—insulin like growth factor 2 (IGF2). Thus, in β cells with the maternal allele deleted, the growth suppressing genes are not expressed, whereas the growth stimulating gene is normally expressed, a combination that results in abnormal proliferation of affected β cells. However, these patients have very severe, diazoxide unresponsive HI, and thus, abnormal β cell proliferation alone is not sufficient to explain the clinical phenotype. In all patients reported by de Lonlay et al, the SUR1 and KIR6.2 loci were within the region deleted; however, there is no evidence to suggest that either of these two genes is imprinted, so alternative explanations were needed.
Recently, Ryan et al and Verkarreet al described five patients with focal HI, loss of the maternal chromosome 11p in the lesion, and a heterozygous germline mutation on the paternal SUR1 allele.47 ,48Another seven patients with focal HI, all presenting with a paternally inherited germline mutation, have been subsequently identified (JC Fournet et al, unpublished data, 1999).49 Somatic loss of the maternal allele of chromosome 11p in a patient carrying a SUR1 mutation on the paternal allele results in reduction to hemizygosity or homozygosity for the mutant allele in the affected β cells. Therefore, the β cells within the focal lesion will proliferate, presumably because of unbalanced production of tumour growth suppressing and growth stimulating factors, and will lack functional KATP channels. This will cause unregulated insulin secretion that is unresponsive to drug treatment. Therefore, the combination of a paternally inherited SUR1 mutation, along with a second somatic event causing the loss of chromosome 11p will create a proliferating β cell population that secretes insulin in an unregulated manner, thus completely explaining the clinical phenotype of focal HI.
It is not possible to study the natural history of focal HI directly because the definitive diagnosis of focal disease can only be made at the time of surgical resection. However, a cohort of medically treated patients was recently reported who are heterozygous for SUR1 mutations on the paternal chromosome. The unique genetic characteristics of this cohort suggested that most of the patients have focal disease. Interestingly, these patients appear to enter clinical remission after a mean of about 16 months of medical treatment. A review of histology from three other patients with confirmed focal lesions showed increased β cell apoptosis within the focal lesion. It was hypothesised that, if treated medically, focal lesions will undergo apoptosis and disappear, leaving the patient with normal β cell function.49
The importance of these findings is clear, but how they should be incorporated into the clinical decision making process is less clear. In diffuse HI, all β cells are abnormal. Therefore, partial pancreatectomy invariably leads to persistent hypoglycaemia or early diabetes, depending on the extent of the resection. Most, if not all, of these patients will eventually develop insulin requiring diabetes during childhood or adolescence.50 In sharp contrast, in patients with focal HI, the remaining pancreas is functionally normal. Therefore, selective excision of the focal region results in complete cure.30 However, if patients with focal HI do enter complete remission after medical treatment, as suggested by the above cited study, then perhaps surgery can be avoided, because even under the best circumstances, pancreatic surgery carries with it a certain morbidity and mortality.
In the absence of long term follow up data on medically treated patients with confirmed focal lesions, it seems logical to decide whether or not to operate on a particular patient with focal HI based on the degree of difficulty encountered when attempting medical treatment. This, however requires a preoperative diagnosis of focal HI, and this has proved to be very difficult. Trans-hepatic portal venous sampling of insulin concentrations during induced, controlled hypoglycaemia has been used extensively and appears to be a reliable, albeit a technically very difficult, procedure that is not available in most centres.51 Recently, it was proposed that the insulin response to intravenous tolbutamide could be used to differentiate diffuse from focal disease. The test is based on the hypothesis that patients with focal HI will have a considerable β cell mass that has normal KATP channels and therefore will respond to tolbutamide, whereas in patients with diffuse HI, the entire β cell mass is affected and, hence, no response to tolbutamide will be seen. Although preliminary data suggest that this may in fact be a clinically useful test (CA Stanley and A Grimberg, personal communication), many more patients need to be tested at the time of diagnosis, before this test can be recommended for clinical use. Particularly worrying is the fact that for about 50% of patients the molecular diagnosis is not known, and therefore the response to tolbutamide cannot be predicted. Some patients with insulinomas respond to tolbutamide with a massive output of insulin and severe, prolonged hypoglycaemia. It is not known whether some forms of HI will respond similarly. Despite these difficulties, the search for an effective test is worthwhile, because the ability to diagnose focal HI preoperatively could radically change the clinical approach to patients with HI.
Clinical ramifications of genetic findings
HI is a genetically heterogeneous entity. During the past three years major advances have been made in our understanding of the molecular genetic aetiology of this syndrome. Although these discoveries have provided important new information about β cell physiology, the clinical usefulness of these findings is still limited. However, in some situations, the recent discoveries have already been translated into clinically useful tests providing rapid clinical diagnosis and providing the basis for more precise genetic counselling.
Hyperinsulinsim with hyperammonaemia is a new syndrome, the genetics of which are now known. Previously, the finding of hypoglycaemia and raised concentrations of ammonia suggested a disorder of fatty acid oxidation. In light of these discoveries, the differential diagnosis of patients with neonatal or infantile hypoglycaemia should be extended to include the hyperinsulinsm and hyperammonaemia syndrome. If raised or borderline ammonia concentrations are found, rapid genetic diagnosis is now possible in most cases. In contrast to most patients with HI, children with GDH mutations usually respond to diazoxide treatment, and in most surgery can be avoided. In many, the disease is caused by a de novo mutation, suggesting that the chances that a subsequent sibling will be affected are extremely small. In other families, autosomal dominant inheritance can be documented, meaning that future siblings have a 50% chance of being affected.
The clinical usefulness of genetic evaluation in patients with HI without hyperammonaemia is more complicated. In as many as 50% of these patients, no mutation may be found in the coding regions of SUR1 and KIR6.2. The genetic diagnosis in these cases is still totally unknown. Even in patients with SUR1 or KIR6.2 mutations, in most populations no founder mutation has been described. Because the gene is very large (39 exons), screening of the entire coding sequence using conventional techniques is not feasible in the clinical setting. In sharp contrast, in populations in which a founder mutation is known, genetic analysis can provide very important clinical information. In the Ashkenazi Jewish population, we routinely use mutation analysis to aid the clinical diagnosis. Frequently, a genetic diagnosis can be made even before results on hormone analysis are obtained. Furthermore, genetic analysis is now used routinely for genetic counselling in this population.
Because of the unique clinical characteristics of focal HI described above, and because modern, non-invasive imaging techniques are not useful in diagnosing this entity, rapid genetic diagnosis would be very valuable clinically. Unfortunately, because of the large number of possible mutations in the SUR1 gene, this is not yet possible in most populations. In populations with founder mutations, genetic analysis can provide evidence for, but not definitive proof of, the presence of this entity. For example, in Ashkenazi patients, if two mutant alleles are identified, or if a single mutation is identified on the maternal allele, focal HI can be excluded. In the latter case, the presence of a novel, unidentified mutation on the paternal allele is postulated. If a single mutation is identified on the paternal allele, then focal HI is suggested; however, given current technology, the presence of another, novel mutation on the maternal allele cannot be entirely excluded. It is in this instance that studies of the acute insulin response to different stimuli might be the key to preoperative diagnosis and might alter the management of the patients (see discussion above).
Nomenclature
Since its initial description, the clinical syndrome of hyperinsulinaemic hypoglycaemia of infancy has been referred to by a large number of different descriptive names. Now that the aetiology of the disease is becoming known, we believe that it is time to standardise the nomenclature used to describe the syndrome. We suggest using the term “hyperinsulinism (HI)” as a general term. When other clinical or histological characteristics are known these should be stated, such as HI/HA for the hyperinsulinaemia hyperammonaemia syndrome and focal HI for focal disease. If the genetic aetiology is known, then the mutated gene can be added to the name, such as HI-SUR1, HI-KIR6.2, HI-GK, or HI-GLUD1 for patients with confirmed mutations in the subunits of the β cell KATP channel, GK, or GDH genes, respectively. Using this nomenclature it will become easier to identify the precise clinical, biochemical, and physiological characteristics of each specific disease. Comparison of clinical and other data from different centres will be made easier. Furthermore, this approach might facilitate the identification of subgroups of patients with novel forms of this syndrome
Summary
The genetic analysis of HI has provided important new information about β cell physiology. The clinical ramifications of these findings are still limited, although in specific populations genetic analysis is already making a considerable impact on the clinical management of these patients. Given the recent advances in rapid sequence analysis technology, genetic analysis of the entire SUR1 and KIR6.2 genes might be feasible in the clinical setting in the near future, thus making the clinical usefulness of genetic analysis available to all populations. However, it is important to emphasise that for as many as 50% of sporadic HI cases, genes other than the four described above might be responsible. For these patients especially, further genetic studies are needed to define the pathophysiology of disease more accurately.
Acknowledgments
This manuscript is the product of a collaboration supported by grant BMH4-CT98–3284, awarded as part of the European Commission Biomed 2 programme. Updated information on neonatal hyperinsulinism is available on the European Network for Research into Hyperinsulinism (ENRHI) website: http://www.phhi.u-net.com.