Iodine-124: A Promising Positron Emitter for Organic PET Chemistry

Abstract
:1. Introduction
2. Production, processing, and PET imaging of 124I
2.1. 124I production routes

{kind=link}
{kind=link}
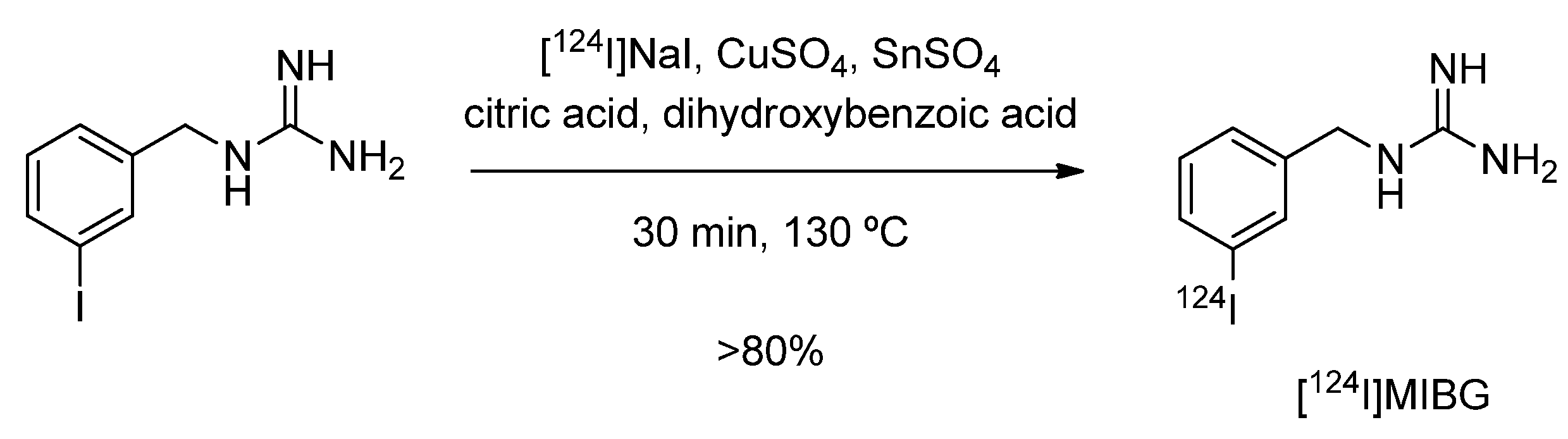{kind=link}
{kind=link}
{kind=link}
{kind=link}
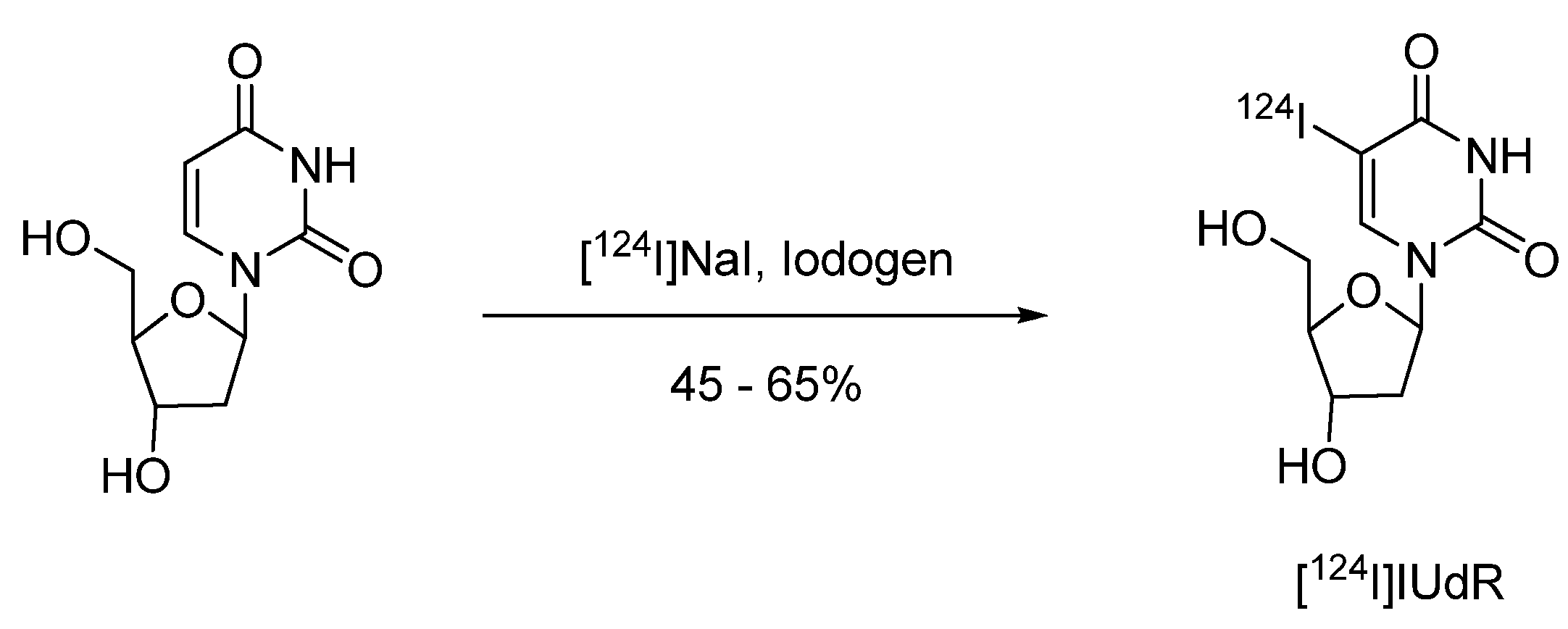{kind=link}
{kind=link}
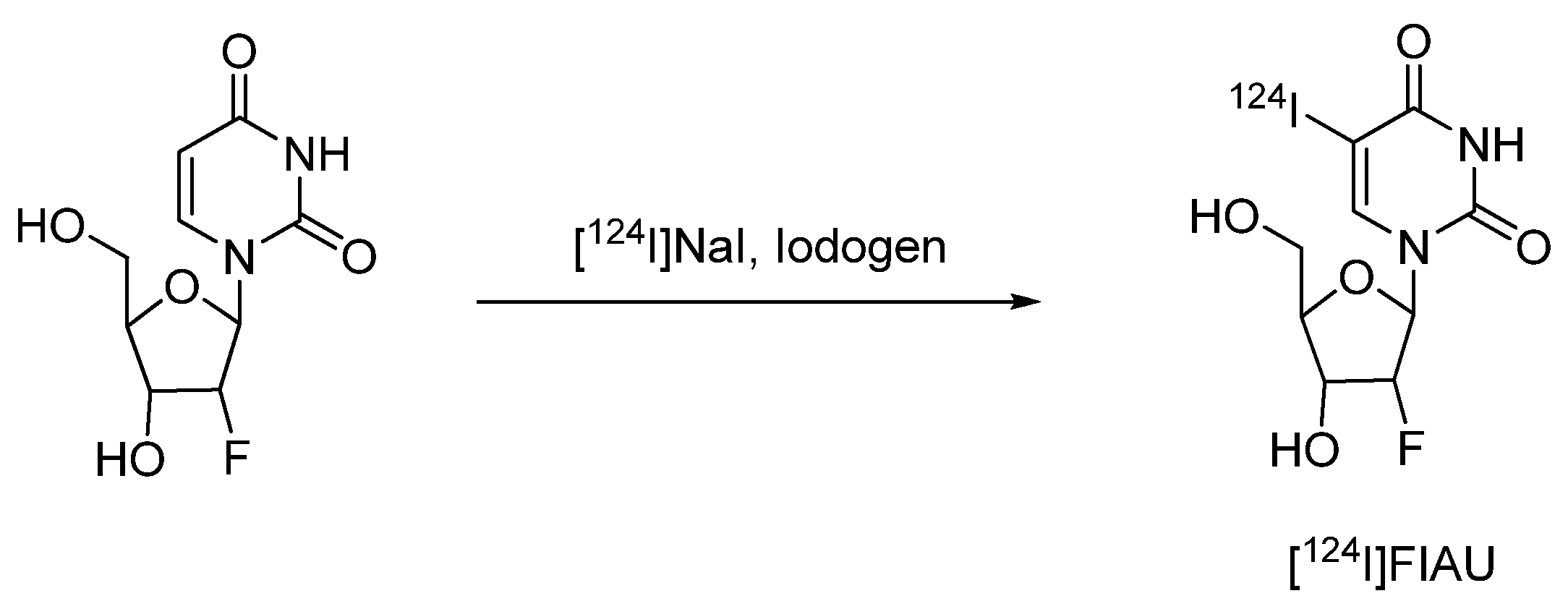{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nuclear reaction | Effective Energy [MeV] | Targetmaterial | Enrichment [%] | Yield [MBq/μAh] | Radioiodine impurities [%] | Reference |
|---|---|---|---|---|---|---|
| 124Te(p,n)124I | 13→9 | Te | 99.51 | 20 a | 123I(41) | [17] |
| 12.2→0 | TeO2 | 99.8 | 13 | 123I(10.039), 125I(0.018), 126I(0.041), 130I(0.379) | [15] | |
| 13.5→9 | TeO2 / 5% Al2O3 | 99.8 | 5.8 | 125I(0.01), 126I(<0.0001) | [18] | |
| 12.5→5 | TeO2 | 99.3 | 9.0 ± 1.0 | 125I(0.053 ± 0.015) | [19] | |
| 11→2.5 | TeO2 / 6% Al2O3 | 99.5 | 6.40 ± 0.44 | 125I(<0.02), 126I(<0.001) | [20] | |
| 14→7 | TeO2 / 5% Al2O3 | 99.86 | 21.1 | 125I(0.03), 126I(0.007) | [21] | |
| 125Te(p,2n)124I | 20.1→10.5 | TeO2 | 93 | 43.3 | 123I(8), 125I(5) | [25] |
| 22→4 | Te | 98.3 | 111a | 125I(0.89) | [26] | |
| 21→15 | Te | 98.3 | 81 a | 123I(7.4), 125I(0.9) | [24] | |
| 22 | TeO2 | 98.5 | 104 | 123I(<1) | [27] | |
| 126Te(p,3n)124I | 36.8→33.6 | Te | Nat | 67 a | -- | [28] |
| 38→28 | Te | > 98 | 148 a | 123I(84), 125I(1.5), 126I(1.4) | [29] | |
| 123Te(d,n)124I | 11→6 | Te | 91.0, 85.4 | 2.8 a | 123I(3321 b) | [30] |
| 124Te(d,2n)124I | 15→0 | Te | 95 | 20.4 ± 2.2 | 126I(0.5) | [12] |
| 15→8 | Te | 91.7 | 18.9 | 125I(0.35 b), 126I(0.39 b), 131I(0.08 b) | [13] | |
| 16→6 | Te | 96.7 | 23.7 a | -- | [14] | |
| 14→0 | TeO2 | 89.6 | 15 | 123I(1.16), 125I(1.41), 126I(1.16), 130I (7.87), 131I(0.31) | [15] | |
| 14→10 | Te | 99.8 | 17.5 a | 125I(1.7) | [16] | |
| natSb(α,xn)124I | 22→13 | Sb | Nat | 1.02 a | 123I(892 b), 125I(13 b), 126I(0.16 b) | [31] |
| 121Sb(α,n)124I | 22→13 | Sb | 99.45 | 2.11 a | 123I(891 b), 125I(<0.2), 126I(<0.2) | [31] |
| natSb(3He,xn)124I | 35→13 | Sb | Nat | 0.95 a | 121I(37700 b), 123I(3877 b), 125I(0.6), 126I(0.6) | [32] |
2.2. Thermal design and irradiation considerations
2.3. Processing: Dry distillation of the 124I
2.4. PET imaging of 124I
3. Basic Principles of Radiochemistry with Radioiodine Isotopes
3.1. Nucleophilic substitution reactions
3.2. Electrophilic substitution reactions
3.3. Radioiodination of peptides and proteins
4. Examples for 124I-labeled Compounds
4.1. 124I-labeled compounds prepared via nucleophilic exchange reactions
4.1.1. meta-[124I]Iodobenzylguanidine ([124I]MIBG)

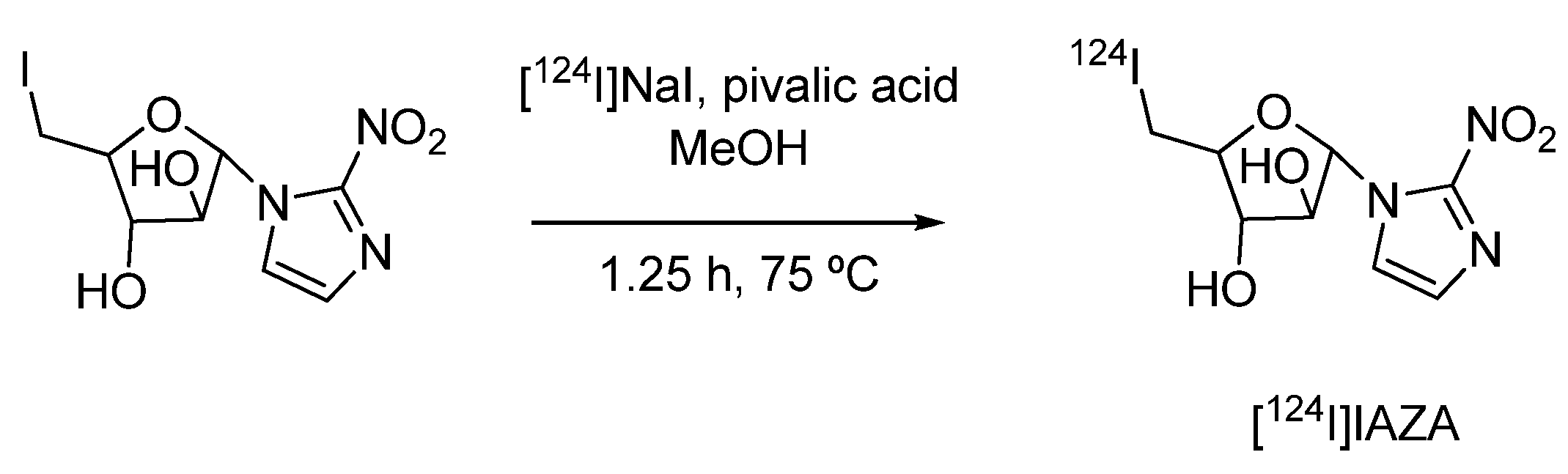
4.1.2. 1-α-D-(5-deoxy-5-[124I]iodo-arabinofuranosyl)-2-nitroimidazole ([124I]IAZA)

4.1.3. [124I]Iodo-azomycin-galactopyranoside ([124I]IAZG)

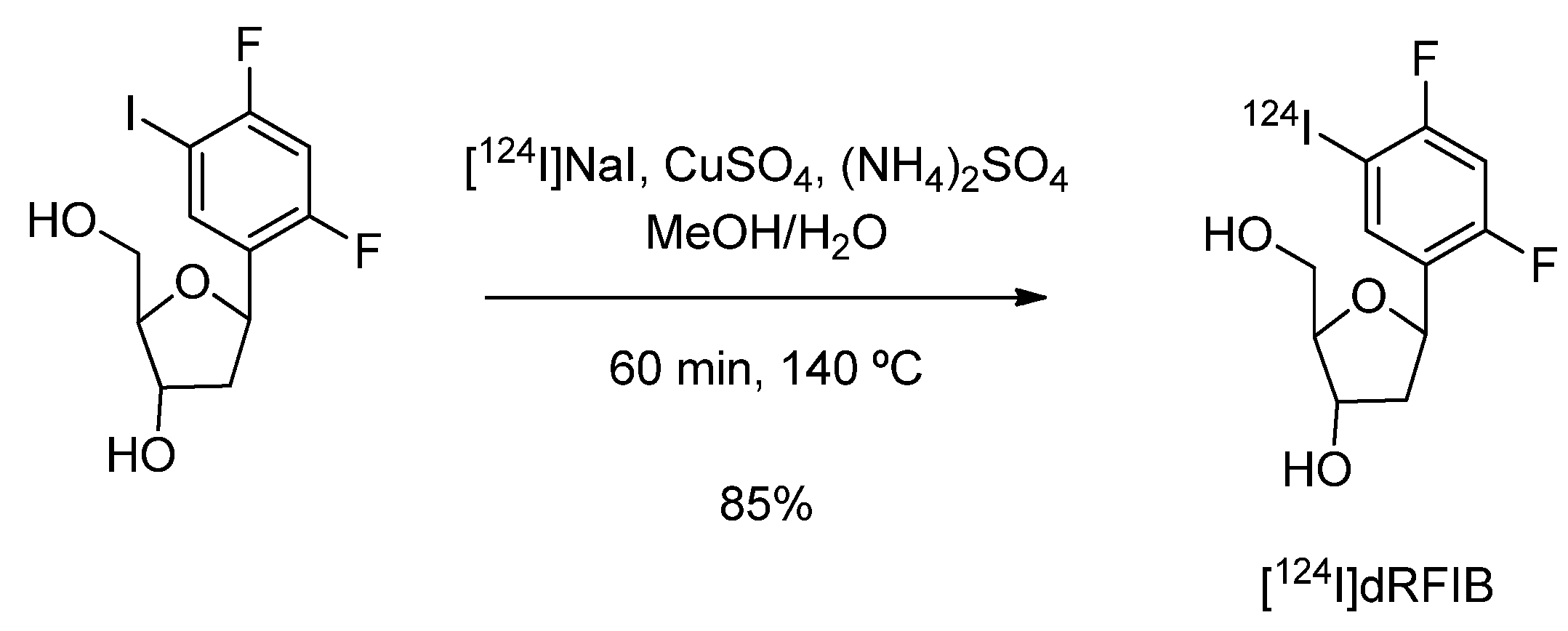
4.1.4. 1-(2-Deoxy--β-D-ribofuranosyl)-2,4-difluoro-5-[124I]iodobenzene ([124I]dRFIB)

4.2. 124I-labeled compounds prepared via electrophilic substitution reactions
4.2.1. Direct electrophilic substitution on activated aromatic systems
4.2.1.1. 5-[124I]Iodo-2′-deoxyuridine ([124I]IUdR)

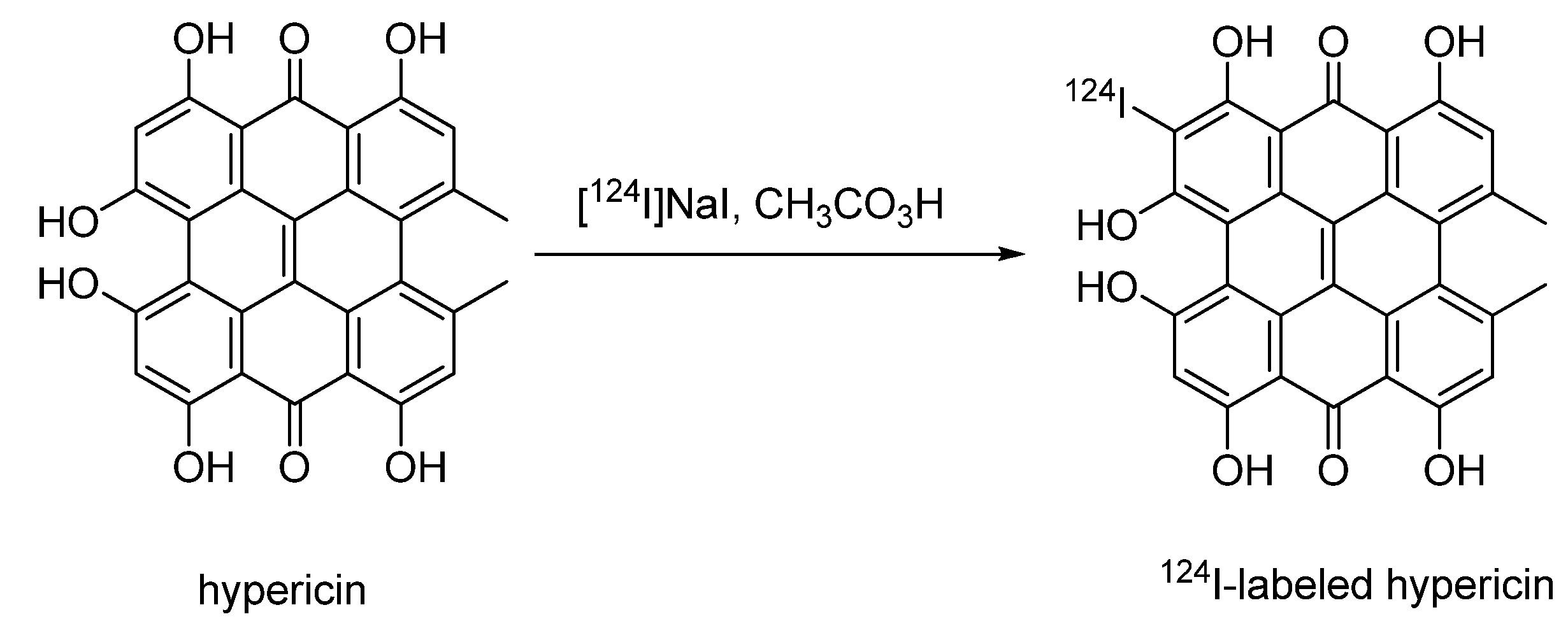
4.2.1.2. 124I-labeled hypericin

4.2.1.3. 2'-Fluoro-2'-deoxy-1-β-D-arabinofuranosyl-5-[124I]iodouracil ([124I]FIAU)

4.2.2. Electrophilic radioiodo-destannylation reactions
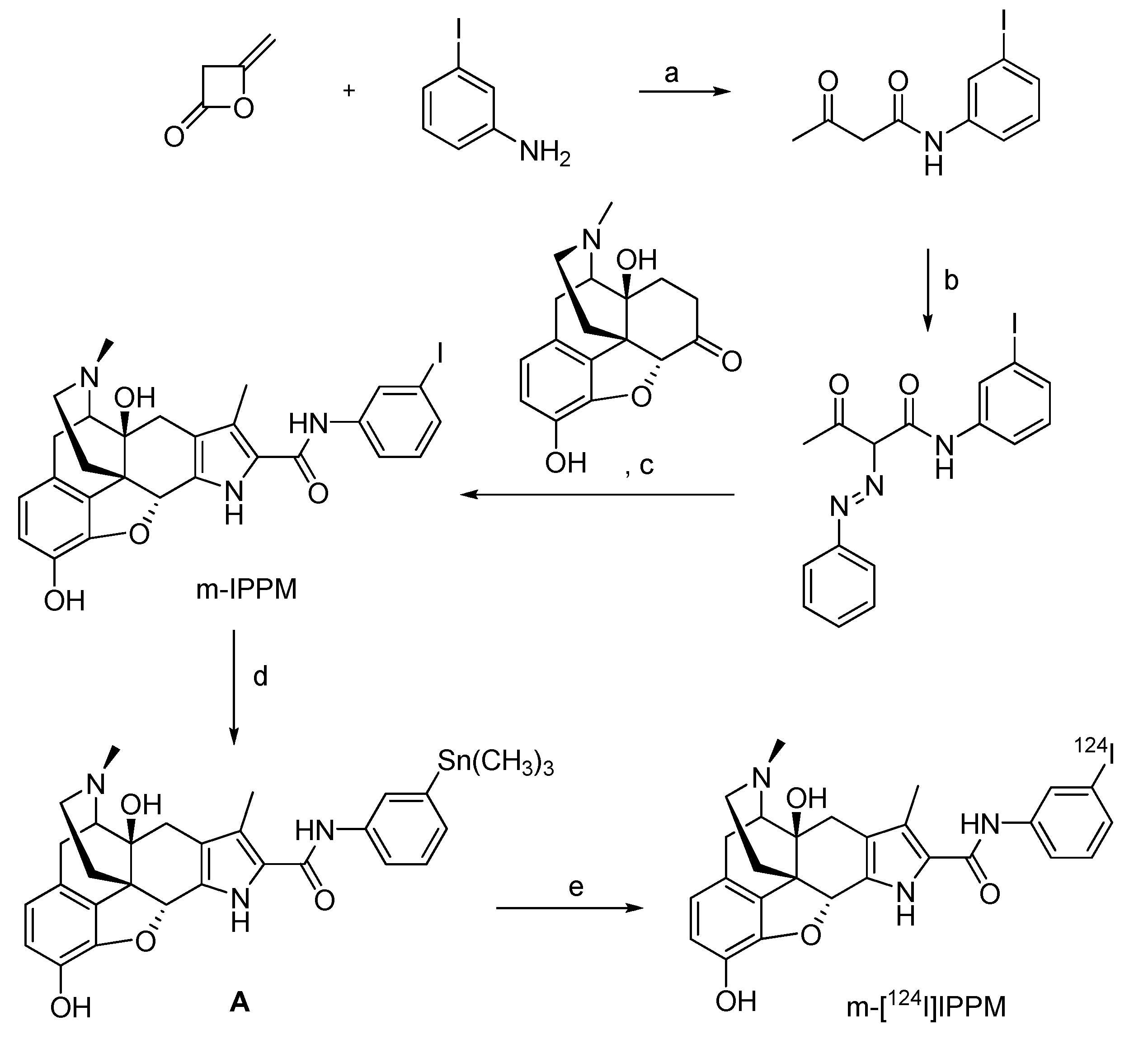
4.2.2.1. 124I-labeled m-iodophenylpyrrolomorphinan (m-[124I]IPPM)

4.2.2.2. [4-(3-[124I]iodoanilino)-quinazolin-6-yl]amide-(3-morpholino-4-ylpropyl)amide (mor-pholino-[124I]IPQA)

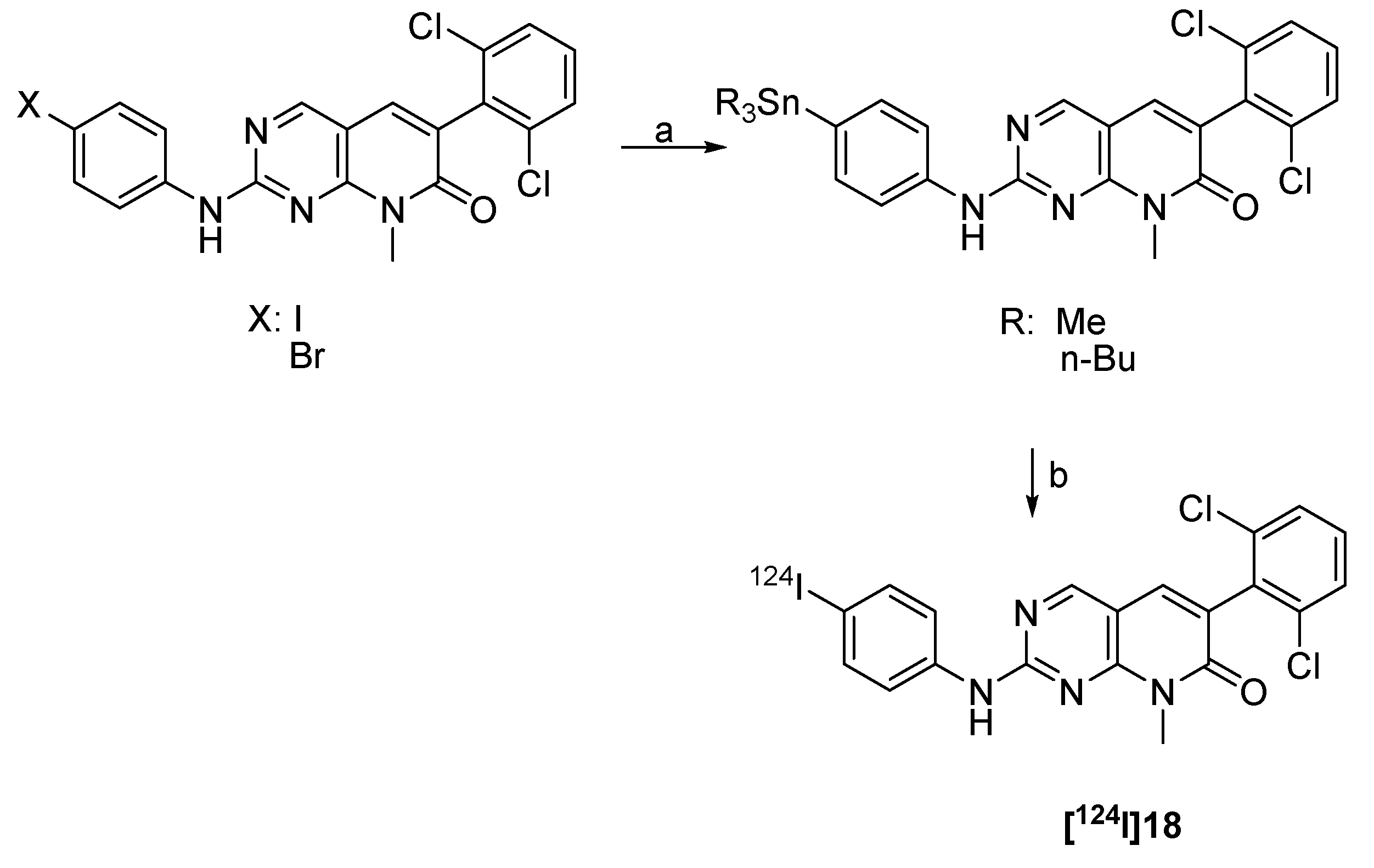
4.2.2.3. 124I-labeled 6-anilino-quinazoline derivatives as EGFR inhibitors

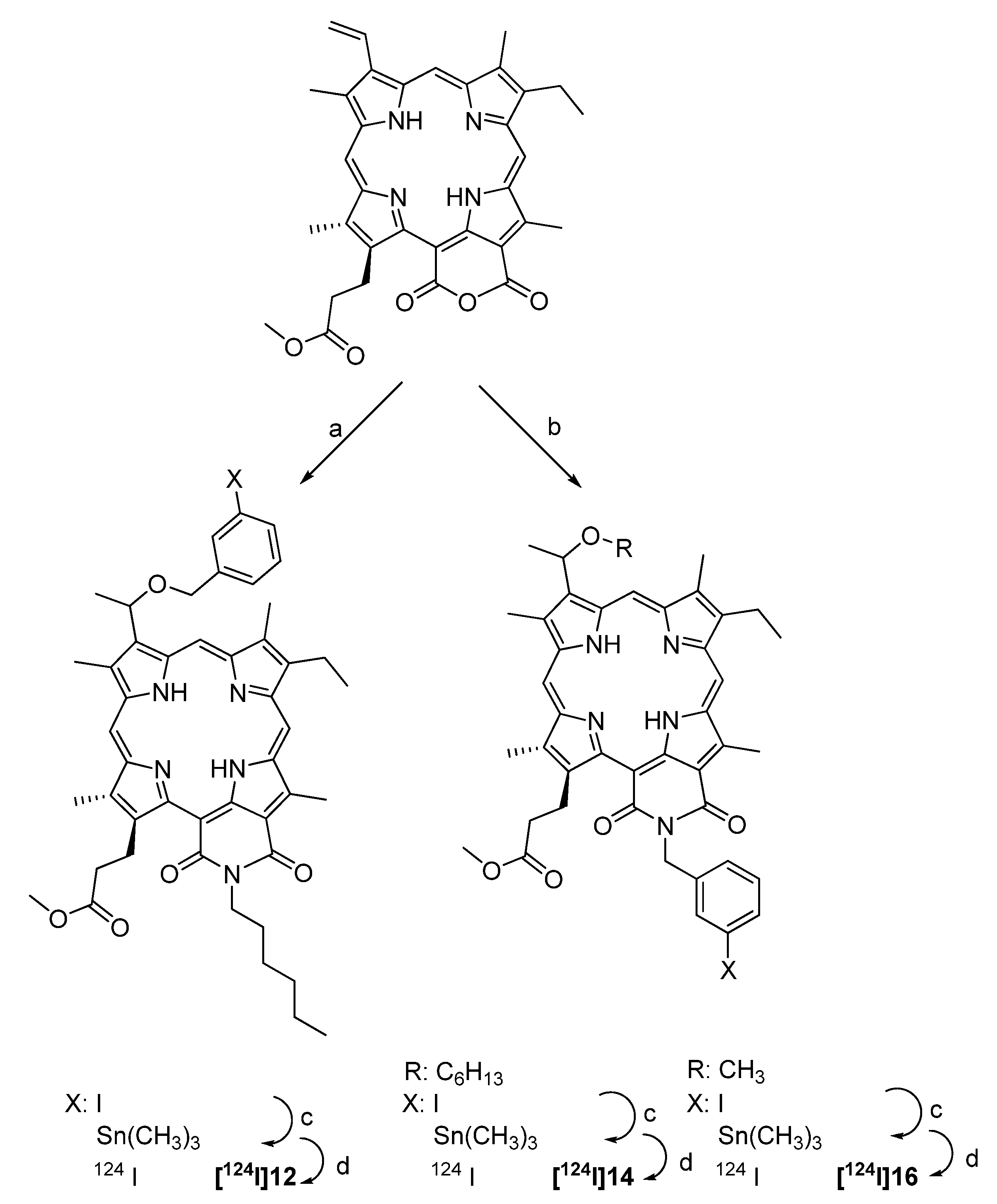
4.2.2.4. Synthesis of 124I-labeled purpurinimide derivatives

4.2.2.5. Synthesis of 124I-labeled 2-(4-iodophenylamino) pyrido[2,3-d]pyrimidin-7-one

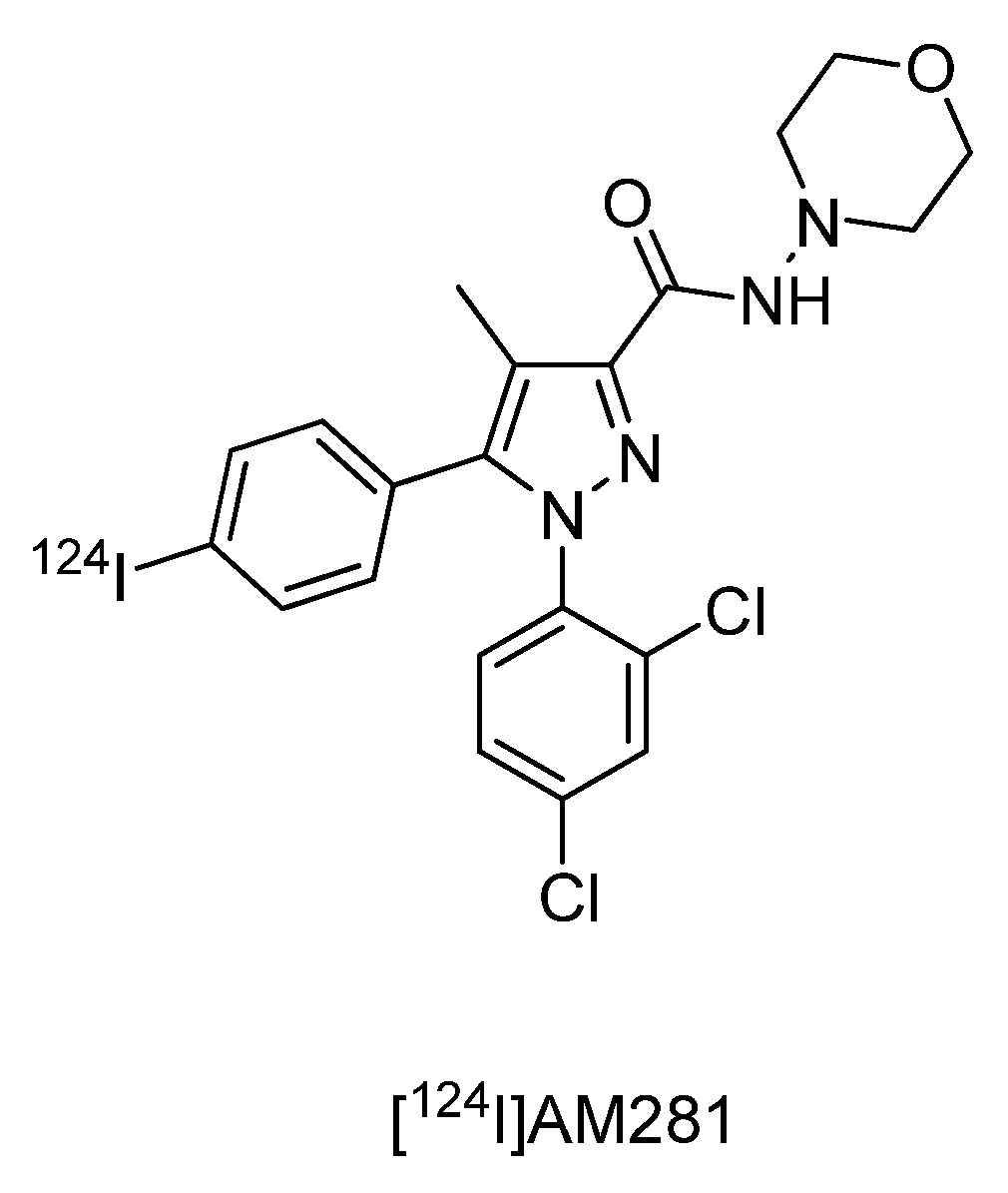
4.2.2.6. Synthesis of n-(morpholin-4-yl)-1-(2,4-dichlorophenyl)-5-(4-[124I]iodophenyl)-4-methyl-1h-pyrazole-3-carboxamide ([124I]AM281)

4.2.2.7. Synthesis of 124I-labeled CDK4/6 inhibitors

4.3. Radioiodination of peptides and proteins
4.3.1. Direct labeling of peptides and proteins with 124I
| Method | Imaging target | Antibody | Results | Reference |
|---|---|---|---|---|
| Iodogen® | HER2 | Anti-HER2 | RCY: 13.8–18.8%; | [98] |
| AS: 33-102 MBq/mg, | ||||
| positive tumours | Diabody | RCP: >93.5% | ||
| Apoptotic cells | Annexin V | RCY: 20–70% | [99,100] | |
| Annexin V | RCY: 6–12% | [101,102,103] | ||
| RCP: 95% | ||||
| MBP-Annexin V | [104] | |||
| CD44v6 | cMAb U36 | RCY: 72% | [105,106] | |
| positive cells | RCP: >97% | |||
| AS: 32.6 MBq/mg | ||||
| squamous cell | ||||
| carcinoma | ||||
| CEA | Anti-CEA | RCY: 33–88% | [107] | |
| mini- and diabodies | ||||
| CAT-method | Apoptotic cells | Annexin V | RCY: 4-6%, | [103] |
| RCP: >99%, | ||||
| AS: 8.6 GBq/µmol | ||||
| Human glioma | 3F8 mAb | RCY: >90% | [108,109,110] | |
| A33 mAb | RCY: 55% | [110] | ||
| Colon cancer | huA33 mAb | RCY: 40%, | [111] | |
| RCP >98%, | ||||
| AS: 7.4 MBq/mg | ||||
| CEA | mAb CE-25, | RCY: 70–80%, | [112] | |
| CE 4-8-13 | ||||
| Ovarian cancer | MX35 or MH99 | RCY: >87% | [113] | |
| NBS-method | Breast cancer | C-erbB2 | RCY: 96% | [114] |
| HMFG1 | [115] |
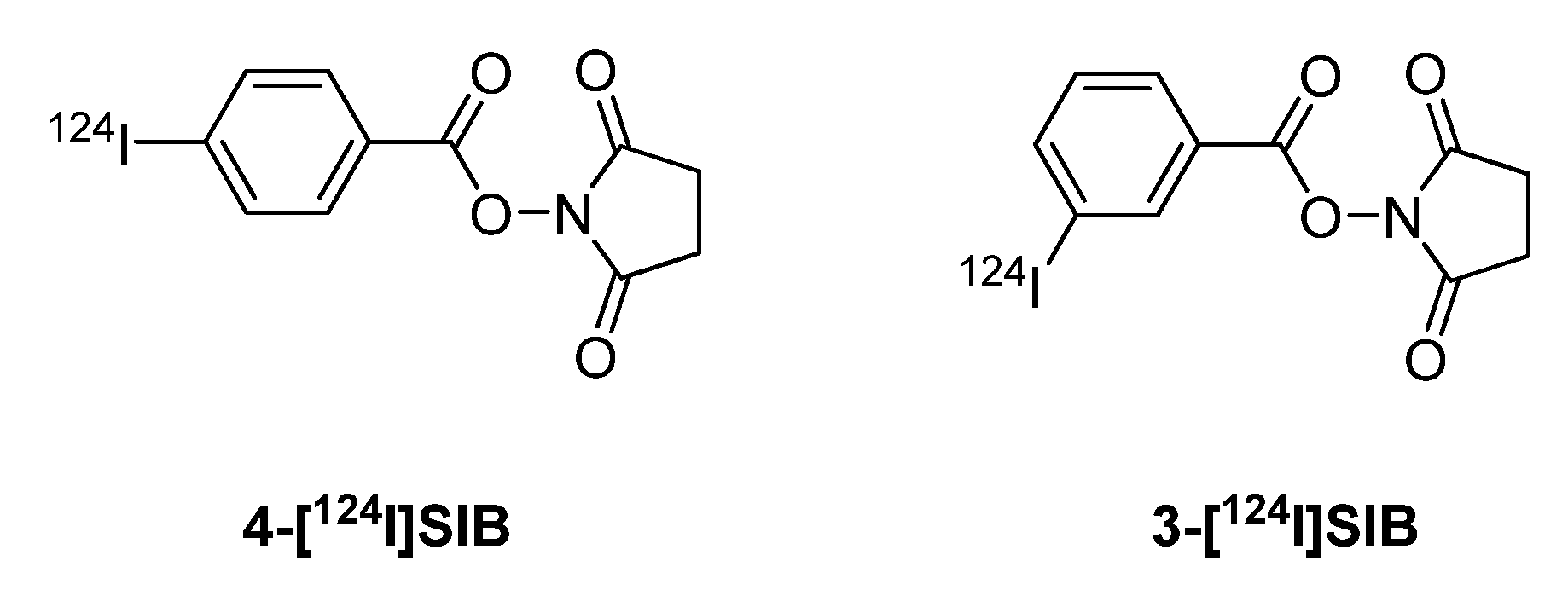
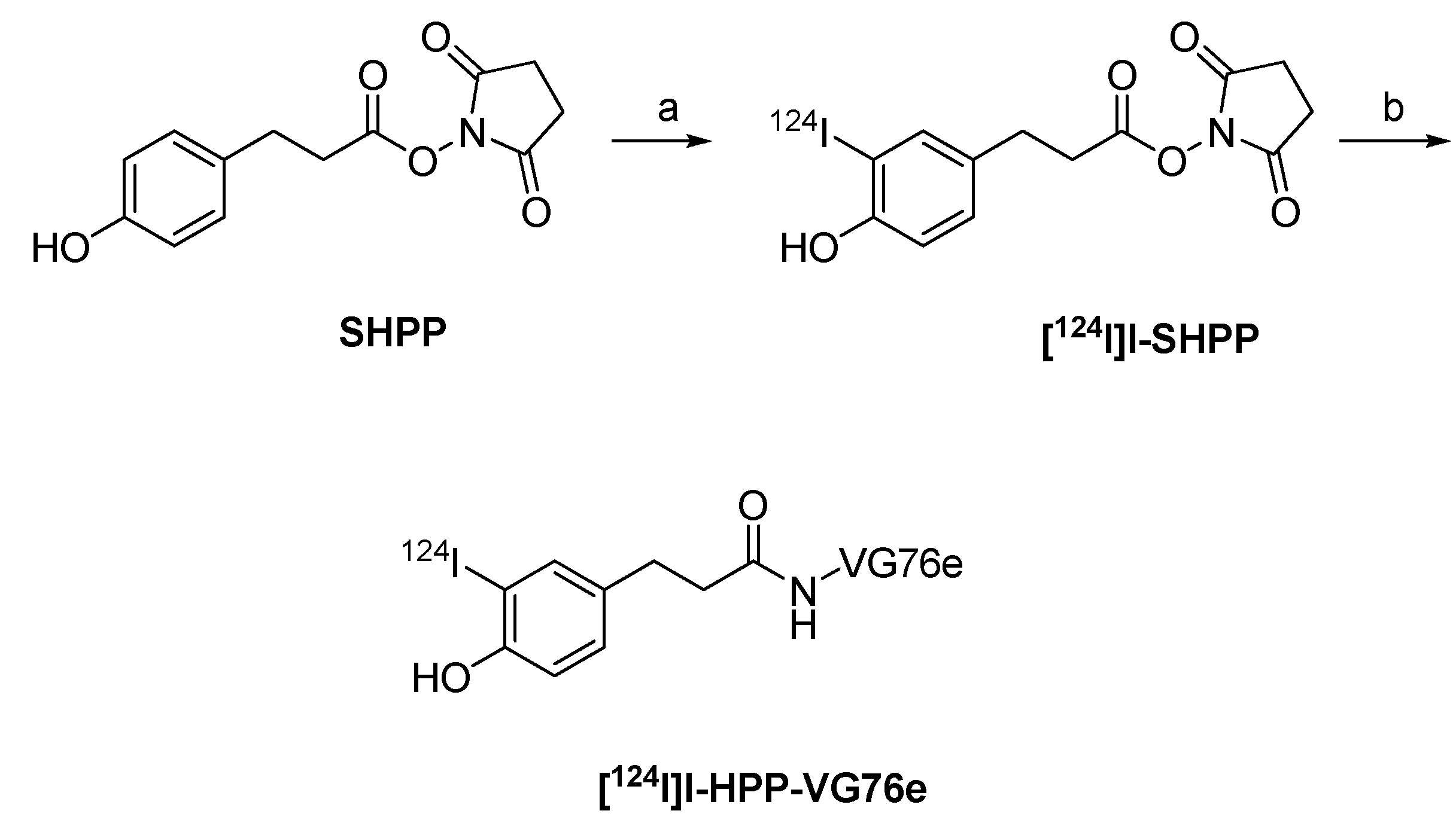
4.3.2. 124I-labeled prosthetic groups for radiolabeling of peptides and proteins




5. Summary and Conclusions
References
- Weissleder, R.; Pittet, M.J. Imaging in the era of molecular oncology. Nature 2008, 452, 580–589. [Google Scholar] [CrossRef]
- Blasberg, R.G. Molecular imaging and cancer. Mol. Cancer Ther. 2003, 2, 335–343. [Google Scholar]
- Palladino, F.; Canadè, A.; Bianchi, A.; Lesti, G.; Antoniol, O.M.; Macis, G.; Marano, P. Molecular imaging: state of the art. Rays 2003, 28, 45–61. [Google Scholar]
- Phelps, M.E. PET: the merging of biology and imaging into molecular imaging. J. Nucl. Med. 2000, 41, 661–681. [Google Scholar]
- Phelps, M.E. Inaugural article: positron emission tomography provides molecular imaging of biological processes. Proc. Natl. Acad. Sci. USA 2000, 97, 9226–9233. [Google Scholar] [CrossRef]
- Paans, A.M.; van Waarde, A.; Elsinga, P.H.; Willemsen, A.T.; Vaalburg, W. Positron emission tomography: the conceptual idea using a multidisciplinary approach. Methods 2002, 27, 195–207. [Google Scholar] [CrossRef]
- Willemsen, A.T.; van den Hoff, J. Fundamentals of quantitative PET data analysis. Curr. Pharm. Des. 2002, 16, 1513–1526. [Google Scholar]
- van den Hoff, J. Principles of quantitative positron emission tomography. Amino Acids 2005, 29, 341–353. [Google Scholar] [CrossRef]
- Welch, M.J.; Laforest, R.; Lewis, J.S. Production of non-standard PET radionuclides and the application of radiopharmaceuticals labeled with these nuclides. Ernst Schering Res. Found. Workshop 2007, 62, 159–181. [Google Scholar]
- McQuade, P.; Rowland, D.J.; Lewis, J.S.; Welch, M.J. Positron-emitting isotopes produced on biomedical cyclotrons. Curr. Med. Chem. 2005, 12, 807–818. [Google Scholar] [CrossRef]
- Fujibayashi, Y.; Suzuki, K.; Fukumura, T.; Mori, T.; Kasamatsu, S. Non-standard radionuclide production for PET in Japan. Q. J. Nucl. Med. Mol. Imaging 2008, 52, 140–144. [Google Scholar]
- Lambrecht, R.M.; Sajjad, M.; Qureshi, M.A.; Al-Yanbawi, S.J. Production of iodine-124. J. Radioanal. Nucl. Chem. Lett. 1988, 127, 143–150. [Google Scholar]
- Sharma, H.L.; Zweit, J.; Downey, S.; Smith, A.M.; Smith, A.G. Production of 124I for positron emission tomography. J. Labelled Compd. Rad. 1988, 26, 165–167. [Google Scholar]
- Firouzbakht, M.L.; Schlyer, D.J.; Finn, R.D.; Laguzzi, G.; Wolf, A.P. Iodine-124 production: excitation functions for the 124Te(d,2n)124I and 124Te(d,3n)123I reactions from 7 to 24 MeV. Nucl. Instrum. Meth. B 1993, 79, 909–910. [Google Scholar] [CrossRef]
- Knust, E.J.; Weinreich, R. Yields and impurities in several production reactions for 124I. In Proc. 7th Workshop on Targetry and Target Chemistry, Heidelberg, Germany, June 8–11, 1997; pp. 253–262.
- Bastian, Th.; Coenen, H.H.; Qaim, S.M. Excitation functions of 124Te(d,xn)124,125I reactions from threshold up to 14 MeV: comparative evaluation of nuclear routes for the production of 124I. Appl. Radiat. Isot. 2001, 55, 303–308. [Google Scholar] [CrossRef]
- Scholten, B.; Kovács, Z.; Tárkányi, F.; Qaim, S.M. Excitation functions of 124Te(p, xn)124,123I reactions from 6 to 31 MeV with special reference to the production of 124I at a small cyclotron. Appl. Radiat. Isot. 1995, 46, 255–259. [Google Scholar] [CrossRef]
- Qaim, S.M.; Hohn, A.; Bastian, Th.; El-Azoney, K.M.; Blessing, G.; Spellerberg, S.; Scholten, B.; Coenen, H.H. Some optimization studies relevant to the production of high-purity 124I and 120gI at a small-sized cyclotron. Appl. Radiat. Isot. 2003, 58, 69–78. [Google Scholar] [CrossRef]
- Glaser, M.; Mackay, D.B.; Ranicar, A.S.O.; Waters, S.L.; Brady, F.; Luthra, S.K. Improved targetry and production of iodine-124 for PET studies. Radiochim. Acta. 2004, 92, 951–956. [Google Scholar] [CrossRef]
- Nye, J.A.; Avila-Rodriguez, M.A.; Nickles, R.J. Production of [124I]-iodine on an 11 MeV cyclotron. Radiochim. Acta 2006, 94, 213–216. [Google Scholar] [CrossRef]
- Sajjad, M.; Bars, E.; Nabi, H.A. Optimization of 124I production via 124Te(p,n)124I reaction. Appl. Radiat. Isot. 2006, 64, 965–970. [Google Scholar] [CrossRef]
- Weinreich, R; Knust, E.J. Quality control of 124I. In Proc. 6th Workshop on Targetry and Target Chemistry, Vancouver, Canada, August 17–19, 1995; pp. 84–86.
- 2009. Available online: http://www.nndc.bnl.gov/).
- Hohn, A.; Nortier, F.M.; Scholten, B.; van der Walt, T.N.; Coenen, H.H.; Qaim, S.M. Excitation functions of 125Te(p,xn)-reactions from their respective thresholds up to 100 MeV with special reference to the production of 124I. Appl. Radiat. Isot. 2001, 55, 149–156. [Google Scholar] [CrossRef]
- Vaidyanathan, G.; Wieland, B.W.; Larsen, R.H.; Zweit, J.; Zalutsky, M.R. High-yield production of iodine-124 using the 125Te(p,2n)124I reaction. In Proc. 6th Workshop on Targetry and Target Chemistry, Vancouver, Canada, August 17–19, 1995; pp. 87–88.
- Qaim, S.M.; Hohn, A.; Nortier, F.M.; Blessing, G.; Schroeder, I.W.; Scholten, B.; van der Walt, T.N; Coenen, H.H. Production of 124I at small and medium sized cyclotrons. In Proc. 8th Workshop on Targetry and Target Chemistry, St. Louis, MO, June 23–26, 1999; pp. 131–133.
- Kim, J.; Lee, J.S.; Lee, T.; Park, H.; Chun, K. Optimized studies for the production of 124I and 64Cu radioisotopes at KIRAMS. In Proc. 11th Workshop on Targetry and Target Chemistry, Cambridge, UK, August 28–31, 2006; pp. 58–62.
- Zweit, J.; Bakir, M.A.; Ott, R.J.; Sharma, H.L.; Cox, M.; Goodall, R. Excitation functions of proton induced reactions in natural tellurium: Production of no-carrier added iodine-124 for PET applications. In Proc. 4th Workshop on Targetry and Target Chemistry, Villigen, Switzerland, September 9–12, 1991; pp. 76–78.
- Scholten, B.; Spellerberg, S.; Hassan, K.F.; Qaim, S.M.; Coenen, H.H. Comparison of production routes of 124I with emphasis on the 124Te(p,n)124I reaction. In Proc. 11th Workshop on Targetry and Target Chemistry, Cambridge, UK, August 28–31, 2006; pp. 90–91.
- Scholten, B.; Takács, S.; Kovács, Z.; Tárkányi, F.; Qaim, S.M. Excitation functions of deuteron induced reactions on 123Te: Relevance to the production of 123I and 124I at low and medium sized cyclotrons. Appl. Radiat. Isot. 1997, 48, 267–271. [Google Scholar]
- Hassan, K.F.; Qaim, S.M.; Saleh, Z.A.; Coenen, H.H. Alpha-particle-induced reactions on natsb and 121sb with particular reference to the production of the medically interesting radionuclide 124I. Appl. Radiat. Isot. 2006, 64, 101–109. [Google Scholar]
- Hassan, K.F.; Qaim, S.M.; Saleh, Z.A.; Coenen, H.H. 3He-particle-induced reactions on natSb for production of 124I. Appl. Radiat. Isot. 2006, 64, 409–413. [Google Scholar]
- Nye, J.A.; Avila-Rodriguez, M.A.; Nickles, R.J. A new binary compound for the production of 124I via the 124Te(p,n)124I reaction. Appl. Radiat. Isot. 2007, 65, 407–412. [Google Scholar] [CrossRef]
- Čomor, J.J.; Stevanović, Ž.; Rajčević, M.; Košutić, D. Modeling of thermal properties of a TeO2 target for radioiodine production. Nucl. Instrum. Meth. A. 2004, 521, 161–170. [Google Scholar] [CrossRef]
- Kudelin, B.K.; Gromova, E.A.; Gavrilina, L.V.; Solin, L.M. Purification of recovered tellurium dioxide for re-use in iodine radioisotope production. Appl. Radiat. Isot. 2001, 54, 383–386. [Google Scholar]
- Runz, A.; Wolber, G.; Eisenhut, M.; Semmler, W. The production of I-121 at the vertical beam-line of the MC32NI cyclotron at the DKFZ Heidelberg. In Proc. 9th Workshop on Targetry and Target Chemistry, Turku, Finland, May 23–25, 2002; pp. 64–66.
- Sheh, Y.; Koziorowski, J.; Balatoni, J.; Lom, C.; Dahl, J.R.; Finn, R.D. Low energy cyclotron production and chemical separation of “no carrier added” iodine-124 from a reusable, enriched tellurium-124 dioxide/aluminum oxide solid solution target. Radiochim. Acta 2000, 88, 169–173. [Google Scholar] [CrossRef]
- Brown, D.J.; Mckay, D.B.; Coleman, J.; Luthra, S.K.; Brady, F.; Waters, S.L.; Pike, V.W. A facility for the safe recovery of high activities of iodine-124 produced by the 124Te(p,n)124I reaction. In Proc. 8th Workshop on Targetry and Target Chemistry, St. Louis, MO, 1999; pp. 134–136.
- Der, B.; Sader, J.; McQuarrie, S.; Wilson, J. Heat modeling of water cooled target plates. In Proc. 11th Workshop on Targetry and Target Chemistry, Cambridge, August 28–31, 2006; pp. 20–21.
- Gagnon, K.; Avila-Rodriguez, M.A.; McQuarrie, S.A. Thermal modeling of an 124I solid cyclotron target. In Proc. 12th Workshop on Targetry and Target Chemistry, Seattle, WA, USA, July 21–24, 2008; p. 44.
- Avila-Rodriguez, M.A.; Sader, J.A.; McQuarrie, S.A. 3D modeling and simulation of the thermal performance of solid cyclotron targets. In Proceedings of the COMSOL Conference, Genoble, France, 23-27 October 2007; pp. 359–363.
- Rajec, P.; Reich, M.; Szöllős, O.; Baček, D.; Vlk, P.; Kováč, P.; Čomor, J.J. Production of 124I on an 18/9 MeV cyclotrone. In Proc. 7th International conference on nuclear and radiochemistry, Budapest, Hungary, August 24–29; 2008. [Google Scholar]
- Nye, J.A.; Dick, D.W.; Avila-Rodriguez, M.A.; Nickles, R.J. Radiohalogen targetry at the University of Wisconsin. Nucl. Instrum. Meth. B. 2005, 241, 693–696. [Google Scholar] [CrossRef]
- Standardized high current solid targets for cyclotron production of diagnostic and therapeutic radionuclides. In IAEA Technical Report Series No. 432; IAEA: Vienna, Austria, 2004.
- McCarthy, T.J.; Laforest, R.; Downer, J.B.; Lo, A.-R.; Margenau, W.H.; Hughey, B.; Shefer, R.E.; Klinkowskein, R.E.; Welch, M.J. Investigation of I-124, Br-76, and Br-77 production using a small biomedical cyclotron –Can induction furnaces help in the preparation and separation of targets? In Proc. 8th Workshop on Targetry and Target Chemistry, St. Louis, MO, USA, June 23–26, 1999; pp. 127–130.
- Rowland, D.J.; Laforest, R.; McCarthy, T.J.; Hughey, B.J.; Welch, M.J. Conventional and induction furnace distillation procedures for the routine production of Br-76,77 and I-124 on disk and slanted targets. J. Labelled Compd. Rad. 2001, 44 Suppl. 1. [Google Scholar]
- Sadeghi, M.; Dastan, M.; Ensaf, M.R.; Tehrani, A.A.; Tenreiro, C.; Avila, M. Thick tellurium electrodeposition on nickel-coated copper substrate for 124I production. Appl. Radiat. Isot. 2008, 66, 1281–1286. [Google Scholar] [CrossRef]
- Al-Yanbawi, S.; Melibari, S.; Al-Otabi, F.; Rahma, S.; Al Jammaz, I. Standardized high current solid Te-124 targets for cyclotron production of diagnostic radionuclides. In Proc. 11th Workshop on Targetry and Target Chemistry, Cambridge, UK, August 28–31, 2006; p. 1.
- Van den Bosch, R.; De Goeij, J.J.M.; van der Heide, J.A.; Tertoolen, J.F.W.; Theelen, H.M.J.; Zegers, C. A new approach to target chemistry for the iodine-123 production via the 124Te(p,2n) reaction. Int. J. Appl. Radiat. Isot. 1977, 28, 255–261. [Google Scholar] [CrossRef]
- Alekseev, I.E.; Darmograi, V.V.; Marchenkov, N.S. Development of diffusion-thermal methods for preparing 67Cu and 124I for radionuclide therapy and positron emission tomography. Radiochemistry 2005, 47, 460–466. [Google Scholar]
- Knust, E.J.; Dutschka, K.; Weinreich, R. Preparation of 124I solutions after thermodistillation of irradiated 124TeO2 targets. Appl. Radiat. Isot. 2000, 52, 181–184. [Google Scholar] [CrossRef]
- Weinreich, R.; Knust, E.J. Quality assurance of iodine-124 produced via the nuclear reaction 124Te(d,2n)124I. J. Radioanal. Nucl. Chem. Lett. 1996, 213, 253–261. [Google Scholar] [CrossRef]
- Stevenson, N.R.; Buckley, K.; Gelbart, W.Z.; Hurtado, E.T.; Johnson, R.R.; Ruth, T.J.; Zeisler, S.K. On-line production of radioiodines with low energy accelerators. In Proc. 6th Workshop on Targetry and Target Chemistry, Vancouver, Cananda, August 17–19, 1995; pp. 82–83.
- Zyuzin, A.; Johnson, R.; van Lier, E. Production and extraction of radioiodine isotopes using low melting temperature tellurium alloys. In Proc. 11th Workshop on Targetry and Target Chemistry, Cambridge, UK, August 28–31, 2006; pp. 128–129.
- Johnson, R.R.; Watt, R.; Kovac, B.; Zyuzin, A.; Van Lier, E.; Erdman, K.L.; Gyles, Wm.; Sabaiduc, V.; McQuarrie, S.A.; Wilson, J.; Backhouse, C.; Gelbart, Wm.; Kuo, T. Advances in intense beams, beam delivery, targetry, and radiochemistry at advanced cyclotron systems. Nucl. Instrum. Meth. B. 2007, 261, 803–808. [Google Scholar] [CrossRef]
- Oberdorfer, F.; Hellus, F.; Maier-Borst, W. Experiences in the routine production of 123I via the 124Te(p,2n)123I reaction with a low energy cyclotron. J. Radioanal. Chem. 1981, 65, 51–56. [Google Scholar] [CrossRef]
- Ylimaki, R.J.; Kiselev, M.Y.; Čomor, J.J.; Beyer, G.-J. Development of target delivery and recovery system for commercial production of high purity iodine-124. In Proc. 10th Workshop on Targetry and Target Chemistry, Madison, WI, USA, August 13–15, 2004.
- Pentlow, K.S.; Graham, M.C.; Lambrecht, R.M.; Cheung, N.-K.V.; Larson, S.M. Quantitative imaging of I-124 using positron emission tomography with applications to radioimmunodiagnosis and radioimmunotherapy. Med. Phys. 1991, 18, 357–366. [Google Scholar] [CrossRef]
- Beattie, B.J.; Finn, R.D.; Rowland, D.J.; Pentow, K.S. Quantitative imaging of bromine-76 and yttrium-86 with PET: A method for the removal of spurious activity introduced by cascade gamma rays. Med. Phys. 2003, 30, 2410–2423. [Google Scholar] [CrossRef]
- Vandenberghe, S. Three-dimensional positron emission tomography imaging with 124I and 86Y. Nucl. Med. Commun. 2006, 27, 237–245. [Google Scholar] [CrossRef]
- Lubberink, M.; Schneider, H.; Bergström, M.; Lundqvist, H. Quantitative imaging and correction for cascade gamma radiation of 76Br with 2D and 3D PET. Phys. Med. Biol. 2002, 47, 3519–3534. [Google Scholar]
- Sandström, M.; Tolmachev, V.; Kairemo, K.; Lundqvist, H.; Lubberink, M. Performance of coincidence imaging with long-lived positron emitters as an alternative to dedicated PET and SPECT. Phys. Med. Biol. 2004, 49, 5419–5432. [Google Scholar] [CrossRef]
- Laforest, R.; Liu, X. Cascade removal and microPET imaging with 76Br. Phys. Med. Biol. 2009, 54, 1503–1531. [Google Scholar] [CrossRef]
- Lubberink, M.; van Schie, A.; de Jong, H.W.A.M.; van Dongen, G.A.M.S.; Teule, G.J.J. Acquisition settings for PET of 124I administered simultaneously with therapeutic amounts of 131I. J. Nucl. Med. 2006, 47, 1375–1381. [Google Scholar]
- Gregory, R.A.; Hooker, C.A.; Partridge, M.; Flux, G.D. Optimization and assessment of quantitative 124I imaging on a Philips Gemini dual GS PET/CT system. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 1037–1048. [Google Scholar]
- Trotter, D.E.G.; Manjeshwar, R.M.; Doss, M.; Shaller, C.; Robinson, M.K.; Tandon, R.; Adams, G.P.; Alder, L.P. Quantitation of small-animal 124I activity distributions using a clinical PET/CT scanner. J. Nucl. Med. 2004, 45, 1237–1244. [Google Scholar]
- Liu, X.; Laforest, R. Quantitative small animal PET imaging with nonconventional nuclides. Nucl. Med. Biol. 2009, 36, 551–559. [Google Scholar] [CrossRef]
- Adam, M.J.; Wilbur, D.S. Radiohalogens for imaging and therapy. Chem. Soc. Rev. 2005, 34, 153–163. [Google Scholar]
- Seevers, R.H.; Counsell, R.E. Radioiodination techniques for small organic molecules. Chem. Rev. 1982, 82, 575–590. [Google Scholar]
- Moroz, M.A.; Serganova, I.; Zanzonico, P.; Ageyeva, L.; Beresten, T.; Dyomina, E.; Burnazi, E.; Finn, R.D.; Doubrovin, M.; Blasberg, R.G. Imaging hNET reporter gene expression with 124I-MIBG. J. Nucl. Med. 2007, 48, 827–836. [Google Scholar] [CrossRef]
- Reischl, G.; Dorow, D.S.; Cullinane, C.; Katsifis, A.; Roselt, P.; Binns, D.; Hicks, R.J. Imaging of tumor hypoxia with [124I]IAZA in comparison with [18F]FMISO and [18F]FAZA--first small animal PET results. J. Pharm. Pharm. Sci. 2007, 10, 203–211. [Google Scholar]
- Zanzonico, P.; O'Donoghue, J.; Chapman, J.D.; Schneider, R.; Cai, S.; Larson, S.D.; Wen, B.X.; Chen, Y.C.; Finn, R.; Ruan, S.T.; Gerweck, L.; Humm, J.; Ling, C. Iodine-124-labeled iodo-azomycin-galactoside imaging of tumor hypoxia in mice with serial microPET scanning. Eur. J. Nucl. Med. 2004, 31, 117–128. [Google Scholar] [CrossRef]
- Schneider, R.F.; Engelhardt, E.L.; Stobbe, C.C.; Fenning, M.C.; Chapman, J.D. The synthesis and radiolabeling of novel markers of tissue hypoxia of the iodinated azomycin nucleoside class. J. Label. Compd. Radiopharm. 1997, 39, 541–557. [Google Scholar] [CrossRef]
- Riedl, C.C.; Brader, P.; Zanzonico, P.B.; Chun, Y.S.; Woo, Y.H.; Singh, P.; Carlin, S.; Wen, B.X.; Ling, C.C.; Hricak, H.; Fong, Y.M. Imaging hypoxia in orthotopic rat liver tumors with iodine 124-labeled iodoazomycin galactopyranoside PET. Radiology 2008, 248, 561–570. [Google Scholar] [CrossRef]
- Riedl, C.C.; Brader, P.; Zanzonico, P.; Reid, V.; Woo, Y.; Wen, B.; Ling, C.C.; Hricak, H.; Fong, Y.; Humm, J.L. Tumor hypoxia imaging in orthotopic liver tumors and peritoneal metastasis: a comparative study featuring dynamic F-18-MISO and I-124-IAZG PET in the same study cohort. Eur. J. Nucl. Med. 2008, 35, 39–46. [Google Scholar]
- Stahlschmidt, A.; Machulla, H.J.; Reischl, G.; Knaus, E.E.; Wiebe, L.I. Radioiodination of 1-(2-deoxy-beta-D-ribofuranosyl)-2,4-difluoro-5-iodobenzene (dRFIB), a putative thymidine mimic nucleoside for cell proliferation studies. Appl. Radiat. Isot. 2008, 66, 1221–1228. [Google Scholar]
- Guenther, I.; Wyer, L.; Knust, E.J.; Finn, R.D.; Koziorowski, J.; Weinreich, R. Radiosynthesis and quality assurance of 5- I-124 Iodo-2 '-deoxyuridine for functional PET imaging of cell proliferation. Nucl. Med. Biol. 1998, 25, 359–365. [Google Scholar] [CrossRef]
- Blasberg, R.G.; Roelcke, U.; Weinreich, R.; Beattie, B.; von Ammon, K.; Yonekawa, Y.; Landolt, H.; Guenther, I.; Crompton, N.E.A.; Vontobel, P.; Missimer, J.; Maguire, R.P.; Koziorowski, J.; Knust, E.J.; Finn, R.D.; Leenders, K.L. Imaging brain tumor proliferative activity with I-124 iododeoxyuridine. Cancer Res. 2000, 60, 624–635. [Google Scholar]
- Kim, S.W.; Park, J.H.; Yang, S.D.; Hur, M.G.; Choi, C.W.; Yu, K.R. Synthesis and in vitro/vivo Evaluation of Iodine-123/124 Labelled Hypericin Derivatives. Bull. Korean Chem. Soc. 2008, 29, 2023–2025. [Google Scholar] [CrossRef]
- Tjuvajev, J.G.; Finn, R.; Watanabe, K.; Joshi, R.; Oku, T.; Kennedy, J.; Beattie, B.; Koutcher, J.; Larson, S.; Blasberg, R.G. Noninvasive imaging of herpes virus thymidine kinase gene transfer and expression: A potential method for monitoring clinical gene therapy. Cancer Res. 1996, 56, 4087–4095. [Google Scholar]
- Doubrovin, M.; Ponomarev, V.; Beresten, T.; Balatoni, J.; Bornmann, W.; Finn, R.; Humm, J.; Larson, S.; Sadelain, M.; Blasberg, R.; Tjuvajev, J.G. Imaging transcriptional regulation of p53-dependent genes with positron emission tomography in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 9300–9305. [Google Scholar]
- Brust, P.; Haubner, R.; Friedrich, A.; Scheunemann, M.; Anton, M.; Koufaki, O.N.; Hauses, M.; Noll, S.; Noll, B.; Haberkorn, U.; Schackert, G.; Schackert, H.K.; Avril, N.; Johannsen, B. Comparison of F-18 FHPG and I-124/125 FIAU for imaging herpes simplex virus type 1 thymidine kinase gene expression. Eur. J. Nucl. Med. 2001, 28, 721–729. [Google Scholar]
- Tjuvajev, J.G.; Avril, N.; Oku, T.; Sasajima, T.; Miyagawa, T.; Joshi, R.; Safer, M.; Beattie, B.; DiResta, G.; Daghighian, F.; Augensen, F.; Koutcher, J.; Zweit, J.; Humm, J.; Larson, S.M.; Finn, R.; Blasberg, R. Imaging herpes virus thymidine kinase gene transfer and expression by positron emission tomography. Cancer Res. 1998, 58, 4333–4341. [Google Scholar]
- Akgun, E.; Portoghese, P.S.; Sajjad, M.; Nabi, H.A. Synthesis and I-124-labeling of m-iodophenylpyrrolomorphinan as a potential PET imaging agent for delta opioid (DOP) receptors. J. Label. Compd. Radiopharm. 2007, 50, 165–170. [Google Scholar] [CrossRef]
- Pal, A.; Glekas, A.; Doubrovin, M.; Balatoni, J.; Beresten, T.; Maxwell, D.; Soghomonyan, S.; Shavrin, A.; Ageyeva, L.; Finn, R.; Larson, S.M.; Bornmann, W.; Gelovani, J.G. Molecular imaging of EGFR kinase activity in tumors with I-124-labeled small molecular tracer and positron emission tomography. Mol. Imaging Biol. 2006, 8, 262–277. [Google Scholar]
- Mishani, E.; Abourbeh, G.; Rozen, Y.; Jacobson, O.; Laky, D.; Ben-David, I.; Levitzki, A.; Shaul, M. Novel carbon-11 labeled 4-dimethylamino-but-2-enoic acid [4-(phenylamino)-quinazoline-6-yl]-amides: potential PET bioprobes for molecular imaging of EGFR-positive tumors. Nucl. Med. Biol. 2004, 31, 469–476. [Google Scholar] [CrossRef]
- Ortu, G.; Ben-David, I.; Rozen, Y.; Freedman, N.M.T.; Chisin, R.; Levitzki, A.; Mishani, E. Labeled EGFr-TK irreversible inhibitor (ML03): In vitro and in vivo properties, potential as PET biomarker for cancer and feasibility as anticancer drug. Int. J. Cancer 2002, 101, 360–370. [Google Scholar] [CrossRef]
- Shaul, M.; Abourbeh, G.; Jacobson, O.; Rozen, Y.; Laky, D.; Levitzki, A.; Mishani, E. Novel iodine-124 labeled EGFR inhibitors as potential PET agents for molecular imaging in cancer. Bioorg. Med. Chem. 2004, 12, 3421–3429. [Google Scholar] [CrossRef]
- Pandey, S.K.; Sajjad, M.; Chen, Y.H.; Pandey, A.; Missert, J.R.; Batt, C.; Yao, R.T.; Nabi, H.A.; Oseroff, A.R.; Pandey, R.K. Compared to Purpurinimides, the Pyropheophorbide Containing an Iodobenzyl Group Showed Enhanced PDT Efficacy and Tumor Imaging (I-124-PET) Ability. Bioconjug. Chem. 2009, 20, 274–282. [Google Scholar]
- Veach, D.R.; Namavari, M.; Beresten, T.; Balatoni, J.; Minchenko, M.; Djaballah, H.; Finn, R.D.; Clarkson, B.; Gelovani, J.G.; Bornmann, W.G.; Larson, S.M. Synthesis and in vitro examination of I-124 -, I-125 - and I-131 -2-(4-iodophenylamino) pyrido 2,3-d pyrimidin-7-one radiolabeled Abl kinase inhibitors. Nucl. Med. Biol. 2005, 32, 313–321. [Google Scholar]
- Berding, G.; Schneider, U.; Gielow, P.; Buchert, R.; Donnerstag, F.; Brandau, W.; Knapp, W.H.; Emrich, H.M.; Muller-Vahl, K. Feasibility of central cannabinoid CB1 receptor imaging with I-124 AM281 PET demonstrated in a schizophrenic patient. Psychiatry Res. 2006, 147, 249–256. [Google Scholar] [CrossRef]
- Koehler, L.; Graf, F.; Bergmann, R.; Steinbach, J.; Pietzsch, J.; Wuest, F. Radiosynthesis and radiopharmacological evaluation of cyclin-dependent kinase 4 (Cdk4) inhibitors. Eur. J. Med. Chem. 2010, 45, 727–737. [Google Scholar]
- Toogood, P.L.; Harvey, P.J.; Repine, J.T.; Sheehan, D.J.; VanderWel, S.N.; Zhou, H.R.; Keller, P.R.; McNamara, D.J.; Sherry, D.; Zhu, T.; Brodfuehrer, J.; Choi, C.; Barvian, M.R.; Fry, D.W. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J. Med. Chem. 2005, 48, 2388–2406. [Google Scholar]
- VanderWel, S.N.; Harvey, P.J.; McNamara, D.J.; Repine, J.T.; Keller, P.R.; Quin, J.; Booth, R.J.; Elliott, W.L.; Dobrusin, E.M.; Fry, D.W.; Toogood, P.L. Pyrido 2,3-d pyrimidin-7-ones as specific inhibitors of cyclin-dependent kinase 4. J. Med. Chem. 2005, 48, 2371–2387. [Google Scholar]
- Graf, F.; Koehler, L.; Kniess, T.; Wuest, F.; Mosch, B.; Pietzsch, J. Cell cycle regulation kinase Cdk4 as a potential target for tumor cell treatment and tumor imaging. J. Oncol. 2009, 106378, 1–12. [Google Scholar]
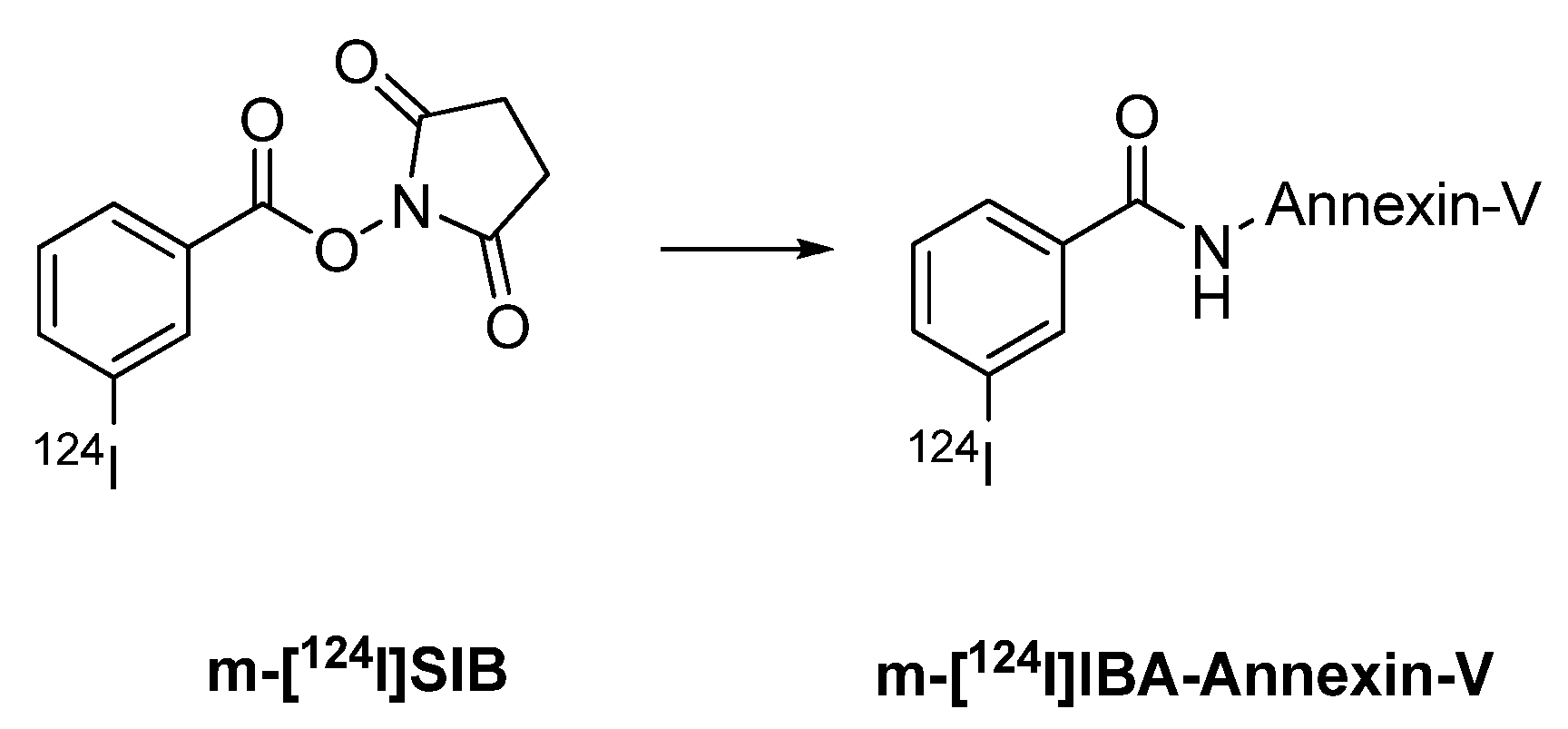
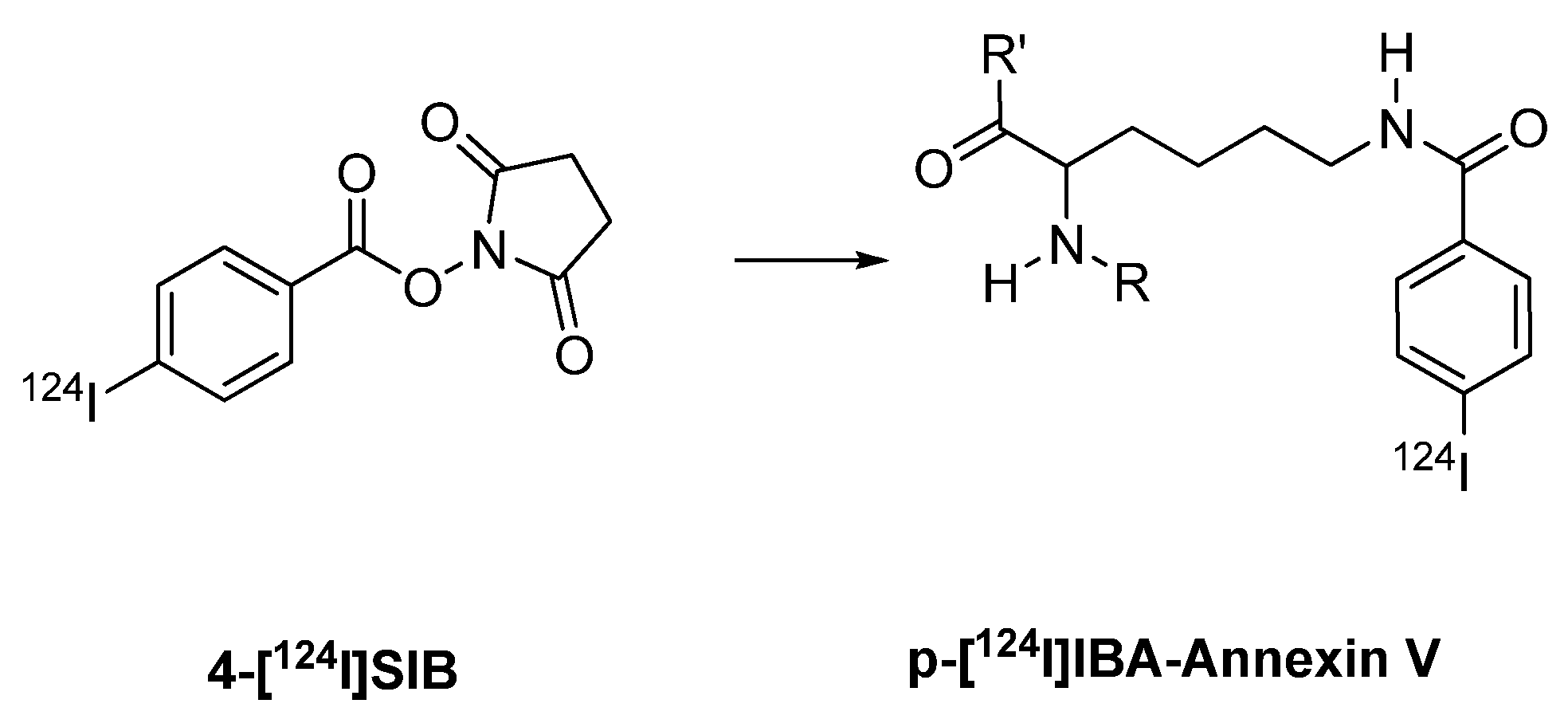
- Glaser, M.; Collingridge, D.R.; Aboagye, E.O.; Bouchier-Hayes, L.; Brown, D.J.; Hutchinson, O.C.; Martin, A.; Price, P.; Luthra, S.K.; Brady, F. Preparation of [124I]IBA-ANNEXIN-V as a potential PET probe for apoptosis. J. Label. Compd. Radiopharm. 2001, 44, S336–S338. [Google Scholar]
- Iozzo, P.; Osman, S.; Glaser, M.; Knickmeier, M.; Ferrannini, E.; Pike, V.W.; Camici, P.G.; Law, M.P. In vivo imaging of insulin receptors by PET: preclinical evaluation of iodine-125 and iodine-124 labelled human insulin. Nucl. Med. Biol. 2002, 29, 73–82. [Google Scholar] [CrossRef]
- Robinson, M.K.; Doss, M.; Shaller, C.; Narayanan, D.; Marks, J.D.; Adler, L.P.; Trotter, D.E.G.; Adams, G.P. Quantitative immuno-positron emission tomography imaging oil HER2-positive tumor xenografts with an iodine-124 labeled anti-HER2 diabody. Cancer Res. 2005, 65, 1471–1478. [Google Scholar]
- Dekker, B.; Keen, H.; Shaw, D.; Disley, L.; Hastings, D.; Hadfield, J.; Reader, A.; Allan, D.; Julyan, P.; Watson, A.; Zweit, J. Functional comparison of annexin V analogues labeled indirectly and directly with iodine-124. Nucl. Med. Biol. 2005, 32, 403–413. [Google Scholar] [CrossRef]
- Keen, H.G.; Dekker, B.A.; Disley, L.; Hastings, D.; Lyons, S.; Reader, A.J.; Ottewell, P.; Watson, A.; Zweit, J. Imaging apoptosis in vivo using I-124-annexin V and PET. Nucl. Med. Biol. 2005, 32, 395–402. [Google Scholar] [CrossRef]
- Collingridge, D.R.; Glaser, M.; Osman, S.; Barthel, H.; Hutchinson, O.C.; Luthra, S.K.; Brady, F.; Bouchier-Hayes, L.; Martin, S.J.; Workman, P.; Price, P.; Aboagye, E.O. In vitro selectivity, in vivo biodistribution and tumour uptake of annexin V radiolabelled with a positron emitting radioisotope. Br. J. Cancer. 2003, 89, 1327–1333. [Google Scholar] [CrossRef]
- Glaser, M.; Collingridge, D.R.; Aboagye, E.O.; Bouchier-Hayes, L.; Hutchinson, O.C.; Martin, S.J.; Price, P.; Brady, F.; Luthra, S.K. Iodine-124 labelled Annexin-V as a potential radiotracer to study apoptosis using positron emission tomography. Appl. Radiat. Isot. 2003, 58, 55–62. [Google Scholar] [CrossRef]
- Glaser, M.; Brown, D.J.; Law, M.P.; Iozzo, P.; Waters, S.L.; Poole, K.; Knickmeier, M.; Camici, P.G.; Pike, V.W. Preparation of no-carrier-added I-124 A(14)-iodoinsulin as a radiotracer for positron emission tomography. J. Label. Compd. Radiopharm. 2001, 44, 465–480. [Google Scholar]
- Dekker, B.; Keen, H.; Lyons, S.; Disley, L.; Hastings, D.; Reader, A.; Ottewell, P.; Watson, A.; Zweit, J. MBP-annexin V radiolabeled directly with iodine-124 can be used to image apoptosis in vivo using PET. Nucl. Med. Biol. 2005, 32, 241–252. [Google Scholar] [CrossRef]
- Verel, I.; Visser, G.W.M.; Boerman, O.C.; van Eerd, J.E.M.; Finn, R.; Boellaard, R.; Vosjan, M.; Walsum, M.S.V.; Snow, G.B.; van Dongen, G. Long-lived positron emitters zirconium-89 and iodine-124 for scouting of therapeutic radioimmunoconjugates with PET. Cancer Biother. Radiopharm. 2003, 18, 655–661. [Google Scholar] [CrossRef]
- Verel, I.; Visser, G.W.M.; Vosjan, M.; Finn, R.; Boellaard, R.; van Dongen, G. High-quality I-124-labelled monoclonal antibodies for use as PET scouting agents prior to I-131-radioimmunotherapy. Eur. J. Nucl. Med. 2004, 31, 1645–1652. [Google Scholar] [CrossRef]
- Sundaresan, G.; Yazaki, P.J.; Shively, J.E.; Finn, R.D.; Larson, S.M.; Raubitschek, A.A.; Williams, L.E.; Chatziioannou, A.F.; Gambhir, S.S.; Wu, A.M. I-124-labeled engineered Anti-CEA minibodies and diabodies allow high-contrast, antigen-specific small-animal PET imaging of xenografts in athymic mice. J. Nucl. Med. 2003, 44, 1962–1969. [Google Scholar]
- Daghighian, F.; Pentlow, K.S.; Larson, S.M.; Graham, M.C.; Diresta, G.R.; Yeh, S.D.J.; Macapinlac, H.; Finn, R.D.; Arbit, E.; Cheung, N.K.V. Development of a methode to measure kinetics of radiolabeled monoclonal antibody in human tumor with applications to microdosimetry - Positron emission tomography studies of I-124 labeled 3F8 monoclonal-antibody in glioma. Eur. J. Nucl. Med. 1993, 20, 402–409. [Google Scholar]
- Larson, S.M.; Pentlow, K.S.; Volkow, N.D.; Wolf, A.P.; Finn, R.D.; Lambrecht, R.M.; Graham, M.C.; Diresta, G.; Bendriem, B.; Daghighian, F.; Yeh, S.D.J.; Wang, G.J.; Cheung, N.K.V. PET scanning of iodine-124-3F8 as an approach to tumor dosimetry during treatment planning for radioimmunotherapy in a child with neuroblastoma. J. Nucl. Med. 1992, 33, 2020–2023. [Google Scholar]
- Finn, R.; Cheung, N.K.V.; Divgi, C.; St. Germain, J.; Graham, M.; Pentlow, K.; Larson, S.M. Technical challenges associated with the radiolabeling of monoclonal-antibodies utilizing short-lived, positron emitting radionuclides. Int. J. Rad. Appl. Instrum. B 1991, 18, 9–13. [Google Scholar]
- Lee, F.T.; Hall, C.; Rigopoulos, A.; Zweit, J.; Pathmaraj, K.; O'Keefe, G.J.; Smyth, F.E.; Welt, S.; Old, L.J.; Scott, A.M. Immune-PET of human colon xenograft-bearing BALB/c nude mice using I-124-CDR-grafted humanized A33 monoclonal antibody. J. Nucl. Med. 2001, 42, 764–769. [Google Scholar]
- Westera, G.; Reist, H.W.; Buchegger, F.; Heusser, C.H.; Hardman, N.; Pfeiffer, A.; Sharma, H.L.; Vonschulthess, G.K.; Mach, J.P. Radioimmuno Positron Emission Tomography with monoclonal-antibodies - a new approach to quantifying invivo tumor concentrations and biodistribution for radioimmunotherapy. Nucl. Med. Commun. 1991, 12, 429–437. [Google Scholar] [CrossRef]
- Rubin, S.C.; Kairemo, K.J.A.; Brownell, A.L.; Daghighian, F.; Federici, M.G.; Pentlow, K.S.; Finn, R.D.; Lambrecht, R.M.; Hoskins, W.J.; Lewis, J.L.; Larson, S.M. High-resolution positron emission tomography of human ovarian-cancer in nude rats using I-124 labeled monoclonal-antibodies. Gynecol. Oncol. 1993, 48, 61–67. [Google Scholar] [CrossRef]
- Bakir, M.A.; Eccles, S.A.; Babich, J.W.; Aftab, N.; Styles, J.M.; Dean, C.J.; Ott, R.J. C-ERB2 protein overexpression in breast cancer as a target for PET using iodine-124-labeled monoclonal antibodies. J. Nucl. Med. 1992, 33, 2154–2160. [Google Scholar]
- Wilson, C.B.; Snook, D.E.; Dhokia, B.; Taylor, C.V.J.; Watson, I.A.; Lammertsma, A.A.; Lambrecht, R.; Waxman, J.; Jones, T.; Epenetos, A.A. Quantitative measurement of monoclonel-antibody distribution and blood-flow using positron emission tomography and I-124 in patients with breast cancer. Int. J. Cancer 1991, 47, 344–347. [Google Scholar] [CrossRef]
- Glaser, M.; Carroll, V.A.; Collingridge, D.R.; Aboagye, E.O.; Price, P.; Bicknell, R.; Harris, A.L.; Luthra, S.K.; Brady, F. Preparation of the iodine-124 derivative of the Bolton-Hunter reagent ( I-124 I-SHPP) and its use for labelling a VEGF antibody as a PET tracer. J. Label. Compd. Radiopharm. 2002, 45, 1077–1090. [Google Scholar] [CrossRef]
- Collingridge, D.R.; Carroll, V.A.; Glaser, M.; Aboagye, E.O.; Osman, S.; Hutchinson, O.C.; Barthel, H.; Luthra, S.K.; Brady, F.; Bicknell, R.; Price, P.; Harris, A.L. The development of I-124 iodinated-VG76e: A novel tracer for imaging vascular endothelial growth factor in vivo using positron emission tomography. Cancer Res. 2002, 62, 5912–5919. [Google Scholar]
- Chaturvedi, R.; Heimburg, J.; Yan, J.; Koury, S.; Sajjad, M.; Abdel-Nabi, H.H.; Rittenhouse-Olson, K. Tumor immunolocalization using I-124-iodine-labeled JAA-F11 antibody to Thomsen-Friedenreich alpha-linked antigen. Appl. Radiat. Isot. 2008, 66, 278–287. [Google Scholar] [CrossRef]
- Koziorowski, J.; Henssen, C.; Weinreich, R. A new convenient route to radioiodinated N-succinimidyl 3- and 4-iodobenzoate, two reagents for radioiodination of proteins. Appl. Radiat. Isot. 1998, 49, 955–959. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Koehler, L.; Gagnon, K.; McQuarrie, S.; Wuest, F. Iodine-124: A Promising Positron Emitter for Organic PET Chemistry. Molecules 2010, 15, 2686-2718. https://doi.org/10.3390/molecules15042686
Koehler L, Gagnon K, McQuarrie S, Wuest F. Iodine-124: A Promising Positron Emitter for Organic PET Chemistry. Molecules. 2010; 15(4):2686-2718. https://doi.org/10.3390/molecules15042686
Chicago/Turabian StyleKoehler, Lena, Katherine Gagnon, Steve McQuarrie, and Frank Wuest. 2010. "Iodine-124: A Promising Positron Emitter for Organic PET Chemistry" Molecules 15, no. 4: 2686-2718. https://doi.org/10.3390/molecules15042686