



Abstract
There is increasing interest in using exogenous labels such as bromodeoxyuridine (BrdU) or superparamagnetic iron oxide nanoparticles (SPION) to label cells to identify transplanted cells and monitor their migration by fluorescent microscopy or in vivo magnetic resonance imaging (MRI), respectively. Direct implantation of cells into target tissue can result in >80% cell death due to trauma or apoptosis. Bystander uptake of labeled cells by activated macrophages (AM) can confound the interpretation of results. This study investigated the frequency of BrdU or SPION uptake by AM using the Boyden chamber model of inflammation. SPION/BrdU-labeled bone marrow stromal cells or HeLa cells, AM, and mouse fibroblasts (MF) or human fibroblasts (HF) were mixed in various ratios in Matrigel in the upper chamber and incubated for up to 96 hours. The AM were chemotactically induced to migrate to the lower chamber. Fluorescence-activated cell sorting analysis of AM from lower and upper chambers, in the presence of either MF or HF using anti-CD68, anti-BrdU, anti-dextran antibodies, revealed 10%–20% dextran-positive or 10% BrdU-positive AM after 96 hours of incubation. Transfer of iron to AM accounted for <10% of the total iron in labeled cells. The uptake of BrdU and SPION was dependent on the ratio of labeled cells to inflammatory cells and microenvironmental conditions. Direct implantation of BrdU/SPION-labeled cells into target tissue can result in uptake of label by AM; therefore, care should be taken to validate by histology transplanted cells for bystander cell markers and correlation with MRI results.
Disclosure of potential conflicts of interest is found at the end of this article.
Introduction
Cell therapy is increasingly used for tissue regeneration and repair, especially for the treatment of autoimmune diseases and malignancy [1, 2]. Stem cells have emerged as a potential new therapeutic treatment approach that can be delivered by direct implantation or i.v. or intra-arterial injection [3–5]. To exploit their therapeutic potential, new cell labeling techniques are being developed for in vivo visualization and tracking the fate of transplanted cells, in both animal models and clinical trials.
Bromodeoxyuridine (BrdU) labeling is a commonly used method for monitoring cell migration [6]. BrdU is a thymidine analog that incorporates into DNA of dividing cells during the S-phase of the cell cycle [7]. BrdU labeling has allowed for identification of sites of neurogenesis in the adult and neonatal brain and has also been used as a label to track migration and differentiation of transplanted stem cells in various animal models [8–12].
Recently, stem cells and other cells have been labeled by complexing two Food and Drug Administration-approved agents, ferumoxides (FE), a dextran-coated superparamagnetic iron oxide nanoparticle (SPION), and protamine sulfate (Pro), to noninvasively monitor temporal and spatial migration and tissue distribution of the labeled cells by in vivo magnetic resonance imaging (MRI) [13–21]. Direct transplantation of cells into target tissue as part of cellular therapy can result in more than 50%–80% of the cells undergoing immediate cell death or apoptosis-induced cell death in areas of inflammation [22, 23]. However, there is a perception and concern by some investigators that exogenous or endogenous cellular markers (i.e., SPION, Quantum dots, and BrdU) may be transferred from implanted cells to local, endogenous bystander cells, such as activated macrophages (AM), in areas of damage and inflammation, potentially confounding the interpretation of noninvasive cellular imaging or microscopy [24–29]. Although multiple reports have cautioned against overinterpreting the results from labeled transplanted cells to host cells, none have quantified the number of AM engulfing the exogenous label [22, 30].
The objective of this study was to determine the frequency of transfer of cellular labels, such as BrdU and SPION, from labeled cells to human AM in a modified Boyden chamber (BC) as a localized model of inflammation. In this model we used primary AM as inflammatory cells, labeled bone marrow stromal cells (BMSCs) or HeLa cells as exogenous cells, and mouse or human fibroblasts as a surrogate for endogenous host cells that could be present in an in vivo models. Quantifying the percentage of macrophages that incorporate the iron oxide nanoparticles or BrdU label from viable, apoptotic, or dead cells will aid in the interpretation of the results of vivo cellular imaging and microscopic tracking analysis.
Materials and Methods
Cell Culture and Labeling with FE-Pro and BrdU
Human BMSCs, which are a subset of multipotent skeletal stem cells, were obtained as described below [31]. Briefly, fragments of bone with marrow were obtained from volunteers undergoing bone marrow biopsy under institutional review board-approved procedures, in accordance with NIH regulations governing the use of human subjects. Fragments of trabecular bone and marrow were scraped gently with a steel blade into cold modified α-Minimum Essential Medium (α-MEM) (Life Technologies, Grand Island, NY, http://www.lifetech.com) and pipetted repeatedly. The released marrow cells were passed consecutively through 16- and 20-gauge needles and filtered through a 70-μm pore size nylon cell strainer (Becton, Dickinson and Company, Franklin Lakes, NJ, http://www.bd.com) to remove cell aggregates. Bone marrow single-cell suspensions were plated at a high cell density of 1.0 × 107 nucleated cells per 75-cm2 flask (Becton Dickinson). Growth medium included α-MEM, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin sulfate (Biofluids, Rockville, MD, http://www.invitrogen.com), and 20% fetal bovine serum (Equitech-Bio, Kerrville, TX, http://www.equitech-bio.com). Cells were cultured at 37°C in a 95% air/5% CO2 atmosphere. For generating large numbers of BMSCs, high-density cultures were prepared in triplicate; the medium was replaced on day 6 or 7. The cultures were first passaged on day 12 with two consecutive applications of 1 × trypsin-EDTA (Life Technologies) for 5–10 minutes each at room temperature. The cells harvested from each 75-cm2 flask were counted separately using a Coulter counter (Beckman Coulter, Hialeah, FL, http://www.beckmancoulter.com and plated into a separate 175-cm2 flask (Becton Dickinson). The subsequent passages were performed at 4–7-day intervals. BMSCs were used at passages 3–5 for this study.
Primary human skin fibroblast cultures were established from biopsies taken from healthy volunteers. Cells were cultured in α-MEM (Life Technologies), 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin sulfate (Biofluids, Rockville, MD), and 20% fetal bovine serum (Equitech-Bio) under 5% CO2 and at 37°C. For all experiments, cells from passages 1–5 were used after harvesting by trypsinization.
Human cervical carcinoma (HeLa) cells (CCL-2) (American Type Culture Collection, Manassas, VA, http://www.atcc.org) were grown in standard culture medium at 37°C and in a 95% air/5% CO2 atmosphere. Cells were allowed to grow until they reached 90% confluence in 75-cm2 culture flasks prior to labeling.
Primary monocytes, isolated from healthy donors and obtained from the NIH cell processing section Department of Transfusion Medicine, were plated in 175-cm2 culture flasks at a density of 1.5 × 106 cells per milliliter. Monocytes were cultured overnight with macrophage colony-stimulating factor (M-CSF) (50 ng/ml; Peprotech, Rocky Hill, NJ, http://www.peprotech.com). The following day, lipopolysaccharide (LPS) (500 ng/ml) was added to monocyte cultures for 4 hours to allow differentiation of monocytes into activated macrophages. The activation state of macrophages was determined by flow cytometry measurements of intracellular cytokine interleukin-6 (IL-6) using BD Cytofix/Cytoperm Plus Kit (with GolgiStop containing monensin) (BD Biosciences, San Jose, CA, http://www.bdbiosciences.com). Briefly, at the time of LPS treatment, a protein transport inhibitor was added to monocyte cultures. After 4 hours of coincubation with LPS, macrophages were washed with sterile phosphate-buffered saline (PBS), scraped, fixed, and permeabilized. Cells were stained with anti-CD68-fluorescein isothiocyanate (FITC), anti-IL-6-phycoerithrin (PE), and isotype controls (all from BD Biosciences) and analyzed by flow cytometry (FACSCalibur; Becton Dickinson).
FE (Feridex IV; Berlex Laboratories, Wayne, NJ, http://berlex.bayerhealthcare.com) is a dextran-coated SPION approximately 120–150 nm in size and is provided at a total iron content of 11.2 mg/ml. Pro (American Pharmaceuticals Partner, Schaumburg, IL, http://www.apppharma.com), supplied at 10 mg/ml, was prepared as a fresh stock solution of 1 mg/ml in sterile distilled water immediately before labeling. Ferumoxides at a concentration of 100 μg/ml was put into a 50-ml conical tube containing serum-free RPMI 1640 (Biosource, Camarillo, CA, http://www.invitrogen.com) with 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), MEM nonessential amino acids, sodium pyruvate, and l-glutamine. Protamine sulfate was added to the solution at 6 μg/ml and mixed for 2 minutes with intermittent hand shaking. Culture medium was aspirated from the flasks containing BMSCs or HeLa cells and replaced with medium containing FE-Pro complexes. After 2 hours of incubation at 37°C, an equal amount of relevant complete medium was added to HeLa cells and BMSCs for final concentrations of FE and Pro of 50 and 3 μg/ml, respectively. Cells were incubated overnight, approximately 16 hours, and washed three times with sterile PBS containing 10 U/ml heparin sulfate (American Pharmaceuticals Partner). Complete medium was added to each flask, and labeled cells were kept in culture for 2 days to ensure that all FE-Pro complexes were endocytosed.
HeLa cells labeled with FE-Pro were incubated overnight with 500 ng/ml TNF-related apoptosis inducing ligand (TRAIL) (Chemicon, Temecula, CA, http://www.chemicon.com) to induce apoptosis and cell death. The viability of cells undergoing apoptosis was determined by trypan blue exclusion test and the Vybrant Apoptosis Assay Kit #2-Alexa Fluor 488 annexin V/ propidium iodide (Invitrogen, Carlsbad, CA, http://www.invitrogen.com) according to the manufacturer's instructions. Mouse skeletal muscle fibroblast cells (CLL-197; American Type Culture Collection) were grown in Dulbecco's modified Eagle's medium supplemented with 20% fetal bovine serum in a 95% air/5% CO2 atmosphere at 37°C.
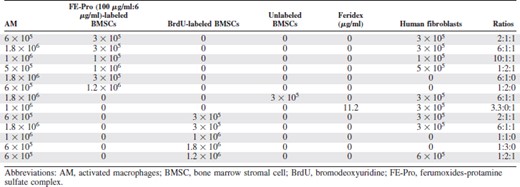
After reaching 60% confluence, BMSCs were labeled with BrdU by adding 100 μg of BrdU (BD Biosciences) to culture flasks and incubating for 4 days. After labeling, cells were washed three times with sterile PBS, trypsinized, and used in the BC experiments. In the BC experiments we used unlabeled BMSCs as a control and BMSCs that were labeled with either BrdU or FE-Pro only, FE-Pro-labeled HeLa cells, and unlabeled fibroblasts.
Prussian Blue Staining and Labeling Efficiency
To visualize the iron within FE-Pro-labeled cells, Prussian blue (PB) staining was performed. After overnight labeling and 2 days of post-labeling culture, BMSCs and HeLa cells were trypsinized and transferred to cytospin slides. Cells were fixed with 4% glutaraldehyde, washed, and incubated for 30 minutes with 2% potassium ferric-ferrocyanide (Perl's reagent for staining; Sigma-Aldrich, St. Louis, http://www.sigmaaldrich.com) in 3.7% hydrochloric acid. Cells were washed again, counterstained with nuclear fast red, and evaluated for iron staining using light microscopy (Axioplan Imaging II; Carl Zeiss, Oberkochen, Germany, http://www.zeiss.com) at ×40/0.75 objective lens and Axiovision 4.4 software (Carl Zeiss). The images were processed using Adobe Photoshop 7.0 (Adobe Systems Inc., San Jose, CA, http://www.adobe.com). FE-Pro labeling efficiency was determined by manual counting of PB-stained and unstained cells using a Zeiss microscope (Axioplan Imaging II; Carl Zeiss) at ×100 magnification using a ×100/1.30 oil immersion objective lens. The percentage of labeled cells was determined from the average of five high-powered fields.
MTS Assay
The viability of AM was determined using the CellTiter 96 AQueous Assay, according to the manufacturer's instructions (Promega, Madison, WI, http://www.promega.com). The MTS assay is based on a tetrazolium compound (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt [MTS]) and the electron coupling reagent phenazine methosulfate. MTS is chemically reduced by cells into formazan, the production of which is proportional to the number of living cells and can be measured spectrophotometrically. Briefly, 5 × 104 activated macrophages in 100 μl of medium containing 50 ng/ml M-CSF were plated in triplicate in a 96-well plate. At days 1, 4, and 7, fresh medium was added to a volume of 100 μl per well. Twenty microliters of MTS reagent (Promega) was then added. After a 3-hour incubation at 37°C, the absorbance was measured at 490 nm using an enzyme-linked immunosorbent assay plate reader (MR5000; Dynatech, Chantilly, VA, http://www.dynextechnologies.com). Results are reported as the mean of three A490 readings corrected with a blank.
Modified Boyden Chamber Experiment of Inflammation
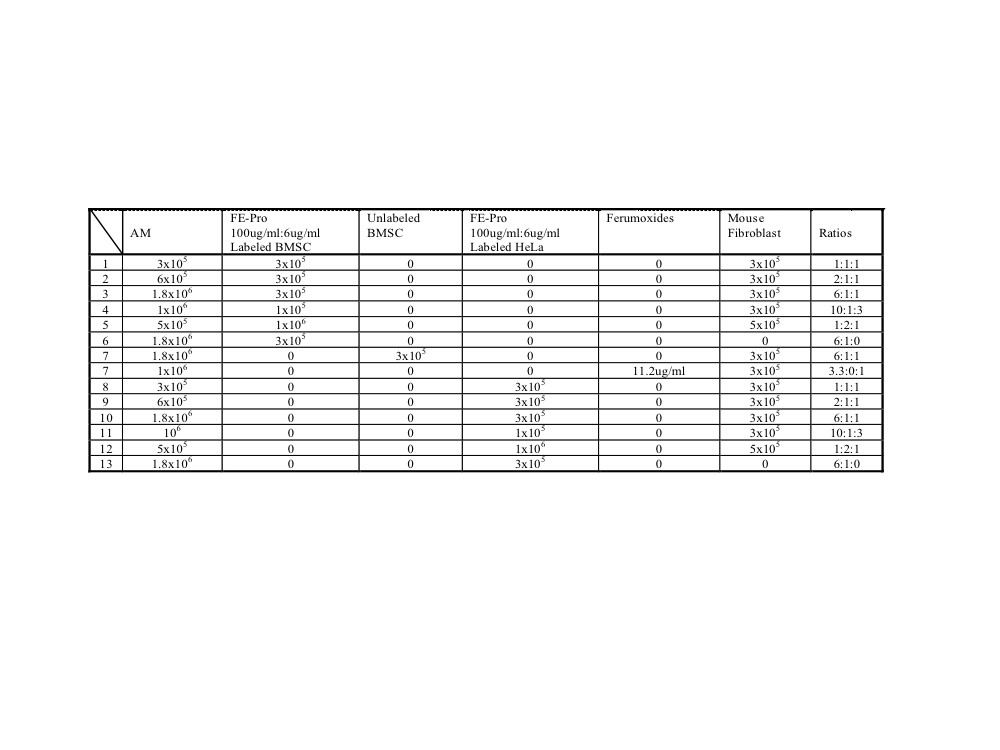
A modified Boyden chamber assay was performed in 24-well plates to simulate a localized area of inflammation. The upper chamber consisted of 24-well MilliCell culture plate inserts (Millipore, Billerica, MA, http://www.millipore.com) with a 1.2-cm2 well area and a 3.0-μm pore size (Fig. 1). Table 1 and supplemental online Table 2 summarize the various ratios of AM, FE-Pro-labeled BMSCs or HeLa cells, human fibroblasts (HF), BrdU-labeled BMSCs, and mouse fibroblasts (MF) that were mixed with 200 μl of Matrigel (extracellular matrix gel from Engelbreth-Holm-Swarm murine sarcoma; Sigma-Aldrich) and placed into the upper chamber. In this new in vitro model of inflammation, AM represented inflammatory cells, whereas labeled BMSCs and HeLa cells were exogenous cells. Mouse or human fibroblasts were used as a surrogate for endogenous, bystander cells that are always present in vivo models. The objective of using MF was to test whether uptake of the label by AM would be enhanced because of potentially higher immunogenicity of MF. The ratios of cells used were based on the report that estimated the number of macrophages per mm3 in an inflammatory lesion [32]. The ratios of AM to BrdU- or SPION-labeled cells to fibroblasts were chosen to cover a range of equal numbers of cells to either excess numbers (i.e., up to 1.8 × 106) of AM to labeled cells in a limited volume to simulate the biological variability that could be observed in vivo. The MF and HF were kept constant to minimize the number of comparisons. As a matched control, unlabeled BMSCs and ferumoxides alone were mixed in Matrigel with the AM and HF.
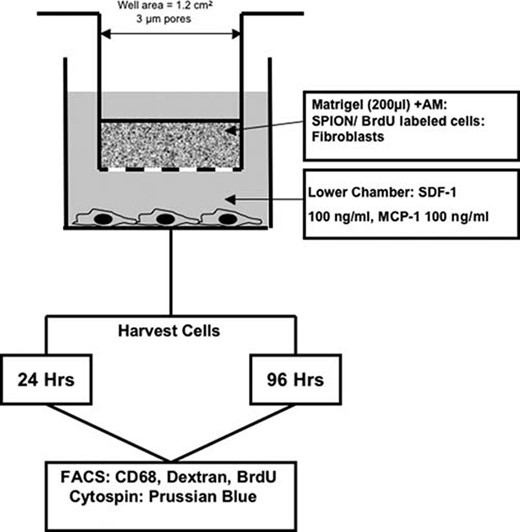
Schema of modified Boyden chamber assay as a model of localized inflammation in the tissues. Abbreviations: AM, activated macrophages; BrdU, bromodeoxyuridine; FACS, fluorescence-activated cell sorting; Hrs, hours; MCP, monocyte chemotactic protein; SDF, stromal cell-derived factor.
The lower chamber contained RPMI 1640 medium (American Type Culture Collection) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Norcross, GA, http://www.atlantabio.com), 100 ng/ml stromal cell-derived factor-1 (SDF-1), and 100 ng/ml monocyte chemotactic protein-1 (MCP-1) (both from Peprotech). SDF-1 and MCP-1 at the concentration used stimulated macrophage migration through the membrane into the lower chamber. Cells were incubated in the BC for 24 and 96 hours. The control migration experiments included upper chambers containing only BMSCs, HeLa cells, or HF/MF cells mixed with Matrigel to show no migration, whereas lower chambers stayed the same. After incubation, cells in the upper chamber were recovered by treatment of Matrigel with dispase for 2 hours at 37°C (BD Biosciences). Activated macrophages in the lower chamber were scraped and collected, and all harvested cells were analyzed by flow cytometric analysis (fluorescence-activated cell sorting [FACS]). Each combination of cells used in the BC experiment was performed in triplicate, and each experiment was repeated three to five times.
Flow Cytometry and Fluorescence Microscopy Analysis
Activated macrophages collected from both chambers were fixed and permeabilized with BD Cytofix/Cytoperm Plus (with GolgiStop containing monensin) kit (BD Biosciences) according to the manufacturer's instructions. Cells were stained for dextran and CD68 using anti-dextran IgG1-FITC (StemCell Technologies, Vancouver, BC, Canada, http://www.stemcell.com) and anti-CD68 IgG2a-PE (BD Biosciences) antibodies. The control experiments included unstained cells and cells stained with isotype controls for each of the antibodies. BrdU was detected in activated macrophages using an FITC-BrdU Flow Kit (BD Biosciences) according to the manufacturer's instructions. Stained cells were analyzed by FACS (FACSCalibur; Becton Dickinson). A total of 15,000 cells were analyzed at the FL1 and FL2 detectors, and the results were presented as the mean percentage of dextran- and CD68-positive or BrdU- and CD68-positive cells from the experiments.
For fluorescence microscopy, cells were collected from both chambers, permeabilized, and stained for BrdU, dextran, and CD68 using the FITC-BrdU Flow Kit (BD Biosciences), anti-dextran IgG1-FITC (StemCell Technologies), and mouse anti-CD68 (AbD Serotec, Raleigh, NC, http://www.ab-direct.com) followed by secondary goat anti-mouse IgG-rhodamine or IgG-Alexa Fluor 594 (Invitrogen), respectively. Stained cells were transferred to cytospin slides, coverslipped, mounted with Vectashield mounting medium with 1.5 μg/ml 4,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA, http://www.vectorlabs.com), and analyzed by fluorescence microscopy (Axioplan Imaging II; Carl Zeiss) at ×40/0.75 objective lens and using Axiovision 4.4 software (Carl Zeiss). The images were processed using Adobe Photoshop 7.0.
Iron Content Measurements
Cells from three upper chambers or three lower chambers were combined and digested overnight in 1 ml of 5 N hydrochloric acid. The digested solution (500 μl) was mixed 1:1 with 5% potassium ferrocyanide (Sigma-Aldrich) and incubated at room temperature for 15–20 minutes. Absorbance was measured at 670 nm using a standard spectrophotometer (UV-1601; Shimadzu, Columbia, MD, http://www.shimadzu.com). The amount of iron per sample was determined by calibration to a standard curve, which was generated using ferrous chloride standards ranging from 0 to 0.011 mg/ml of iron. Iron content of labeled HeLa cells and BMSCs was also determined prior to BC experiments and was reported as picograms of iron per cell.
Statistical Analysis
Each experiment was performed at least three times, and each combination of cells was tested in triplicate. Data are expressed as a mean ± SD. Statistically significant differences were detected using analysis of variance with the Tukey-Kramer honestly significant difference test for mean comparisons of all pairs and Bonferroni correction for multiple comparisons with significant p value <.05 (Prism 4 for Macintosh; GraphPad Software, Inc., San Diego, http://www.graphpad.com) [33]. To represent a biological variability observed in an experimental model, all the data from the multiple combinations in Table 1 and supplemental online Table 2 were pooled, and the statistical analysis was performed on the combined data.
Results
Validation of Cell Labeling, Viability, and Activation of Primary Macrophages
FE-Pro labeling of BMSCs and HeLa cells resulted in approximately 100% of cells being labeled after counting PB-positive cells. Representative photomicrographs of PB-stained HeLa cells and BMSCs revealed an abundant uptake of the FE-Pro complex into the cytoplasm (Fig. 2A, 2B). The iron content of the Hela cells was 13.07 ± 2.55 pg per cell, and for the BMSCs, it was 34.75 ± 0.32 pg per cell. The amount of iron per cell was similar to what was previously reported when labeling cells with FE-Pro [13].
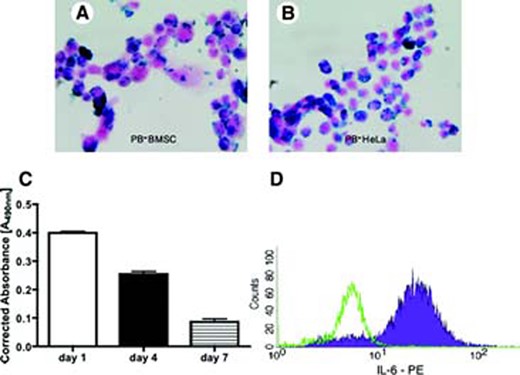
Validation of ferumoxides-protamine sulfate complex (FE-Pro) labeling, viability, and activation of macrophages. Shown are representative PB-stained FE-Pro labeled BMSCs (A), HeLa cells (B); magnification, ×40. (C): A bar graph showing the viability of activated macrophages (AM) up to 7 days in culture measured using the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay. Note that only 20% of AM were alive after 7 days in culture. (D): Flow cytometry histogram showing the activation state of macrophages as measured by IL-6 expression. Green peak indicates AM stained with isotype control antibodies, and the filled purple peak shows AM expressing IL-6. The MTS data are presented as a mean ± SD from three samples. Abbreviations: BMSC, bone marrow stromal cell; IL, interleukin; PB, Prussian Blue; PE, phycoerithrin.
The MTS assay of activated macrophages showed that after 7 days in culture, only 20% of macrophages were viable (Fig. 2C). On the basis of these results, the BC experiments were designed to include time points at 24 and 96 hours of incubation. The MTS data were validated with the trypan blue exclusion test, which showed a similar survival rate of activated macrophages in vitro (data not shown). The FACS of IL-6 expression revealed that the majority of macrophages were activated following LPS treatment (Fig. 2D).
BrdU Uptake by AM
To investigate the frequency of BrdU uptake by macrophages, the modified BC experiment was used (Fig. 1). All combinations of cells were chosen experimentally (Table 1) to cover a concentration range of AM to labeled cells in a limited volume. The flow cytometry analysis revealed that after 96 hours of incubation with various ratios of BrdU-labeled BMSCs and human fibroblasts, 2.37% ± 1.02% to 6.03% ± 2.13% of CD68- and BrdU-positive macrophages were found in the lower chambers and 9.23% ± 2.81% to 18.85% ± 1.62% in the upper chambers (Fig. 3A). The percentage of BrdU-positive macrophages depended on the ratio of macrophages to fibroblasts or BMSCs used (Fig. 3B). To simulate the biological variability associated with inflammation, the FACS results from the different ratios of labeled and bystander cells to AM were pooled together. The mean uptake of BrdU in the upper well was significantly higher (p < .0001) compared with the mean BrdU uptake by AM in the lower chamber (Fig. 3C). The average percentage uptake of BrdU by AM when combining results from both chambers was 8.32% ± 5.10%. The CD68- and BrdU-positive macrophages were also visualized under fluorescence microscope (Fig. 3D). The frequency of BrdU- and CD68-positive cells was comparable to flow cytometry results.
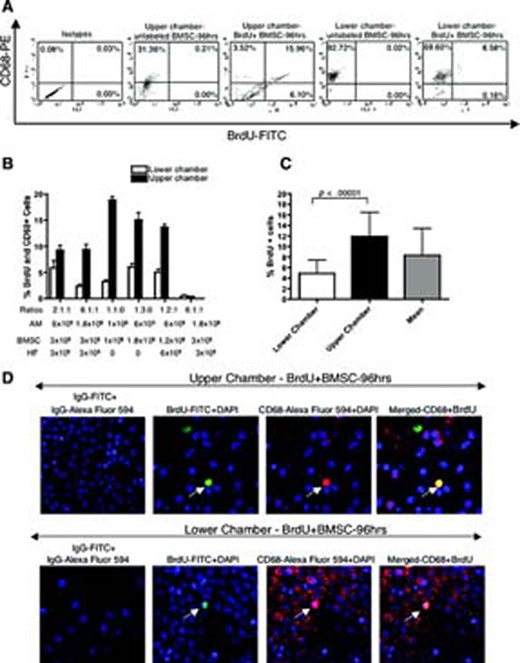
BrdU uptake by activated macrophages (AM) from live BrdU-labeled BMSCs. (A): Representative fluorescence-activated cell sorting dot plots showing 15.96% of CD68-PE- and BrdU-FITC-positive macrophages in the upper chamber and 6.58% of CD68-PE- and BrdU-FITC-positive macrophages in the lower chamber after 96 hrs of incubation. (B): Bar graph showing the percentage of BrdU- and CD68-positive AM collected from the lower and upper chamber after 96 hrs of incubation. (C): Bar graph showing the means of BrdU- and CD68-positive AM after pulling all ratios. Note that the mean uptake of BrdU in the upper chamber was significantly higher (p < .0001) compared with the mean BrdU uptake by AM in the lower chamber. (D): Immunofluorescent detection of BrdU with anti-BrdU-FITC, CD68 with anti-CD68-Alexa Fluor 594 on AM recovered from both chambers (magnification, ×40). On the merged image of the upper chamber, note cells positive for BrdU-FITC only (BrdU-labeled BMSCs), cells positive for CD68-Alexa Fluor 594 only that are AM, and cells positive for BrdU-FITC and CD68-Alexa Fluor 594. Cells that took up BrdU from BrdU-labeled BMSCs are AM. On the merged image of the lower chamber, note cells positive for CD68-Alexa Fluor 594 only that are AM and cells positive for BrdU-FITC and CD68-Alexa Fluor 594 (AM that took up BrdU from BrdU-labeled BMSCs). Data are presented as a mean ± 1 SD from nine samples. Abbreviations: AM, activated macrophages; BMSC, bone marrow stromal cell; BrdU, bromodeoxyuridine; DAPI, 4,6-diamidino-2-phenylindole; FITC, fluorescein isothiocyanate; HF, human fibroblasts; hrs, hours; IL, interleukin; PE, phycoerithrin.
FE-Pro Uptake by AM from Live or Dead/Apoptotic FE-Pro-Labeled Cells
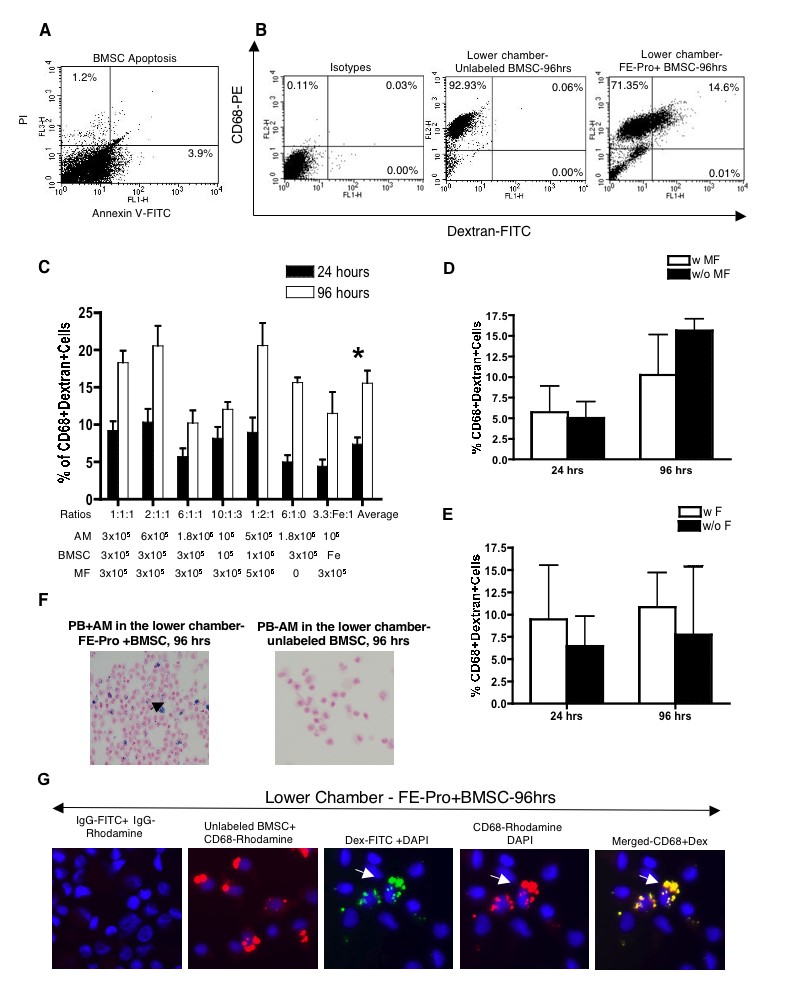
Various amounts of FE-Pro-labeled, live BMSCs, AM, and HF were layered into the Matrigel placed in the upper chamber and incubated for 24 and 96 hours (Table 1). The percentage of dead or apoptotic FE-Pro-labeled BMSCs in the upper chamber after 96 hours of incubation without TRAIL was <5.1% (supplemental online Fig. 1a). The FACS analysis of AM from the lower chambers showed from 4.95% ± 2.68% to 27.39% ± 4.59% of CD68- and dextran-positive macrophages and from the upper chambers from 12.87% ± 9.01% to 29.66% ± 31.14% after 24 hours of incubation (Fig. 4A, 4B). After 96 hours of incubation with various ratios of AM, FE-Pro-labeled BMSCs, and HF, 1.18% ± 2.13% to 21.70% ± 2.61% cells were CD68- and dextran-positive in the lower chambers and 5.88% ± 3.83% to 22.38% ± 3.49% in the upper chambers (Fig. 4A, 4C). The statistical analysis of the pooled results from the six different ratios of labeled and bystander cells to AM showed that the mean uptake of FE-Pro in the upper chamber was significantly higher (p < .004) compared with the mean FE-Pro uptake by AM in the lower chamber only after the 24-hour time point (Fig. 4D). The presence of bystander HF had no significant effect on FE-Pro uptake by AM in both chambers (p = not significant; Fig. 4E). The CD68- and dextran-positive macrophages were visualized by fluorescence microscopy (Fig. 5A). The frequency of dextran- and CD68-positive cells was comparable to FACS results and to the number of cells positive for iron by PB staining (Fig. 5B).
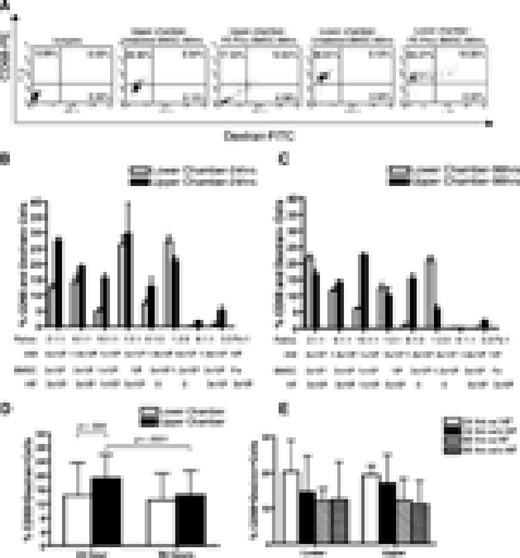
FE-Pro uptake by AM from live, FE-Pro-labeled human BMSCs. (A): Representative fluorescence-activated cell sorting dot plots showing 10.64% of CD68-PE- and dextran-fluorescein isothiocyanate (Dex-FITC)-positive macrophages in the upper chamber and 19.35% of CD68-PE- and dextran-FITC-positive macrophages in the lower chamber after 96 hrs of incubation. (B, C): Bar graph showing the percentage of bromodeoxyuridine (BrdU)- and CD68-positive AM collected from the lower and upper chambers after 24 (B) and 96 (C) hrs of incubation. (D): Bar graph showing the means of BrdU- and CD68-positive AM in the lower and upper chambers after pulling all ratios. Note that the mean uptake of FE-Pro in the upper chamber was significantly higher (p < .004) compared with the mean FE-Pro uptake by AM in the lower chamber only after 24-hr time point. (E): Bar graph showing luck of influence of bystander fibroblasts on FE-Pro uptake by AM after pooling all ratios from the upper and lower chambers. Data are presented as mean ± 1 SD from six samples. Abbreviations: AM, activated macrophages; BMSC, bone marrow stromal cell; FE-Pro, ferumoxide-protamine sulfate complex; HF, human fibroblasts; hrs, hours; IL, interleukin; w/, with; w/o, without; PE, phycoerithrin.
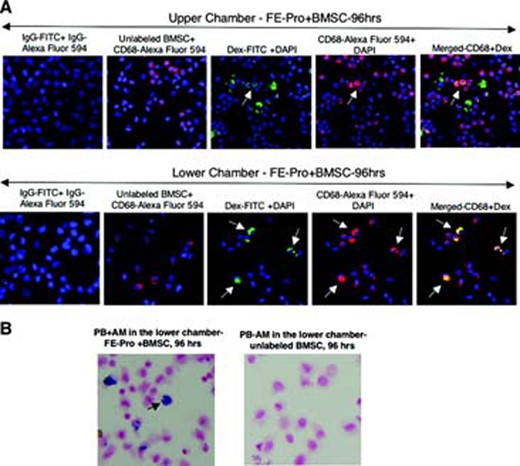
Immunofluorescent and PB staining of AM with transferred FE-Pro from live, FE-Pro-labeled human BMSCs. (A): Representative immunofluorescent images showing detection of Dex with anti-Dex-FITC, CD68 with anti-CD68-Alexa Fluor 594 on AM recovered from both chambers; magnification, ×40. On the merged image of the upper chamber, note cells positive for Dex-FITC only (FE-Pro-labeled BMSCs), cells positive for CD68-Alexa Fluor 594 only that are AM, and cells positive for Dex-FITC and CD68-Alexa Fluor 594. Those cells that took up FE-Pro from FE-Pro-labeled BMSCs are AM. On the merged image of the lower chamber, note cells positive for CD68-Alexa Fluor 594 only that are AM and cells positive for Dex-FITC and CD68-Alexa Fluor 594 (AM that took up FE-Pro from FE-Pro-labeled BMSCs). (B): PB staining of AM recovered from the lower chamber after 96 hrs of incubation with FE-Pro labeled and unlabeled BMSCs. Abbreviations: AM, activated macrophages; BMSC, bone marrow stromal cell; DAPI, 4,6-diamidino-2-phenylindole; Dex, dextran; FE-Pro, ferumoxide-protamine sulfate complex; FITC, fluorescein isothiocyanate; hrs, hours; PB, Prussian Blue.
In the next step, we evaluated whether replacing human fibroblasts with potentially more immunogenic mouse fibroblasts would result in higher FE-Pro uptake by AM. Similar ratios of AM, FE-Pro-labeled BMSCs, and MF were layered into the Matrigel, placed in the upper chamber, and incubated for 24 and 96 hours (supplemental online Table 1). FACS analysis of the lower chamber showed 4.46% ± 1.95% to 10.34% ± 5.29% CD68- and dextran-positive macrophages after 24 hours of incubation and 10.24% ± 4.92% to 20.62% ± 7.35% after 96 hours (supplemental online Fig. 1a, 1b). After pooling all ratios of results from the labeled cells and AM, the comparison of the mean CD68- and dextran-positive cells showed significant differences (p < .001) between the 24- and 96-hour incubation time points. The presence of bystander MF had no significant effect on FE-Pro uptake by AM (supplemental online Fig. 1c). The level of FE-Pro uptake by AM in the presence of MF was comparable to or lower than the mean uptake of the label by AM in the presence of HF. When the ratios from both experiments with MF and HF were combined and separated according to the presence of fibroblasts, there was no difference (p > .05) in the uptake of FE-Pro by AM (supplemental online Fig. 1e).
When the AM were visualized by fluorescence microscopy (supplemental online Fig. 1g), the percentage of dextran- and CD68-positive cells was comparable to FACS results. The PB staining of cells (supplemental online Fig. 1f) from the lower chamber revealed the frequency of iron-containing cells to be comparable to that of dextran-positive macrophages, as determined by FACS analysis.
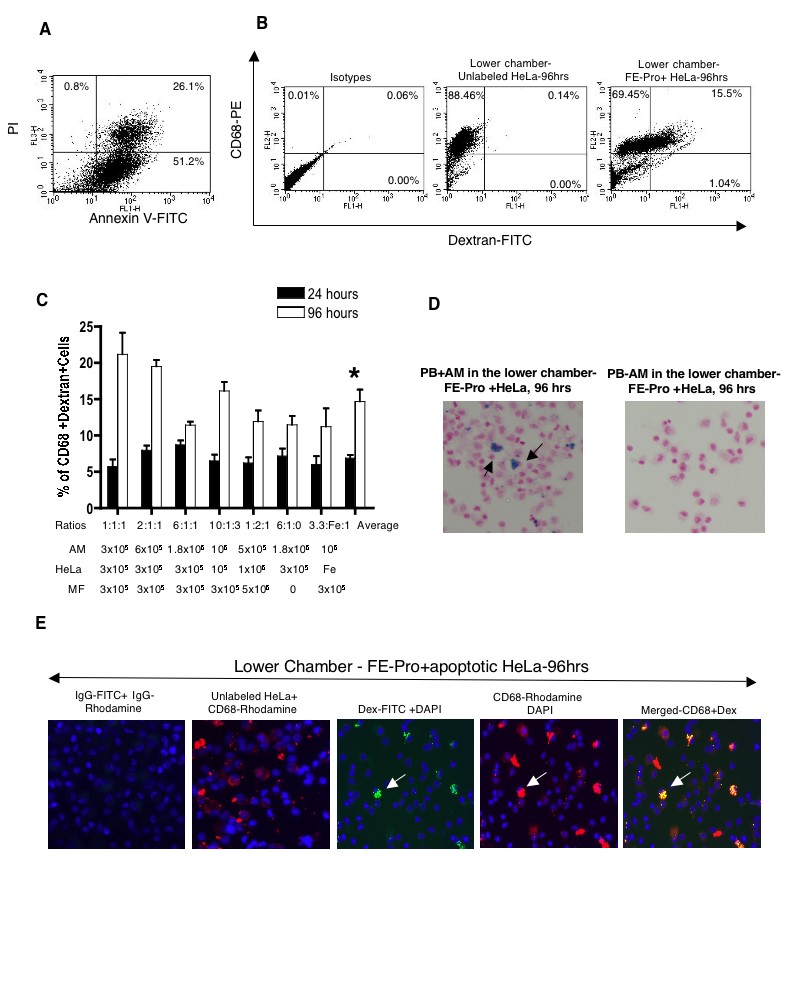
To test whether the uptake of the label from dead/proapoptotic cells is similar to the uptake of the label from live cells, we induced apoptosis in HeLa cells and layered them into the Matrigel together with AM and MF, placed them in the upper chamber, and incubated them for 24 and 96 hours (supplemental online Table 1). After 24 hours of incubation with TRAIL, 78% of HeLa cells were dead (supplemental online Fig. 2a). FACS of the lower chamber revealed 5.7% ± 2.98% to 8.7% ± 1.89% CD68- and dextran-positive macrophages after 24 hours of incubation and 11.43% ± 1.55% to 21.2% ± 8.8% after 96 hours (supplemental online Fig. 2b, 2c). After pooling all ratios of results from the labeled HeLa cells and AM, the comparison of the mean CD68- and dextran-positive cells showed significant differences (p < .01) between the 24- and 96-hour incubation time points (supplemental online Fig. 2c). The AM were also visualized by fluorescence microscopy (supplemental online Fig. 2e). The percentage of dextran- and CD68-positive cells was comparable to FACS results and PB staining (supplemental online Fig. 2d). For comparison, the uptake of FE-Pro not contained in cells by AM harvested from lower chambers was in the range of 1.5% ± 2.57% to 15.7% ± 3.7% at 24 and 96 hours (supplemental online Figs. 4b, 4c, 1c, 2c).
For the control experiments, unlabeled BMSCs or Hela cells were mixed into the Matrigel with AM and HF/MF in the ratio of 6:1:1, and unlabeled BMSCs, HeLa, or MF alone were placed in the upper chamber without AM and incubated for 24 and 96 hours in the presence of SDF-1 and MCP-1 in the lower chamber. BMSCs, Hela cells, or MF/HF did not migrate into the lower chamber, as determined by FACS analysis at each time point (supplemental online Figs. 3a, 4a, 1b, 2b).
Iron Content in AM
To assess the potential impact of FE-Pro uptake by macrophages on MRI, the iron content in the upper and lower chambers was measured in experiments with MF and HF. The iron content in the lower chambers was similar when either FE-Pro-labeled BMSCs or HeLa cells were used. The average iron content was in the range of 3.73 ± 1.04 to 13.78 ± 4.47 μg in the upper chamber and 0.07 ± 0.01 to 0.35 ± 0.21 μg in the lower chamber. Figure 6 summarizes the percentage of the total Fe uptake by AM from both chambers. The transfer of iron was <10% of total iron load of SPION-labeled cells placed in the upper chambers.
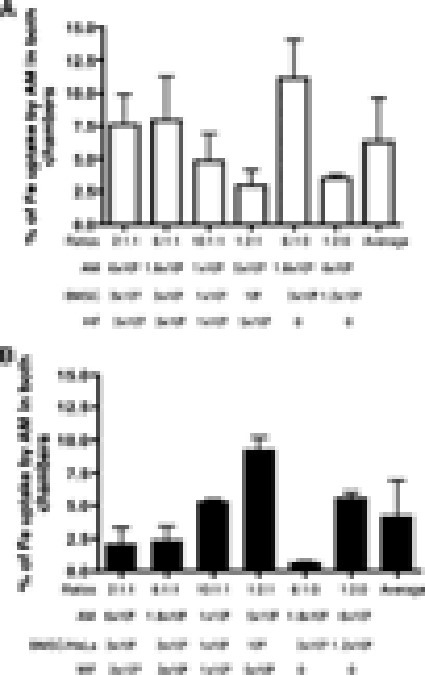
Iron content in AM from the upper and lower chambers of a Boyden chamber. Results are presented as a percentage of total iron that was taken up by AM in the presence of HF (A) and MF (B). Abbreviations: AM, activated macrophages; BMSC, bone marrow stromal cell; HF, human fibroblasts; MF, mouse fibroblasts.
Discussion
The major finding of this study is the quantification of the number of SPION- or BrdU-labeled cells taken up by activated macrophages in an in vitro model of localized inflammation. After 96 hours of incubation, 10%–20% of the labeled cells were taken up by activated macrophages. This modified BC experiment was developed to simulate a localized area of inflammation where labeled cells would be directly transplanted into a site as part of a therapy approach [32, 23]. Circulating monocytes stimulated by antigen (LPS in this study) leave the circulation in areas of inflammation and then phagocytose damaged host or foreign cells. The ratios of AM to transplanted labeled cells (BMSCs or HeLa cells) to bystander cells (HF or MF) used in this study were estimated on the basis of the number of cells used in direct transplantation of cells reported in other studies [32, 23]. We included in our BC assay human or mouse fibroblasts to represent local endogenous cells that are present in vivo in our BC assay. The presence of either human or mouse bystander cells had no effect on the uptake of SPION-labeled cells by macrophages, suggesting a critical role for the ratio of labeled cells to inflammatory cells and microenvironmental conditions. Moreover, potent stimulators of macrophage migration, SDF-1 and MCP-1, were placed in the lower chambers to stimulate migration into and out of damaged inflamed tissue.
Recently, several studies have indicated that exogenous or endogenous label from cells could be transferred to tissue macrophages in vivo following direct transplantation of labeled stem cells. Burns et al. reported that BrdU and other thymidine analogs are transferred from donor transplanted cells to host neural precursor cells and glia in the developing and adult brain [22]. In the rat fluid-percussion model, Molcanyi et al. demonstrated that live green fluorescent protein-labeled embryonic stem cells implanted into the rat brain were phagocytosed by activated macrophages [30]. Lepore et al. showed that following implantation of ferumoxides labeled lineage-restricted neural precursors into an intact spinal cord, some of tissue macrophages took up the iron from grafted cells [28]. Recently, Brekke et al. demonstrated the in vivo transfer of gadolinium rhodamine dextran label from neural stem cells to macrophages in gliomas in rat brain [34]. The above studies only qualitatively assessed the presence of macrophages positive for exogenous label in the regions examined. The results in the current study that 10%–20% of the BrdU- or FE-Pro-labeled cells were taken by AM in the upper or lower chambers potentially provides an estimate to aid in the interpretation of future direct cell transplant studies. For example, in neural transplantation studies, where as much as 90% of transplanted cells into central nervous system may die shortly after transplantation, 10%–15% of false-positive labeled cells may be of great importance and require further validation by additional staining for macrophage markers or markers expressed by other bystander cells [22].
Since less than 10% of the total iron from the FE-Pro-labeled BMSCs were found in the AM, the contribution of iron-containing macrophages to the hypointense regions observed on in vivo cellular MRI in an area of magnetically labeled cell would be considered minimal. It can be inferred from this study that labeling cells with other nanoparticles, such as quantum dots, would also result in the transfer of the label from directly transplanted cells to activated tissue macrophages and contribute minimally to the observed fluorescence on in vivo optical imaging [35].
The quantification of iron uptake by AM was based on FACS analysis of cells stained with anti-dextran antibodies. The results of this study indicate that the FACS analysis is a highly reproducible, reliable, and unbiased method of determining cells containing dextran-coated iron oxide nanoparticles and complements the standard PB staining technique that is commonly used. We observed a similar uptake of AM containing SPION when stained with anti-dextran antibodies and analyzed by FACS compared with counting of PB-stained cells at 96 hours. In the current study we did not carry out the BC experiments past 96 hours because serial viability studies of AM demonstrate that <20% were alive at 120 hours.
Bourrinet et al. showed dextran to begin detaching from ferumoxtran-10, a dextran-coated ultrasmall SPION, within 24 hours [36]. Iron (PB) and dextran staining of AM after 96 hours of incubation revealed similar results indicating dextran to be coupled with iron oxide core and validating the reliability of dextran staining and FACS analysis of FE-Pro-labeled cells up to 96 hours. FACS analysis of FE-Pro-labeled cells at later time points using anti-dextran antibody is under investigation.
A possible limitation of this study is the extrapolation to the in vivo condition from the in vitro model that we have developed. We were unable to predict the biological diversity and number of cells that would be found in inflammatory lesions in which cells would be transplanted. Therefore, we used different ratios of cells in this model of local inflammation. By pooling the results from the different ratios of labeled cells to activate macrophages, we were able to show a limited number of significant differences in the mean uptake of exogenous label between studies involving labeled human BMSCs and HF at 24 and 96 hours of incubation. In addition, we observed no difference in pooled experimental results when AM were incubated with TRAIL-treated HeLa cells versus viable BMSCs, nor did it make a difference whether the bystander cells were human or mouse fibroblasts. There is a need to demonstrate a similar uptake by AM in an in vivo model of controlled or localized inflammation that would be easily accessible for an injection of labeled cells.
Conclusion
In conclusion, we have shown that direct implantation of BrdU/SPION labeled cells can result in the uptake of the label by activated macrophages. Quantifying the percentage of macrophages that incorporate the iron oxide or BrdU label from live, dying, or dead cells will aid in the interpretation of results obtained both microscopically and by in vivo cellular imaging.
Acknowledgements
This research was supported by the Intramural Research Program of the Clinical Center in the National Institutes of Health.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}