
Abstract
The metabolic properties of cancer cells diverge significantly from those of normal cells. Energy production in cancer cells is abnormally dependent on aerobic glycolysis. In addition to the dependency on glycolysis, cancer cells have other atypical metabolic characteristics such as increased fatty acid synthesis and increased rates of glutamine metabolism. Emerging evidence shows that many features characteristic to cancer cells, such as dysregulated Warburg-like glucose metabolism, fatty acid synthesis and glutaminolysis are linked to therapeutic resistance in cancer treatment. Therefore, targeting cellular metabolism may improve the response to cancer therapeutics and the combination of chemotherapeutic drugs with cellular metabolism inhibitors may represent a promising strategy to overcome drug resistance in cancer therapy. Recently, several review articles have summarized the anticancer targets in the metabolic pathways and metabolic inhibitor-induced cell death pathways, however, the dysregulated metabolism in therapeutic resistance, which is a highly clinical relevant area in cancer metabolism research, has not been specifically addressed. From this unique angle, this review article will discuss the relationship between dysregulated cellular metabolism and cancer drug resistance and how targeting of metabolic enzymes, such as glucose transporters, hexokinase, pyruvate kinase M2, lactate dehydrogenase A, pyruvate dehydrogenase kinase, fatty acid synthase and glutaminase can enhance the efficacy of common therapeutic agents or overcome resistance to chemotherapy or radiotherapy.
Similar content being viewed by others


Facts
-
• The metabolic properties of cancer cells are remarkably different from those of normal cells.
-
• Dysregulated cellular metabolism is linked to drug resistance in cancer therapy.
-
• Targeting metabolic enzymes improves the efficacy of cancer therapy.
-
• Targeting metabolic enzymes may overcome therapeutic resistance.
Open Questions
-
• Whether the dysregulated cellular metabolism contributes to therapeutic resistance?
-
• Is inhibition of metabolic enzymes a promising strategy to improve the efficacy of cancer therapy or overcome therapeutic resistance?
-
• Is targeting dysregulated metabolism a selective approach to inhibit cancer cells?
-
• What are the mechanisms by which targeting metabolic enzymes improves the efficacy of cancer therapy or overcomes chemoresistance?
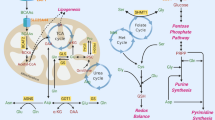
The metabolic properties of cancer cells are different from those of normal cells. Cancer cells are more dependent on aerobic glycolysis, fatty acid synthesis and glutaminolysis for proliferation.1 This difference suggests that targeting metabolic dependence could be a selective approach to treat cancer patients. In 1956, Warburg observed that the rate of glycolysis was abnormally high in cancer cells, yet a smaller fraction of this glucose is broken down by oxidative phosphorylation. This ‘Warburg effect’ indicates that cancer cells prefer glycolytic breakdown of glucose for energy, rather than mitochondrial oxidative phosphorylation.1, 2, 3, 4, 5, 6, 7, 8, 9 Although the molecular mechanisms that define the Warburg effect are not yet fully understood, the increased glycolysis observed in cancer cells is well accepted to be important for the support of malignant phenotypes (Box 1).8
In addition to the dependency on glycolysis, cancer cells exhibit other metabolic characteristics such as increased fatty acid synthesis and glutamine metabolism. Enhanced fatty acid synthesis provides rapidly proliferating tumor cells lipids for membrane biogenesis, conferring both a growth and survival advantage.10 Similarly, cancer cells are extremely sensitive to glutamine deprivation and cannot proliferate in culture without it. ‘Glutamine addiction’ results in enhanced production of byproducts necessary for rapidly proliferating cells, such as amino-acid precursors.11, 12
Recently many review articles on cancer and metabolism13, 14, 15, 16, 17, 18, 19, 20, 21 have been published. However, dysregulated metabolism in therapeutic resistance, a highly clinical relevant area in cancer research, has not been specifically addressed. Here we will discuss the relationship between cellular metabolism and drug resistance in cancer cells and how to improve cancer therapeutics and to overcome drug resistance by targeting dysregulated metabolic enzymes and pathways.
Dysregulated Metabolism has been Linked to Drug Resistance
The ability to reduce chemoresistance would be a significant boon for cancer patients, demonstrating the importance of research into the mechanisms underlying how chemoresistance arises (Box 2). Mounting evidence supports the idea that dysregulated cellular metabolism is linked to drug resistance in cancer therapy.22, 23, 24, 25 In the glycolytic pathway, lactate dehydrogenase A (LDHA) contributes to paclitaxel/trastuzumab resistance in breast cancer and pyruvate dehydrogenase kinase 3 (PDK3) contributes to hypoxia-induced drug resistance in cervical and colon cancer. Fatty acid synthase (FASN), a key complex catalyzing fatty acid synthesis, is linked to acquired docetaxel/trastuzumab/adriamycin resistance in breast cancer or intrinsic gemcitabine and radiation resistance in pancreatic cancer. Finally, glutaminolysis is linked to cisplatin resistance via the activation of mammalian target of rapamycin complex 1 (mTORC1) signaling in gastric cancer (Table 1). In this review, we will discuss the role of these enzymes or processes in drug resistance in detail below.
Targeting Cellular Metabolism to Improve Cancer Therapeutics
Targeting glycolytic enzymes
As a central energetic resource for the cell, glucose metabolism is quite complex. Many enzymes contribute to the series of reactions necessary for the glycolytic breakdown of glucose. Below we will discuss glycolytic inhibition as an anticancer strategy in the context of selected components of the glycolytic pathway, such as glucose transporters (GLUTs), hexokinase (HK), pyruvate kinase M2 (PKM2) and LDHA.
Glucose transporters
The first rate-limiting step of glucose metabolism is the transport of glucose across the plasma membrane. The GLUT family of proteins is responsible for this, and are often found dysregulated or overexpressed in malignant cells.26 The human GLUT family consists of 14 members (GLUT1-14 or SLC2A1-14).26, 27, 28 Here we will focus on targeting GLUT1, GLUT3 and GLUT4 for improving cancer therapy.
WZB117 is an inhibitor of GLUT1 that decreases glucose uptake, intracellular ATP levels and glycolytic enzymes leading to a lowered rate of glycolysis and cellular growth. Exogenous ATP rescues growth of WZB117-treated cancer cells, suggesting that reduction of ATP is an important mechanism of WZB117’s anticancer effect. WZB117 also induces endoplasmic reticulum (ER) stress leading to cell-cycle arrest. The combination of WZB117 and cisplatin or paclitaxel displayed synergistic anticancer effects (Table 1).29, 30 Under hypoxia, the GLUT1 inhibitor phloretin significantly enhances daunorubicin’s anticancer effects (Table 1) and overcomes hypoxia-conferred drug resistance. Inhibition of glucose uptake by phloretin sensitizes P-glycoprotein overexpressed doxorubicin-resistant cells to daunorubicin via enhancing daunorubicin-induced apoptosis only under hypoxia.27
Multiple myeloma (MM) cells are dependent on GLUT4 activity for basal glucose consumption, maintenance of Mcl-1 protein levels, growth and viability. Ritonavir displays off-target inhibitory effects on GLUT4 and inhibits glucose consumption and proliferation by reducing Mcl-1 expression to induce apoptosis. Ritonavir also inhibits viability of primary myeloma cells and increases the sensitivity to doxorubicin (Table 1).28 Temozolomide is used with radiation and chemotherapy to treat glioblastoma, yet nearly all glioblastoma patients develop resistance. Long-term treatment of glioblastoma cells with temozolomide in vitro induces partial resistance in vivo through upregulation of GLUT3, suggesting involvement in temozolomide resistance and that selective targeting of GLUT3 could delay the acquisition of such resistance in glioblastoma cells.31 Inhibiting glucose uptake may potentiate cancer therapeutics or overcome hypoxia/drug-induced resistance.
Hexokinase
HK has important roles in both glycolysis and apoptosis and inhibitors of HK, such as 2-deoxyglucose (2-DG), 3-bromopyruvate (3-BrPA) and lonidamine (LND) are in pre-clinical and early phase clinical trials. The effects of 2-DG, 3-BrPA and LND on cell death in combination with chemotherapy or radiotherapy have been reviewed in detail.17 We will discuss the impact of these inhibitors on cell death and their use to combat drug resistance.
2-DG is a glucose analog that is phosphorylated by HK to 2-DG-phosphate, which cannot be further metabolized. Accumulation of 2-DG inhibits glycolysis causing ATP depletion, cell cycle inhibition and cell death.32, 33 Under normoxic conditions, 2-DG can interfere with N-linked glycosylation and induce an unfolded protein response, leading to subsequent induction of some proapoptotic BH3-only proteins.17, 34 There are no ongoing clinical trials using 2-DG as a single agent as in some systems it does not have a significant effect on tumor growth in vivo.35 However, combining 2-DG with radiation or chemotherapeutic treatments potentiates the tumor-destroying effects and enhances the clinical efficacy.36
Bcl-2 family proteins have an important role in the regulation of apoptosis, tumorigenesis and cellular response to cancer therapeutics. Bcl-2 family proteins are divided into three groups: anti-apoptotic members (Bcl-2, Bcl-XL, Bcl-w, Mcl-1 and A1); pro-apoptotic members (Bax and Bak); and those with only a BH3 domain that promote apoptosis by binding anti-apoptotic proteins (Bad, Bid, Bim, Noxa and Puma).37 BH3-mimetics, such as ABT-737 and ABT-263, are small-molecule inhibitors of Bcl-2, Bcl-XL, Bcl-w, but not Mcl-1. Several recent studies have demonstrated that 2-DG or LND enhances ABT-263/737-induced apoptosis both in vitro and in vivo (Table 1).38, 39, 40 There are two proposed mechanisms explaining the effect of 2-DG on ABT-263/737-induced apoptosis. In the first 2-DG decreases Mcl-1 levels indirectly by inhibiting glycolysis and depleting ATP levels, leading to activation of AMP-activated protein kinase and inhibition of Mcl-1 translation.38, 39, 41 In the second mechanism, 2-DG weakens the interaction between Bak and Mcl-1, which increases the ability of ABT-263/737 to release Bak from the Mcl-1/Bcl-XL/Bak heterotrimer, thus inducing apoptosis.40 Both 2-DG and ABT-737 are well tolerated by patients and in clinical trials, suggesting 2-DG-ABT-737 co-treatment has the potential to be developed in treating ABT-737 resistance.
Trastuzumab is a humanized monoclonal antibody against ErbB2 and has shown efficacy treating ErbB2-positive breast cancer patients, yet acquired trastuzumab resistance occurs in most patients.42, 43, 44, 45, 46, 47, 48 Our previous studies showed that overexpression of ErbB2 promotes glycolysis and increases their sensitivity to glycolytic inhibition.49 Trastuzumab-resistant human cells also have increased glucose uptake and lactate production, indicative of increased glycolysis. Trastuzumab also inhibits glycolysis via downregulation of HSF1 and LDHA in breast cancer (Figure 1).23 We found 2-DG/trastuzumab combination therapy synergistically inhibits growth of both trastuzumab-sensitive and trastuzumab-resistant human breast cancers in vitro and in vivo (Table 1), because of more efficient glycolysis inhibition.23 These results suggest that 2-DG can effectively enhance efficacy of trastuzumab in treating ErbB2-positive human breast cancer cells and overcome trastuzumab resistance.

Dysregulated metabolism affects chemoresistance via multiple cellular pathways. Glycolytic intermediates generated by dysregulated cancer metabolism fuel expanded cellular growth and contribute to clinical resistance. ATP generated by the glycolytic breakdown of glucose fuels the active export of chemotherapeutic agents by the ABC transporters and induces HIF-1α expression. Export of the glycolytic end product, lactate and expression of carbonic anhydrases shift the pH ratio of the interior and exterior of the cell resulting in decreased passive transport of basic drugs. Signaling pathways activated by dysregulated metabolism also contribute to resistance, either via repressing pro-apoptotic signaling or activating compensatory pathways to circumvent drug-induced signal inhibition
3-BrPA is a glycolysis inhibitor that targets HKII and depletes cellular ATP reserves, a key determinant of chemoresistance in certain cancer types.50, 51 In leukemia and MM cells increased glycolysis raises ATP levels, which activates ATP-binding cassette (ABC) transporters and confers drug resistance via enhanced drug efflux activity (Figure 1). 3-BrPA causes ATP depletion, decreasing ABC transporter activity and drug efflux, therefore enhancing drug retention in cells producing preferential cell death in malignant cells. Glycolysis inhibition by 3-BrPA not only enhances the cytotoxic effects of daunorubicin and doxorubicin, but also markedly suppresses tumor growth when used with doxorubicin to treat MM-bearing mice (Table 1).52 In addition to activating ABC transporters, increased ATP levels from elevated glycolysis upregulate HIF-1α and enhance HIF-1α-mediated signaling, which can confer chemoresistance (Figure 1). ATP depletion by 3-BrPA partially reversed the resistant phenotype and resensitized cells to chemotherapeutic agents such as oxaliplatin and 5-fluorouracil (5-FU; Table 1).53 These findings demonstrate that glycolysis inhibition by 3-BrPA causes ATP depletion, which can improve cancer therapeutics or overcome chemoresistance.
Most treatment failure in childhood acute lymphoblastic leukemia (ALL) is ascribed to glucocorticoid (e.g., prednisolone) resistance. Increased glycolysis is directly associated to glucocorticoid resistance and inhibition of glycolysis by 2-DG, 3-BrPA or LND increases prednisolone-induced toxicity in leukemia cells (Table 1).54 Importantly, 2-DG can reverse glucocorticoid resistance in primary leukemia cells isolated from pediatric ALL patients.54
Pyruvate kinase M2
Pyruvate kinase (PK) is the last rate-limiting enzyme in the glycolytic pathway and catalyzes the conversion of phosphoenolpyruvate and ADP into pyruvate and ATP. There are four isoforms of PK in mammals (M1, M2, L and R), which are expressed in different cell types.14, 55 PKM2 is expressed predominantly in tumor cells56 and is important for cancer metabolism and tumor growth.57 Several studies showed a negative correlation between PKM2 expression and drug resistance.58, 59, 60 Decreased PKM2 protein and activity is linked to cisplatin resistance while suppression of PKM2 expression by siRNA increased cisplatin resistance.60 Both PKM2 mRNA and protein levels are downregulated in oxaliplatin-resistant cells and PKM2 mRNA levels are inversely correlated with oxaliplatin resistance in a panel of eight colorectal cancer cell lines. Low PKM2 mRNA levels in patients are associated with high p53 protein levels and predict poor response to oxaliplatin.59 In contrast, PKM2 levels are significantly upregulated in secreted proteins of the 5-FU-resistant colon cancer cell line. Moreover, increased PKM2 is also observed in sera and tissues from colorectal cancer patients with poor response to 5-FU. These findings suggested that upregulation of PKM2 is linked to 5-FU resistance in colon cancer.61
Changes in PKM2 expression are associated with drug resistance in different tumor. This indicates that PKM2 is a potential target for adjuvant cancer therapy. For example, shRNA targeting PKM2 improves the therapeutic efficacy of cisplatin by increasing apoptosis and inhibiting proliferation (Table 1).62 Silencing of PKM2 enhances the efficacy of docetaxel because of increased inhibition of proliferation and apoptosis-inducing activity both in vitro and in vivo (Table 1).63 A possible mechanism for the sensitization of lung cancer cells to docetaxel is that shPKM2 decreases ATP levels leading to intracellular accumulation of docetaxel.63 These results indicated that targeting PKM2 can effectively improve the efficacy of chemotherapeutic drugs.
Lactate dehydrogenase A
LDHA catalyzes the final step in the glycolytic pathway, the conversion of pyruvate and NADH to lactate and NAD+, and has a critical role in tumor maintenance. Knockdown of LDHA in tumor cells produces increased mitochondrial respiration, decreased cellular ability to proliferate under hypoxic conditions, and suppressed tumorigenicity.64 LDHA knockdown in the fumarate hydratase knockdown background results in increased apoptosis via ROS production, resulting in a reduction in tumor growth and indicating that LDHA might be a promising therapeutic target.65 Inhibition of LDHA by siRNA or FX11 treatment reduces ATP levels and induces significant oxidative stress resulting in cell death.66 Importantly, combining FX11 with FK866, an NAD+ synthesis inhibitor, induces lymphoma regression in a xenograft model (Table 1).66
Paclitaxel (taxol) is a widely used chemotherapeutic agent in the treatment of a variety of human cancers (Table 1). LDHA expression and activity is higher in taxol-resistant breast cancer cells than in taxol-sensitive cells, and downregulation of LDHA resensitizes taxol-resistant cells to taxol. Taxol-resistant cells are more sensitive to oxamate, a pyruvate analog that inhibits glycolysis by inhibiting the conversion of pyruvate to lactate. These results indicate that LDHA and lactate metabolism have an important role in the resistance to paclitaxel. Moreover, combination of paclitaxel with oxamate shows synergistic inhibitory effect on taxol-resistant cells (Table 1) by promoting cellular apoptosis.24
Heat shock factor 1 (HSF1) is the master regulator of the heat shock response in eukaryotes. HSF1 functions primarily to coordinate the response to heat shock, but recent studies demonstrate HSF1 exhibiting non-heat shock functions important for cancer development.67, 68, 69 Dai et al70 reported that HSF1 increases glucose uptake, lactate production and LDH activity. Our previous study showed that ErbB2 promotes glycolysis partially through upregulation of HSF1 and LDHA (Figure 1), whereas downregulation of HSF1 leads to decreased glycolysis.49 Our recent studies showed that trastuzumab-resistant cells have significantly higher HSF1 protein levels than trastuzumab-sensitive cells. Moreover, we found that inhibition of HSF1 sensitizes cells to trastuzumab and overexpression of HSF1 increased trastuzumab resistance, demonstrating that HSF1 can have an important role in resistance to trastuzumab.23
We reported that increased glycolysis via HSF1 and LDHA contributes to trastuzumab resistance. Importantly, we found that combination of trastuzumab and oxamate synergistically inhibits growth of both trastuzumab-sensitive and trastuzumab-resistant cancer both in vitro and in vivo (Table 1), because of more efficient glycolysis inhibition.23 Overall, high-rate glycolysis confers chemoresistance and HSF1 and LDHA may potentially act as excellent targets for overcoming this resistance in cancer patients.
Targeting PDK
Pyruvate dehydrogenase (PDH) is responsible for the rate-limiting conversion of pyruvate to acetyl-CoA, which enters the tricarboxylic acid (TCA) cycle to generate ATP. PDK phosphorylates PDH and inhibits its enzymatic activity. Four isotypes of PDK (PDK1–4) have been identified with PDK3 demonstrating the highest activity coupled with a lack of inhibition in response to high concentrations of pyruvate.71 Hypoxia induces PDK3 expression via upregulation of HIF-1α, which binds to the promoter of PDK3, resulting in a switch from mitochondrial respiration to glycolysis for energy production. Hypoxia-mediated PDK3 induction or forced PDK3 overexpression significantly inhibits cell apoptosis and increases resistance to cisplatin or paclitaxel (Figure 1). Knockdown of PDK3 inhibited hypoxia-induced glycolysis and increases susceptibility of cancer cells to anticancer drugs such as cisplatin, paclitaxel or oxaliplatin (Table 1).71, 72 Moreover, PDK3 levels are elevated and correlated with the HIF-1α level in patient colon cancer tissues and strongly correlates with the severity of the cancer while predicting poor disease-free survival outcomes.72 These findings indicate that PDK3 contributes to hypoxia-induced drug resistance and is potentially a novel target for improving chemotherapy or overcoming drug resistance.
Dichloroacetate (DCA) inactivates PDK leading to reactivation of PDH and a metabolic switch from glycolysis to mitochondrial respiration.55, 73 The preclinical trials on DCA have shown its effectiveness in a variety of tumors via induction of apoptosis.74, 75, 76, 77, 78 However, its effect as a solitary agent is limited in the ongoing clinical trials.79, 80 Combinational therapy has displayed more effectiveness; cotreatment with DCA and omeprazole exhibits synergistic antitumor activity (Table 1).79 Cotreatment of DCA, omeprazole and tamoxifen completely blocks the proliferation of fibrosarcoma cells (Table 1), whereas the same combination does not affect the proliferation of human normal fibroblast cells. Moreover, these three drugs were prescribed to a cholangiocarcinoma patient and successfully blocked the disease progression for 3 months.80 Owing to its low price, low toxicity, oral administration, long history of clinical use and ability to overcome cancer cells apoptosis resistance DCA serves as a potential metabolic-targeting molecule for sensitizing cancer cells to chemotherapy or radiotherapy.76 DCA potentiates the anticancer effects of 5-FU (Table 1) via inducing more mitochondrial-mediated apoptosis.81 Sulindac, a FDA-approved non-steroidal anti-inflammatory drug, has anticancer activity. The combination of DCA and sulindac enhances killing of lung and squamous cell carcinoma cells (Table 1), but not normal cells. The selective killing mechanism involves ROS production, loss of mitochondrial membrane potential, JNK-mediated signaling and apoptotic death.82 DCA can also increase the sensitivity to radiotherapy.75 Cao et al75 reported that DCA sensitizes both wild-type and Bcl-2-overexpressing cancer cells to radiation (Table 1) by potentiating the apoptotic machinery via interaction with Bcl-2. In summary, targeting PDK can sensitize cancer cells to chemotherapy, radiotherapy or overcome drug resistance.
Targeting FASN
The fatty acid biosynthesis pathway catalyzes lipid synthesis from basic metabolites like acetyl- and malonyl-CoA. The FASN complex facilitates lipogenesis by synthesizing palmitate from its base components. FASN expression in normal adult tissues is generally very low or undetectable, and it is significantly upregulated and correlates with poor prognosis in many types of cancer. The metabolic products of the FASN complex are rapidly consumed by actively dividing cells and recent data demonstrates that FASN expression is important for tumor growth and survival, suggesting that FASN is a metabolic oncogene.83
FASN has an active role in ErbB2-induced breast cancer chemoresistance to docetaxel,84 while trastuzumab-resistant breast cancer cells gain high sensitivity to FASN inhibition indicating that FASN is also important in ErbB2-induced resistance in breast cancers.85 FASN is overexpressed and its activity is increased in the multidrug-resistant breast cancer cell line MCF7/AdVp3000.22 Increased palmitic acid production from ectopic FASN overexpression is also shown to decrease adriamycin and mitoxantrone-induced apoptosis.22 In pancreatic cancer, there is also a positive correlation between FASN expression and resistance to chemo- or radiotherapy. FASN expression is significantly upregulated in pancreatic cancer and inhibition of FASN by siRNA or the FAS inhibitor orlistat reduces gemcitabine resistance, whereas ectopic overexpression of FASN contributes to intrinsic resistance to gemcitabine and radiation. FASN-induced radiation resistance may result from decrease in radiation-mediated ceramide production, leading to reduced caspase 8-induced apoptosis. However, the mechanism of FASN-induced gemcitabine resistance remains to be elucidated.86
To date, several FASN inhibitors have shown antitumor activity including cerulenin, C75, orlistat, C93, GSK 837149A and natural plant-derived polyphenols. Both cerulenin and C75 are early small-molecule FASN inhibitors. Cerulenin is a natural compound isolating from Cephalosporium caerulens and contains an epoxy group that reacts with FASN to inhibit its activity. C75 is derived from cerulenin and interacts with FASN to inhibit its activity.83 Both cerulenin and C75 induce cancer cell apoptosis by similar mechanism including malonyl-CoA accumulation,87 p53 accumulation,88 induction of ER stress89 and suppression of DNA replication.90 FASN blockade by cerulenin synergistically enhances the efficacy of docetaxel against ErbB2-overexpressing and docetaxel-resistant SKBR3 cells (Table 1) in part via decreasing ErbB2 expression.84 Inhibition of FASN activity with cerulenin/C75 or by siRNA upregulates the expression of PEA3, a transcriptional repressor of ErbB2, leading to downregulation of ErbB2 in ErbB2-overexpressing breast and ovarian cancer cells.84 A combination of the FASN inhibitor cerulenin and trastuzumab synergistically downregulates ErbB2 expression, leading to more effective tumor growth inhibition (Table 1). Furthermore, inhibition of FASN activity synergistically enhances trastuzumab-induced apoptosis in ErbB2-overexpressing breast cancer cells.91 The model proposed by Menendez et al91 describes crosstalk between FASN and ErbB2 and suggests that FASN has a role in regulation of proliferation and cell survival by assisting in the maintenance of the cancerous phenotype. FASN inhibition affects the phospholipid partitioning and the formation of lipid rafts, which may result in the internalization and degradation of ErbB2 rather than successfully migration to the cell surface. This depletion of cell surface-associated ErbB2 could enhance the antitumor effects of trastuzumab (Table 1).92 In addition to enhancing the efficacy of docetaxel and trastuzumab, cerulenin increases 5-FU-induced growth inhibition (Table 1).93 Similarly, C75 and trastuzumab synergistically decrease ErbB2 expression and enhance apoptotic cell death (Table 1).85
Orlistat is a β-lactone compound and an irreversible inhibitor of FASN. Orlistat induces cell cycle G1/S arrest by downregulating Skp2, a component of the E3 ubiquitin ligase that controls the turnover of p27Kip1, leading to activation of the retinoblastoma protein pathway.94 Orlistat inhibits endothelial cell proliferation and angiogenesis.95 In addition to cytostatic effects, orlistat also has cytotoxic effects through activation of caspase-8-mediated apoptosis because of negative regulation of the mTOR pathway by upregulation of DNA damage-inducible transcript 4.96 FASN inhibition with orlistat increases sensitivity to adriamycin and mitoxantrone in FASN-overexpressing breast cancer cells (Table 1) but not in the normal mammary epithelial cell line.22 Orlistat treatment of pancreatic cancer cells increases the response to gemcitabine (Table 1).86 In summary, FASN is a promising anticancer target that may result in chemosensitization or enhanced efficacy when FASN function is disrupted as part of a combinatorial treatment regimen.
Targeting glutaminolysis
Glutamine has an important role in cell growth and energy metabolism. Glutaminolysis, consists of two steps: the first is catalyzed by glutaminase (GLS) and converts glutamine to glutamate, whereas the second is catalyzed by glutamate dehydrogenase (GDH) and converts glutamate to α-ketoglutarate (α-KG).97 There are two types of GLS in mammalian cells, kidney-type GLS (GLS1) and liver-type GLS (GLS2).98 Metabolic flux experiments tracking 13C show that cancer cells exhibiting Warburg-like metabolism do not stop utilizing the TCA cycle – instead these cells come to rely on glutamine as the carbon source for the TCA cycle.99 This allows the intermediates generated by the TCA cycle to feed other biosynthetic pathways as precursors.98 Therefore, cancer cells are dependent on glutamine to maintain the TCA cycle. Glutaminolysis co-induced by glutamine and leucine activates mTORC1 signaling, which triggers cell growth and inhibits autophagy.97 The mTOR pathway is involved in cisplatin resistance in highly malignant AFP-producing gastric cancer (AFPGC).100 This indicates that elevated glutaminolysis is linked to drug resistance.
Bis-2-[5-phenylacetamido-1,2,4-thiadiazol-2-yl] ethyl sulfide (BPTES), an inhibitor of GLS, caused decreased aerobic cell proliferation and hypoxic cell death.101 Inhibition of GLS by siRNA or BPTES slows the growth of glioblastoma cells with an isocitrate dehydrogenase 1 (IDH1) mutation. BPTES treatment inhibits GLS activity, lowers glutamate and α-KG levels and increases glycolytic intermediates, suggesting that simultaneous inhibition of GLS and glycolysis may be a more efficient strategy to treat mutant IDH1 patients.102 An inhibitor of Rho GTPase-dependent cellular transformation, named 968, was found to block the growth of human breast cancer and B lymphoma cells without affecting normal cells. 968 Targets GLS C, a specific carboxy-terminal splice variant form of GLS1. Elevated levels of basal GLS activity has been shown to be dependent on Rho GTPases and NF-κB activity in transformed fibroblasts and breast cancer cells, which is blocked by 968.25 This demonstrates that oncogenic transformation can be inhibited by targeting GLS activity, a potential therapeutic strategy against human malignancies.11, 16, 25
Rapamycin, a mTORC1 inhibitor, enhances the antitumor effect of cisplatin in AFPGC both in intro and in vivo.100 Inhibition of mTORC1 by NVP-BEZ235, a dual PI3K/mTOR inhibitor, synergizes with chemotherapeutic agents such as cyclophosphamide, cytarabine and dexamethasone in T-cell ALL cell lines. Moreover, NVP-BEZ235 can sensitize vincristine-resistant Jurkat cells, indicating that inhibition of mTORC1 activity may revert chemoresistance.103 Glutaminolysis activates mTORC1 signaling and inhibition of glutaminolysis via GLS inhibitors (DON and BPTES) or siRNA-targeting GLS or GDH, prevents mTORC1 activation.97 It is reasonable to predict that targeting glutaminolysis or GLS may sensitize cancer cells to common chemotherapeutic agents by reducing mTORC1 activity.
Conclusions
Cancer cells reprogram their metabolism in order to satisfy their bioenergetic and biosynthetic requirements. Increased aerobic glycolysis, fatty acid synthesis and glutamine metabolism has been linked to therapeutic resistance in cancer. We speculate that deregulated cancer metabolism promotes cell proliferation because of increased energy production and metabolite synthesis, which decreases drug-induced apoptosis, conferring therapeutic resistance. Molecular mechanisms of drug resistance are complex and include increased drug efflux, drug inactivation, enhanced DNA damage repair and activation of pro-survival signaling (Figure 1). Increased glycolysis produces higher ATP and NADPH levels. Chemotherapeutic drugs display antitumor effects in part by inducing oxidative damage. NADPH is a critical antioxidant and high levels maintained through increased glycolysis in cancer cells may contribute to chemoresistance. ATP exerts two effects on drug resistance: elevated ATP levels can activate ABC transporters leading to increased drug efflux and upregulate HIF-1α signaling inducing hypoxia-associated drug resistance. Both increased drug efflux and upregulation of HIF-1α signaling result in therapeutic resistance.
HIF-1α-mediated resistance occurs through a variety of mechanisms. Upregulation of the enzymes necessary for glycolysis facilitates a metabolic shift that enhances non-mitochondrial mechanisms of ATP production.104 Reduced reliance on mitochondria results in less reactive oxygen species, which prevents the DNA damage that would activate both DNA repair and stress response pathways, steps that help set the stage for the induction of apoptotic pathways.105, 106
The increase in glycolytic metabolism also results in the production of lactate, whose export results in the acidification of the extracellular environment. The resulting extracellular acidification coupled with HIF-1α-induced expression of carbonic anhydrases causes a significant change in the pH ratio between the intracellular and extracellular environments.107, 108, 109 This pH shift decreases the passive absorption of many drugs that would otherwise accumulate at a greater concentration within the cell. Active drug efflux is also fueled by glycolytic ATP production and HIF-1α-induced transporter overexpression resulting in a significant decrease in the cytoplasmic retention of many anticancer agents.110, 111
HIF-1α overexpression also induces cellular compensations that can bypass the mechanisms on which common drugs rely. Inhibitors of EGFR family signaling may demonstrate reduced effect under high HIF-1α expression because of an upregulation of c-MET, which allows alternative signaling networks to produce similar phenotypic effects in the presence of reduced EGFR family signaling.112, 113 In addition, HIF-1α induces a shift in β-tubulin isoform expression, undermining the effect of microtubule destabilizing agents.114, 115 Downregulation of other drug targets, such as topoisomerase II or estrogen receptor α (ERα), can occur when HIF-1α expression is high and reduces the effect of drugs such as tamoxifen and etoposide.116, 117, 118
Finally, HIF-1α induces expression of genes that promote survival through anti-apoptotic signaling (survivin, Bcl-XL, Mcl-1) or other survival mechanisms such as autophagy (BNIP3, BNIP3L).119, 120, 121, 122, 123 HIF-1α expression also decreases pro-apoptotic signaling by inducing the expression of decoy receptors (such as DcR2) that compete for pro-apoptotic signaling factors like tumor necrosis factor-related apoptosis-inducing ligand, thereby decreasing productive signaling through apoptosis inducing receptors including DR4 and DR5.124, 125 Attenuation of pro-apoptotic signaling allows cells to tolerate a higher level of chemotherapeutic insult before inducing cellular death pathways. HIF-1α signaling works with glycolytic metabolism to trigger a variety of anti-drug mechanisms that generate in vitro and clinical resistance (Figure 1). We have provided examples of how disrupting the cancer metabolism can short circuit the feedback loops that provide protection from anticancer agents.
Targeting key metabolic enzymes enhances therapeutic efficacy or combats drug resistance by promoting drug-induced apoptosis of cancer cells. ATP depletion by glycolytic inhibitors promotes intracellular drug accumulation, leading to increased drug sensitization. However, the molecular mechanisms by which targeting metabolism could impair chemoresistance is not fully understood and deserves further investigation. Combining chemotherapeutic agents with targeted disruption of dysregulated cellular metabolism represents a promising strategy to overcome drug resistance and improve the efficacy of current chemotherapeutic agents in cancer patients. Although therapeutic resistance can arise by multiple mechanisms, the examples listed above demonstrate that targeting a common feature across multiple types of cancer – dysregulated metabolism – can result in reduction of chemoresistance in a wide array of cancer types. Further investigation into the workings of cancer metabolism and resistance will help us to design more selective metabolic inhibitors allowing for a wide array of options and a more individually tailored response to chemoresistance.
Abbreviations
- ABC:
-
ATP-binding cassette
- AFPGC:
-
AFP-producing gastric cancer
- ALL:
-
acute lymphoblastic leukemia
- BPTES:
-
bis-2-[5-phenylacetamido- 1,2,4-thiadiazol-2-yl] ethyl sulfide
- 3-BrPA:
-
3-bromopyruvate
- DCA:
-
dichloroacetate
- 2-DG:
-
2-deoxyglucose
- ER:
-
endoplasmic reticulum
- FASN:
-
fatty acid synthase
- 5-FU:
-
5-fluorouracil
- GDH:
-
glutamate dehydrogenase
- GLS:
-
glutaminase
- GLUTs:
-
glucose transporters
- HK:
-
hexokinase
- HSF1:
-
heat shock factor 1
- α-KG:
-
α-ketoglutarate
- LDHA:
-
lactate dehydrogenase A
- LND:
-
lonidamine
- MM:
-
multiple myeloma
- mTORC1:
-
mammalian target of rapamycin complex 1
- PDH:
-
pyruvate dehydrogenase
- PDK:
-
pyruvate dehydrogenase kinase
- PKM2:
-
pyruvate kinase M2
References
Vander Heiden MG, Cantley LC, Thompson CB . Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324: 1029–1033.
Warburg O . On respiratory impairment in cancer cells. Science 1956; 124: 269–270.
Kim JW, Dang CV . Cancer's molecular sweet tooth and the Warburg effect. Cancer Res 2006; 66: 8927–8930.
Chen Z, Lu W, Garcia-Prieto C, Huang P . The Warburg effect and its cancer therapeutic implications. J Bioenerget Biomembranes 2007; 39: 267–274.
Gatenby RA, Gillies RJ . Glycolysis in cancer: a potential target for therapy. Int J Biochem Cell Biol 2007; 39: 1358–1366.
Kroemer G, Pouyssegur J . Tumor cell metabolism: cancer's Achilles' heel. Cancer Cell 2008; 13: 472–482.
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB . The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metabol 2008; 7: 11–20.
Gillies RJ, Robey I, Gatenby RA . Causes and consequences of increased glucose metabolism of cancers. J Nucl Med 2008; 49 (Suppl 2): 24S–42S.
Hsu PP, Sabatini DM . Cancer cell metabolism: Warburg and beyond. Cell 2008; 134: 703–707.
Pandey PR, Liu W, Xing F, Fukuda K, Watabe K . Anti-cancer drugs targeting fatty acid synthase (FAS). Recent Patents Anti-Cancer Drug Discovery 2012; 7: 185–197.
Erickson JW, Cerione RA . Glutaminase: a hot spot for regulation of cancer cell metabolism? Oncotarget 2010; 1: 734–740.
Wise DR, Thompson CB . Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci 2010; 35: 427–433.
Dang CV . Links between metabolism and cancer. Genes Dev 2012; 26: 877–890.
Munoz-Pinedo C, El Mjiyad N, Ricci JE . Cancer metabolism: current perspectives and future directions. Cell Death Disease 2012; 3: e248.
Birsoy K, Sabatini DM, Possemato R . Untuning the tumor metabolic machine: targeting cancer metabolism: a bedside lesson. Nat Med 2012; 18: 1022–1023.
Dang CV, Hamaker M, Sun P, Le A, Gao P . Therapeutic targeting of cancer cell metabolism. J Mol Med (Berlin, Germany) 2011; 89: 205–212.
El Mjiyad N, Caro-Maldonado A, Ramirez-Peinado S, Munoz-Pinedo C . Sugar-free approaches to cancer cell killing. Oncogene 2010; 30: 253–264.
Hamanaka RB, Chandel NS . Targeting glucose metabolism for cancer therapy. J Exp Med 2012; 209: 211–215.
Jones NP, Schulze A . Targeting cancer metabolism--aiming at a tumour’s sweet-spot. Drug Discovery Today 2012; 17: 232–241.
Tennant DA, Duran RV, Gottlieb E . Targeting metabolic transformation for cancer therapy. Nat Rev 2010; 10: 267–277.
Vander Heiden MG . Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov 2011; 10: 671–684.
Liu H, Liu Y, Zhang JT . A new mechanism of drug resistance in breast cancer cells: fatty acid synthase overexpression-mediated palmitate overproduction. Mol Cancer Therapeutics 2008; 7: 263–270.
Zhao Y, Liu H, Liu Z, Ding Y, Ledoux SP, Wilson GL et al. Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res 2011; 71: 4585–4597.
Zhou M, Zhao Y, Ding Y, Liu H, Liu Z, Fodstad O et al. Warburg effect in chemosensitivity: targeting lactate dehydrogenase-A re-sensitizes taxol-resistant cancer cells to taxol. Mol Cancer 2010; 9: 33.
Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010; 18: 207–219.
Macheda ML, Rogers S, Best JD . Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol 2005; 202: 654–662.
Cao X, Fang L, Gibbs S, Huang Y, Dai Z, Wen P et al. Glucose uptake inhibitor sensitizes cancer cells to daunorubicin and overcomes drug resistance in hypoxia. Cancer Chemotherapy Pharmacol 2007; 59: 495–505.
McBrayer SK, Cheng JC, Singhal S, Krett NL, Rosen ST, Shanmugam M . Multiple myeloma exhibits novel dependence on GLUT4, GLUT8, and GLUT11: implications for glucose transporter-directed therapy. Blood 2012; 119: 4686–4697.
Liu Y, Cao Y, Zhang W, Bergmeier S, Qian Y, Akbar H et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol Cancer Therapeutics 2012; 11: 1672–1682.
Monti E, Gariboldi MB . HIF-1 as a target for cancer chemotherapy, chemosensitization and chemoprevention. Curr Mol Pharmacol 2011; 4: 62–77.
Le Calve B, Rynkowski M, Le Mercier M, Bruyere C, Lonez C, Gras T et al. Long-term in vitro treatment of human glioblastoma cells with temozolomide increases resistance in vivo through up-regulation of GLUT transporter and aldo-keto reductase enzyme AKR1C expression. Neoplasia (New York), NY 2010; 12: 727–739.
Maher JC, Krishan A, Lampidis TJ . Greater cell cycle inhibition and cytotoxicity induced by 2-deoxy-D-glucose in tumor cells treated under hypoxic vs aerobic conditions. Cancer Chemother Pharmacol 2004; 53: 116–122.
Pelicano H, Martin DS, Xu RH, Huang P . Glycolysis inhibition for anticancer treatment. Oncogene 2006; 25: 4633–4646.
Kurtoglu M, Gao N, Shang J, Maher JC, Lehrman MA, Wangpaichitr M et al. Under normoxia, 2-deoxy-D-glucose elicits cell death in select tumor types not by inhibition of glycolysis but by interfering with N-linked glycosylation. Mol Cancer Therapeutics 2007; 6: 3049–3058.
Maschek G, Savaraj N, Priebe W, Braunschweiger P, Hamilton K, Tidmarsh GF et al. 2-deoxy-D-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Res 2004; 64: 31–34.
Dwarakanath B, Jain V . Targeting glucose metabolism with 2-deoxy-D-glucose for improving cancer therapy. Fut Oncol (London, England) 2009; 5: 581–585.
Youle RJ, Strasser A . The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008; 9: 47–59.
Coloff JL, Macintyre AN, Nichols AG, Liu T, Gallo CA, Plas DR et al. Akt-dependent glucose metabolism promotes Mcl-1 synthesis to maintain cell survival and resistance to Bcl-2 inhibition. Cancer Res 2011; 71: 5204–5213.
Meynet O, Beneteau M, Jacquin MA, Pradelli LA, Cornille A, Carles M et al. Glycolysis inhibition targets Mcl-1 to restore sensitivity of lymphoma cells to ABT-737-induced apoptosis. Leukemia 2012; 26: 1145–1147.
Yamaguchi R, Janssen E, Perkins G, Ellisman M, Kitada S, Reed JC . Efficient elimination of cancer cells by deoxyglucose-ABT-263/737 combination therapy. PloS one 2011; 6: e24102.
Pradelli LA, Beneteau M, Chauvin C, Jacquin MA, Marchetti S, Munoz-Pinedo C et al. Glycolysis inhibition sensitizes tumor cells to death receptors-induced apoptosis by AMP kinase activation leading to Mcl-1 block in translation. Oncogene 2010; 29: 1641–1652.
Esteva FJ, Valero V, Booser D, Guerra LT, Murray JL, Pusztai L et al. Phase II study of weekly docetaxel and trastuzumab for patients with HER-2-overexpressing metastatic breast cancer. J Clin Oncol 2002; 20: 1800–1808.
Lan KH, Lu CH, Yu D . Mechanisms of trastuzumab resistance and their clinical implications. Ann N Y Acad Sci 2005; 1059: 70–75.
Hudis CA . Trastuzumab--mechanism of action and use in clinical practice. N Engl J Med 2007; 357: 39–51.
Zhang H, Berezov A, Wang Q, Zhang G, Drebin J, Murali R et al. ErbB receptors: from oncogenes to targeted cancer therapies. J Clin Invest 2007; 117: 2051–2058.
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001; 344: 783–792.
Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 2004; 6: 117–127.
Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ . Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Practice 2006; 3: 269–280.
Zhao YH, Zhou M, Liu H, Ding Y, Khong HT, Yu D et al. Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene 2009; 28: 3689–3701.
Geschwind JF, Georgiades CS, Ko YH, Pedersen PL . Recently elucidated energy catabolism pathways provide opportunities for novel treatments in hepatocellular carcinoma. Expert Rev Anticancer Therapy 2004; 4: 449–457.
Ko YH, Pedersen PL, Geschwind JF . Glucose catabolism in the rabbit VX2 tumor model for liver cancer: characterization and targeting hexokinase. Cancer Lett 2001; 173: 83–91.
Nakano A, Tsuji D, Miki H, Cui Q, El Sayed SM, Ikegame A et al. Glycolysis inhibition inactivates ABC transporters to restore drug sensitivity in malignant cells. PloS One 2011; 6: e27222.
Zhou Y, Tozzi F, Chen J, Fan F, Xia L, Wang J et al. Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells. Cancer Res 2012; 72: 304–314.
Hulleman E, Kazemier KM, Holleman A, VanderWeele DJ, Rudin CM, Broekhuis MJ et al. Inhibition of glycolysis modulates prednisolone resistance in acute lymphoblastic leukemia cells. Blood 2009; 113: 2014–2021.
Zhao Y, Liu H, Riker AI, Fodstad O, Ledoux SP, Wilson GL et al. Emerging metabolic targets in cancer therapy. Front Biosci 2011; 16: 1844–1860.
Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K et al. Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal 2009; 2: ra73.
Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008; 452: 230–233.
Li SL, Ye F, Cai WJ, Hu HD, Hu P, Ren H et al. Quantitative proteome analysis of multidrug resistance in human ovarian cancer cell line. J Cell Biochem 2010; 109: 625–633.
Martinez-Balibrea E, Plasencia C, Gines A, Martinez-Cardus A, Musulen E, Aguilera R et al. A proteomic approach links decreased pyruvate kinase M2 expression to oxaliplatin resistance in patients with colorectal cancer and in human cell lines. Molr Cancer Therapeutics 2009; 8: 771–778.
Yoo BC, Ku JL, Hong SH, Shin YK, Park SY, Kim HK et al. Decreased pyruvate kinase M2 activity linked to cisplatin resistance in human gastric carcinoma cell lines. Int J Cancer 2004; 108: 532–539.
Shin YK, Yoo BC, Hong YS, Chang HJ, Jung KH, Jeong SY et al. Upregulation of glycolytic enzymes in proteins secreted from human colon cancer cells with 5-fluorouracil resistance. Electrophoresis 2009; 30: 2182–2192.
Guo W, Zhang Y, Chen T, Wang Y, Xue J, Zhang Y et al. Efficacy of RNAi targeting of pyruvate kinase M2 combined with cisplatin in a lung cancer model. J Cancer Res Clin Oncol 2011; 137: 65–72.
Shi HS, Li D, Zhang J, Wang YS, Yang L, Zhang HL et al. Silencing of pkm2 increases the efficacy of docetaxel in human lung cancer xenografts in mice. Cancer Sci 2010; 101: 1447–1453.
Fantin VR, St-Pierre J, Leder P . Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 2006; 9: 425–434.
Xie H, Valera VA, Merino MJ, Amato AM, Signoretti S, Linehan WM et al. LDH-A inhibition, a therapeutic strategy for treatment of hereditary leiomyomatosis and renal cell cancer. Mol Cancer Therapeutics 2009; 8: 626–635.
Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM et al. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci USA 2010; 107: 2037–2042.
Khaleque MA, Bharti A, Gong J, Gray PJ, Sachdev V, Ciocca DR et al. Heat shock factor 1 represses estrogen-dependent transcription through association with MTA1. Oncogene 2008; 27: 1886–1893.
Khaleque MA, Bharti A, Sawyer D, Gong J, Benjamin IJ, Stevenson MA et al. Induction of heat shock proteins by heregulin beta1 leads to protection from apoptosis and anchorage-independent growth. Oncogene 2005; 24: 6564–6573.
Min JN, Huang L, Zimonjic DB, Moskophidis D, Mivechi NF . Selective suppression of lymphomas by functional loss of Hsf1 in a p53-deficient mouse model for spontaneous tumors. Oncogene 2007; 26: 5086–5097.
Dai C, Whitesell L, Rogers AB, Lindquist S . Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 2007; 130: 1005–1018.
Lu CW, Lin SC, Chen KF, Lai YY, Tsai SJ . Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem 2008; 283: 28106–28114.
Lu CW, Lin SC, Chien CW, Lin SC, Lee CT, Lin BW et al. Overexpression of pyruvate dehydrogenase kinase 3 increases drug resistance and early recurrence in colon cancer. Am J Pathol 2011; 179: 1405–1414.
Kato M, Li J, Chuang JL, Chuang DT . Distinct structural mechanisms for inhibition of pyruvate dehydrogenase kinase isoforms by AZD7545, dichloroacetate, and radicicol. Structure 2007; 15: 992–1004.
Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007; 11: 37–51.
Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S et al. Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate 2008; 68: 1223–1231.
Michelakis ED, Webster L, Mackey JR . Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 2008; 99: 989–994.
Sun RC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC . Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat 2010; 120: 253–260.
Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I . Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecologic Oncol 2008; 109: 394–402.
Ishiguro T, Ishiguro M, Ishiguro R, Iwai S . Cotreatment with dichloroacetate and omeprazole exhibits a synergistic antiproliferative effect on malignant tumors. Oncol Lett 2012; 3: 726–728.
Ishiguro T, Ishiguro R, Ishiguro M, Iwai S . Co-treatment of dichloroacetate, omeprazole and tamoxifen exhibited synergistically antiproliferative effect on malignant tumors: in vivo experiments and a case report. Hepato-gastroenterology 2012; 59: 994–996.
Tong J, Xie G, He J, Li J, Pan F, Liang H . Synergistic antitumor effect of dichloroacetate in combination with 5-fluorouracil in colorectal cancer. J Biomed Biotechnol 2011; 2011: 740564.
Ayyanathan K, Kesaraju S, Dawson-Scully K, Weissbach H . Combination of sulindac and dichloroacetate kills cancer cells via oxidative damage. PloS One 2012; 7: e39949.
Flavin R, Peluso S, Nguyen PL, Loda M . Fatty acid synthase as a potential therapeutic target in cancer. Fut Oncol (London, England) 2010; 6: 551–562.
Menendez JA, Lupu R, Colomer R . Inhibition of tumor-associated fatty acid synthase hyperactivity induces synergistic chemosensitization of HER -2/ neu -overexpressing human breast cancer cells to docetaxel (taxotere). Breast Cancer Res Treatment 2004; 84: 183–195.
Vazquez-Martin A, Colomer R, Brunet J, Menendez JA . Pharmacological blockade of fatty acid synthase (FASN) reverses acquired autoresistance to trastuzumab (Herceptin by transcriptionally inhibiting 'HER2 super-expression' occurring in high-dose trastuzumab-conditioned SKBR3/Tzb100 breast cancer cells. Int J Oncol 2007; 31: 769–776.
Yang Y, Liu H, Li Z, Zhao Z, Yip-Schneider M, Fan Q et al. Role of fatty acid synthase in gemcitabine and radiation resistance of pancreatic cancers. Int J Biochem Mol Biol 2011; 2: 89–98.
Thupari JN, Pinn ML, Kuhajda FP . Fatty acid synthase inhibition in human breast cancer cells leads to malonyl-CoA-induced inhibition of fatty acid oxidation and cytotoxicity. Biochem Biophys Res Commun 2001; 285: 217–223.
Li JN, Gorospe M, Chrest FJ, Kumaravel TS, Evans MK, Han WF et al. Pharmacological inhibition of fatty acid synthase activity produces both cytostatic and cytotoxic effects modulated by p53. Cancer Res 2001; 61: 1493–1499.
Little JL, Wheeler FB, Fels DR, Koumenis C, Kridel SJ . Inhibition of fatty acid synthase induces endoplasmic reticulum stress in tumor cells. Cancer Res 2007; 67: 1262–1269.
Pizer ES, Chrest FJ, DiGiuseppe JA, Han WF . Pharmacological inhibitors of mammalian fatty acid synthase suppress DNA replication and induce apoptosis in tumor cell lines. Cancer Res 1998; 58: 4611–4615.
Menendez JA, Vellon L, Mehmi I, Oza BP, Ropero S, Colomer R et al. Inhibition of fatty acid synthase (FAS) suppresses HER2/neu (erbB-2) oncogene overexpression in cancer cells. Proc Natl Acad Sci USA 2004; 101: 10715–10720.
Menendez JA, Vellon L, Lupu R . Targeting fatty acid synthase-driven lipid rafts: a novel strategy to overcome trastuzumab resistance in breast cancer cells. Med Hypotheses 2005; 64: 997–1001.
Vazquez-Martin A, Ropero S, Brunet J, Colomer R, Menendez JA . Inhibition of fatty acid synthase (FASN) synergistically enhances the efficacy of 5-fluorouracil in breast carcinoma cells. Oncol Rep 2007; 18: 973–980.
Knowles LM, Axelrod F, Browne CD, Smith JW . A fatty acid synthase blockade induces tumor cell-cycle arrest by down-regulating Skp2. J Biol Chem 2004; 279: 30540–30545.
Browne CD, Hindmarsh EJ, Smith JW . Inhibition of endothelial cell proliferation and angiogenesis by orlistat, a fatty acid synthase inhibitor. Faseb J 2006; 20: 2027–2035.
Knowles LM, Yang C, Osterman A, Smith JW . Inhibition of fatty-acid synthase induces caspase-8-mediated tumor cell apoptosis by up-regulating DDIT4. J Biol Chem 2008; 283: 31378–31384.
Duran RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E et al. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell 2012; 47: 349–358.
Deberardinis RJ, Sayed N, Ditsworth D, Thompson CB . Brick by brick: metabolism and tumor cell growth. Curr Opin Genet Dev 2008; 18: 54–61.
DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S et al. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA 2007; 104: 19345–19350.
Kamata S, Kishimoto T, Kobayashi S, Miyazaki M, Ishikura H . Possible involvement of persistent activity of the mammalian target of rapamycin pathway in the cisplatin resistance of AFP-producing gastric cancer cells. Cancer Biol Ther 2007; 6: 1036–1043.
Robinson MM, McBryant SJ, Tsukamoto T, Rojas C, Ferraris DV, Hamilton SK et al. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem J 2007; 406: 407–414.
Seltzer MJ, Bennett BD, Joshi AD, Gao P, Thomas AG, Ferraris DV et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res 2010; 70: 8981–8987.
Chiarini F, Grimaldi C, Ricci F, Tazzari PL, Evangelisti C, Ognibene A et al. Activity of the novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 against T-cell acute lymphoblastic leukemia. Cancer Res 2010; 70: 8097–8107.
Gatenby RA, Gillies RJ . Why do cancers have high aerobic glycolysis? Nat Rev 2004; 4: 891–899.
Denko NC . Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev 2008; 8: 705–713.
Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 2008; 283: 10892–10903.
Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med 2004; 10: 858–864.
Greijer AE, de Jong MC, Scheffer GL, Shvarts A, van Diest PJ, van der Wall E . Hypoxia-induced acidification causes mitoxantrone resistance not mediated by drug transporters in human breast cancer cells. Cell Oncol 2005; 27: 43–49.
Wykoff CC, Beasley NJ, Watson PH, Turner KJ, Pastorek J, Sibtain A et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res 2000; 60: 7075–7083.
Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP . Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res 2002; 62: 3387–3394.
Krishnamurthy P, Ross DD, Nakanishi T, Bailey-Dell K, Zhou S, Mercer KE et al. The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J Biol Chem 2004; 279: 24218–24225.
Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316: 1039–1043.
Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM . Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003; 3: 347–361.
Raspaglio G, Filippetti F, Prislei S, Penci R, De Maria I, Cicchillitti L et al. Hypoxia induces class III beta-tubulin gene expression by HIF-1alpha binding to its 3' flanking region. Gene 2008; 409: 100–108.
Zeng L, Kizaka-Kondoh S, Itasaka S, Xie X, Inoue M, Tanimoto K et al. Hypoxia inducible factor-1 influences sensitivity to paclitaxel of human lung cancer cell lines under normoxic conditions. Cancer Sci 2007; 98: 1394–1401.
Kronblad A, Hedenfalk I, Nilsson E, Pahlman S, Landberg G . ERK1/2 inhibition increases antiestrogen treatment efficacy by interfering with hypoxia-induced downregulation of ERalpha: a combination therapy potentially targeting hypoxic and dormant tumor cells. Oncogene 2005; 24: 6835–6841.
Sullivan R, Graham CH . Hypoxia prevents etoposide-induced DNA damage in cancer cells through a mechanism involving hypoxia-inducible factor 1. Mol Cancer Therapeutics 2009; 8: 1702–1713.
Wen W, Ding J, Sun W, Wu K, Ning B, Gong W et al. Suppression of cyclin D1 by hypoxia-inducible factor-1 via direct mechanism inhibits the proliferation and 5-fluorouracil-induced apoptosis of A549 cells. Cancer Res 2010; 70: 2010–2019.
Altieri DC . Survivin, cancer networks and pathway-directed drug discovery. Nat Rev 2008; 8: 61–70.
Chen N, Chen X, Huang R, Zeng H, Gong J, Meng W et al. BCL-xL is a target gene regulated by hypoxia-inducible factor-1{alpha}. J Biol Chem 2009; 284: 10004–10012.
Erler JT, Cawthorne CJ, Williams KJ, Koritzinsky M, Wouters BG, Wilson C et al. Hypoxia-mediated down-regulation of Bid and Bax in tumors occurs via hypoxia-inducible factor 1-dependent and -independent mechanisms and contributes to drug resistance. Mol Cell Biol 2004; 24: 2875–2889.
Flamant L, Notte A, Ninane N, Raes M, Michiels C . Anti-apoptotic role of HIF-1 and AP-1 in paclitaxel exposed breast cancer cells under hypoxia. Mol Cancer 2010; 9: 191.
Piret JP, Minet E, Cosse JP, Ninane N, Debacq C, Raes M et al. Hypoxia-inducible factor-1-dependent overexpression of myeloid cell factor-1 protects hypoxic cells against tert-butyl hydroperoxide-induced apoptosis. J Biol Chem 2005; 280: 9336–9344.
Mayes PA, Campbell L, Ricci MS, Plastaras JP, Dicker DT, El-Deiry WS . Modulation of TRAIL-induced tumor cell apoptosis in a hypoxic environment. Cancer Biol Ther 2005; 4: 1068–1074.
Pei GT, Wu CW, Lin WW . Hypoxia-induced decoy receptor 2 gene expression is regulated via a hypoxia-inducible factor 1alpha-mediated mechanism. Biochem Biophys Res Commun 2010; 391: 1274–1279.
Acknowledgements
We are grateful to the support from the Vincent F Kilborn, Jr Cancer Research Foundation (M Tan), NIH Grant RO1-CA149646 (M Tan), Radiumhospitalets Legater Award Project 334003 (M Tan) and NFSC Project 81272907 (Y Zhao).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by C Munoz-Pinedo
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Zhao, Y., Butler, E. & Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis 4, e532 (2013). https://doi.org/10.1038/cddis.2013.60
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2013.60
Keywords
This article is cited by
-
Histone acetylation: a key determinant of acquired cisplatin resistance in cancer
Clinical Epigenetics (2024)
-
Association between glycolysis markers and prognosis of liver cancer: a systematic review and meta-analysis
World Journal of Surgical Oncology (2023)
-
Targeting IGF1R signaling enhances the sensitivity of cisplatin by inhibiting proline and arginine metabolism in oesophageal squamous cell carcinoma under hypoxia
Journal of Experimental & Clinical Cancer Research (2023)
-
A multimodal atlas of tumour metabolism reveals the architecture of gene–metabolite covariation
Nature Metabolism (2023)
-
Nogo-B receptor increases glycolysis and the paclitaxel resistance of estrogen receptor-positive breast cancer via the HIF-1α-dependent pathway
Cancer Gene Therapy (2023)
