
Abstract
Background:
We examined the potential of metformin (MET) to enhance non-small cell lung cancer (NSCLC) responses to ionising radiation (IR).
Methods:
Human NSCLC cells, mouse embryonic fibroblasts from wild-type and AMP-activated kinase (AMPK) α1/2-subunit−/− embryos (AMPKα1/2−/−-MEFs) and NSCLC tumours grafted into Balb/c-nude mice were treated with IR and MET and subjected to proliferation, clonogenic, immunoblotting, cell cycle and apoptosis assays and immunohistochemistry (IHC).
Results:
Metformin (2.5 μ M–5 mM) inhibited proliferation and radio-sensitised NSCLC cells. Metformin (i) activated the ataxia telengiectasia-mutated (ATM)–AMPK–p53/p21cip1 and inhibited the Akt–mammalian target of rapamycin (mTOR)–eIF4E-binding protein 1 (4EBP1) pathways, (ii) induced G1 cycle arrest and (iii) enhanced apoptosis. ATM inhibition blocked MET and IR activation of AMPK. Non-small cell lung cancer cells with inhibited AMPK and AMPKα1/2−/−-MEFs were resistant to the antiproliferative effects of MET and IR. Metformin or IR inhibited xenograft growth and combined treatment enhanced it further than each treatment alone. Ionising radiation and MET induced (i) sustained activation of ATM–AMPK–p53/p21cip1 and inhibition of Akt–mTOR–4EBP1 pathways in tumours, (ii) reduced expression of angiogenesis and (iii) enhanced expression of apoptosis markers.
Conclusion:
Clinically achievable MET doses inhibit NSCLC cell and tumour growth and sensitise them to IR. Metformin and IR mediate their action through an ATM–AMPK-dependent pathway. Our results suggest that MET can be a clinically useful adjunct to radiotherapy in NSCLC.
Similar content being viewed by others


Main
Lung cancer (LC) is a leading cause of cancer mortality. More than 80% of LC patients are affected by non-small cell LC. Despite high-dose radiotherapy in combination with chemotherapy, NSCLC shows poor local response (Curran, 2003), indicating a need for effective and well-tolerated adjuncts to radiotherapy.
Ionising radiation (IR) activates molecular pathways of tumour radioresistance, such as the protein kinase B (Akt) and mammalian target of rapamycin (mTOR) pathway (Bussink et al, 2008). These are effectors of tyrosine kinase receptors like EGF receptor (EGFR), which activates them through phosphatidylinositol 3-kinase (PI3k) and phosphoinositide-dependent kinase 1, an Akt-T308 kinase (Brachmann et al, 2009). Akt activates mTOR through (i) phosphorylation and inhibition of tuberous sclerosis complex 2 (TSC2), which inactivates the mTOR-activating GTP-binding protein Rheb, and/or (ii) phosphorylation of PRAS40 a member of mTORC1, one of the two functional complexes of mTOR, which includes mLST8/Gbl and the scaffold protein Raptor (Laplante and Sabatini, 2009). Mammalian target of rapamycin stimulates growth of cellular biomass, proliferation and resistance to cytotoxic agents (Petroulakis et al, 2006). It promotes gene expression and translation through phosphorylation-mediated activation of p70S6-kinase and phosphorylation-mediated inhibition of translation initiation inhibitor eIF4E-binding protein 1 (4EBP1; Petroulakis et al, 2006; Laplante and Sabatini, 2009).
Earlier, we showed that IR activates the energy sensor AMP-activated kinase (AMPK) pathway, a key, evolutionally preserved, kinase that mediates a metabolic cell cycle checkpoint (Sanli et al, 2010). AMPK is an effector of liver kinase B 1 (LKB1), a tumour suppressor mutated in Peutz-Jeghers syndrome, which is associated with breast and LC. AMP-activated kinase is a heterotrimeric enzyme of α-, β- and γ-subunits that senses low energy levels through AMP binding on the γ-subunit and is regulated by phosphorylation of the α-subunit Thr172 (Steinberg and Kemp, 2009). AMP-activated kinase inhibits anabolic processes and protein synthesis by inhibiting mTORC1 through (i) Ser1387 phosphorylation and activation of TSC2, leading to enhanced Rheb GTPase activity and mTOR inhibition, or (ii) Raptor phosphorylation. AMP-activated kinase induces p53 and cyclin-dependent kinase inhibitors p21cip1 and p27kip1, leading to cycle arrest (Gwinn et al, 2008; Mihaylova and Shaw, 2011). Our work suggested that AMPK is also a sensor of genomic stress. We suggested that IR activates AMPK acutely in epithelial cancer cell cultures to transduce signals through a DNA damage response (DDR)-mediated ataxia telengiectasia-mutated (ATM)-AMPK-p53-p21cip1 axis, and mediate cell cycle arrest and radiosensitisation (Sanli et al, 2010).
Metformin (1-(diaminomethylidene)-3,3-dimethylguanidine) is a well-tolerated antidiabetic agent used by >120 million patients worldwide (Lord et al, 2003). Retrospective studies suggested that MET may decrease the risk of cancer in patients with type 2 diabetes (Evans et al, 2005; Bowker et al, 2006). Dowling et al (2007) showed that MET activates AMPK and inhibits mTOR, leading to inhibition of breast cancer cell proliferation (Dowling et al, 2007). This was prevented in cells lacking TSC2, LKB1 and cells exposed to the AMPK inhibitor compound C, indicating that LKB1, AMPK and TSC2 are involved in the mechanism of MET action. Huang et al (2008) showed activation of the LKB1–AMPK pathway by MET and delay in tumour development in PTEN-deficient mice (Huang et al, 2008). Reports suggest that MET can amplify chemotherapy-induced AMPK activation and enhances the cytotoxicity of chemotherapy in breast, lung and prostate cancer models (Iliopoulos et al, 2011; Rocha et al, 2011). Retrospective analysis suggested that MET may enhance chemotherapy responses in women with breast cancer (Jiralerspong et al, 2009). Currently, a randomised phase-III trial examines the potential of MET to reduce disease recurrence and improve survival in early-stage breast cancer patients after curative therapy (Goodwin et al, 2011).
In LC cells, we observed that MET enhances IR-induced activation of AMPK and clonogenic death (Sanli et al, 2010). Metformin is being investigated in a number of cancer sites (Goodwin et al, 2009; Goodwin et al, 2011; Taubes, 2012), but has not been examined in LC in combination with radiotherapy. Investigators remain concerned that most in vitro studies demonstrate antitumour activity at millimolar concentrations of MET, which are not clinically achievable (Dowling et al, 2012). The purpose of this study was to: (i) investigate the antiproliferative effects of clinically achievable low-dose MET in NSCLC models and to analyse the molecular pathways mediating its action and (ii) to examine whether MET can sensitise human NSCLC cells and tumours to IR.
Materials and methods
Antibodies and chemicals
Anti-CD31 antibody was purchased from Abcam (Cambridge, MA, USA) and all other antibodies were purchased from Cell Signalling Technology (Danver, MA, USA). Metformin HCL, rapamycin were obtained from Sigma-Aldrich Canada (Oakville, ON, Canada). The ATM inhibitor KU60019 was obtained from Cedarlane Labs (Burlington, ON, Canada), and validated ATM and AMPKα1/2 siRNA were obtained from Qiagen Canada (Toronto, ON, Canada).
Cell culture
Human lung adenocarcinoma A549, H1299 and squamous cell carcinoma SK-MES1 cells were purchased from ATCC (Philadelphia, PA, USA) and cultured in RPMI 1640 media (10% v/v foetal bovine serum (FBS); A549 and H1299) or DMEM media (5 mM glucose, 10% FBS; SK-MES1). Wild-type (WT)-MEFs and AMPKα1/2−/−-MEFs cells were kindly provided by Dr B Viollet, (INSERM, France).
Incubations
After overnight incubations, cells were treated with indicated doses of MET or rapamycin for 24 h before treatment with the indicated doses of IR. In KU60019 and siRNA experiments, cells were preincubated with those agents or vehicles 24 h and 72 h, respectively, before incubation with or without MET for 48 h and/or treatment with IR 24 h later.
Proliferation assay
Cells were seeded (1 × 103 cells per well) in 96-well plates in triplicates, were allowed to adhere overnight and subjected to treatments accordingly. After treatment, cells were washed with phosphate-buffered saline (PBS), fixed with methanol and stained with crystal violet dye. DNA content was quantified with BioTek Plate reader spectrophotometry software (BioTek, Winooski, VT, USA) at 570 nm.
Clonogenic assays
Cells (500–1000) were seeded into 6-well plates in triplicate and maintained at the indicated doses of MET before IR (0, 2 or 8 Gy). After 7 days cells were fixed with methylene blue and viable colonies (>50 cells) were counted. Data were fitted to the linear quadratic model using Graphpad Prism-5 software (GraphPad Prism Software Inc., La Jolla, CA, USA), as previously described (Sanli et al, 2011).
Cell cycle analysis
Cells were seeded at 1 × 106 cells per ml and left to incubate for 48 h. Cells were treated with MET and IR for 24 h and 48 h, respectively, were then washed in PBS, trypsinised, centrifuged at 1500 r.p.m., resuspended in 1 ml of 95% ethanol and stored at −20 °C for 24 h. Then, they were centrifuged and resuspended in 1 ml of propidium iodide staining solution, quantified with a FACS-calibur flow cytometry and the data were analysed using Expo32 ADC v1.1c software, Beckman-Coulter (Mississauga, ON, Canada).
Annexin-V assay
A549 cells (5 × 105) were seeded onto a glass coverslip and treated with MET and IR as indicated. Then cells were rinsed in PBS and stained using an annexin-V-EGFP apoptosis detection kit, as directed (Genscript, Piscataway, NJ, USA). The annexin V-EGFP antibody was visualised under a fluorescent microscope, as described (Mak et al, 2011).
γH2AX foci detection
A549 cells grown on glass coverslips were treated with MET or IR for the indicated times, washed, fixed using 3% paraformaldehyde and blocked in 5% FBS/PBS/0.3%-Triton-X-100. Cells were incubated with anti-γH2AX-monoclonal antibody followed by an anti-mouse-Alexa-568-labelled secondary. γH2AX foci were detected using a fluorescent microscope, as described (Sanli et al, 2012b).
Xenograft model, treatments and tissue handling
Five-week-old Balb/c-nude mice (Charles-River, Mississauga, ON, Canada) were grafted with 1 × 106A549 or H1299 cells by subcutaneous injection into the right flank. When tumour volume reached 100 mm3, animals were divided into: untreated (control), MET alone, IR or combined treatment (MET+IR). Tumour dimensions were measured by calliper every 5 days and volume was estimated as per: V=length × width × height × 0.5236. All experiments were approved by the McMaster University animal ethics board. Metformin treatment was delivered via drinking water at 300 mg kg−1 body weight per day till euthanasia. Water consumption was evaluated daily, MET intake was calculated and concentration in water was adjusted to achieve a daily intake of 300 mg kg−1. Ionising radiation treatments were delivered with a conformal technique (after dosimetric calculations and simulation) using clinical linear accelerators. Xenografts were subjected to either 0 or 10 Gy IR while animals were under gaseous anaesthesia housed inside high-efficiency particulate air filters (Sigma-Aldrich Co, LCC, Oakville, ON, Canada)-equipped Plexiglas tubes.
At euthanasia, extracted tumours were separated in two halves. One half was snap frozen in liquid nitrogen for later homogenisation, protein extraction and analysis by immunoblotting, and the other half was formalin-fixed (10% solution for 24 h) paraffin-embedded for immunohistochemical analysis.
Immunoblotting and densitometry analysis
Twenty micrograms of protein from cell or xenograft lysates were subjected to 8–12% SDS-PAGE gels and transferred onto PVDF membranes, as described (Sanli et al, 2010). Immunoreactive bands were visualised with the ECL method and quantified using the Image J software (National Institute of Health (NIH), Bethesda, MD, USA). Band densities were first normalised for loading against the density of anti-actin immunoblots and then against the densities of untreated controls. For tumour lysates, for each marker-normalised average density values of bands belonging to the six control tumours were used as a master-control, against which all individual marker band densities were normalised.
Immunohistochemistry
Analysis was performed on 4-μm-thick formalin-fixed paraffin-embedded tumour sections as described earlier (Tsakiridis et al, 2008). Tissues were incubated with rabbit anti-P-AMPK (T172; 1 : 200 dilution), anti-CD31 (1 : 100 dilution) or anti-cleaved caspase 3 (CC3) (1 : 800 dilution) antibodies at 4 °C overnight, followed by incubation with Vector anti-rabbit biotinylated secondary antibody, Vector-ABC reagent (Vector Laboratories (Canada) Inc., Burlington, ON, Canada), diaminobenzidine substrate detection, wash, dehydration and mounting.
Statistical analysis
Paired t-test was utilised for statistical analysis of data between treatment groups for all experiments. Further, two-way ANOVA was used to compare treatment groups. Analysis was performed with SPSS software purchased by SPSS Inc. (Chicago, IL, USA), presently owned by IBM (Armonk, NY, USA). Statistical significance was determined as P<0.05. To determine whether the type of interaction between MET and IR was synergistic, additive or antagonistic, we used the combination index (CI) method of Chou and Talalay (1984) and the CompuSyn software (Combo Syn Inc., Paramus, NJ, USA). This method takes into account the potency (median dose or IC50) and the shape of the dose–effect curve (the m value) to calculate the CI(Chou and Talalay, 1984; Chou, 2010). When the CI=1 it indicates an additive effect; a CI<1 indicates synergy and a CI>1 indicates antagonism.
Results
Metformin inhibits growth and sensitises cells to IR
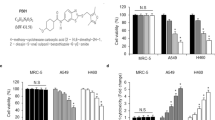
To understand the time course of MET action, we analysed first A549 cells treated for 1–72 h with 5 μ M MET (Figure 1A). We observed evidence of AMPK phosphorylation (T172) within 4 h, which reached high levels by 48–72 h. For that, the majority of subsequent experiments with MET were performed with 48 h incubations, unless otherwise indicated.

Effects of MET and IR on proliferation, clonogenic survival and molecular signals in human LC cells. (A) Time course of MET effects on the pathways of AMPK. Cells were treated with 5 μ M MET doses for 1–72 h, lysed and probed with anti-total and -P-AMPK (T172) and -actin antibodies. Representative immunoblots from three experiments are shown. (B) A549, SK-MES1 and H1299 human LC cells were treated with increasing MET doses (5 μmol–5 mM) for 24 h before treatment with 0, 2 or 8 Gy IR. Cells were fixed 48 h later. Proliferation results (mean±s.e.) of three independent experiments (including six replicates per condition in each experiment) are shown. Statistically significant differences compared with corresponding control cells (not treated with MET) within the 0, 2 and 8 Gy IR treatment groups are shown (*P< 0.05, **P<0.001 for 0 Gy group; #P<0.05, ##P<0.001 for 2 Gy group; xP<0.05 and xxP<0.001 for 8 Gy group respectively). (C) Metformin and IR reduced clonogenic survival of LC cells. A549 and H1299 LC cells were first treated with increasing doses of MET (0–50 μ M) and subsequently with combination of 0, 5 or 25 μ M MET and 0–8 Gy IR dose. Clonogenic survival was determined as outlined in Materials and Methods. Data were fitted into the linear quadratic model. Average results of three independent experiments are shown (* indicates statistical significance at P<0.05). (D) Comparison of MET and rapamycin effects in combination with IR on A549 human LC cell proliferation. Cells were treated with either 5 μ M, 100 μ M or 5 mM MET or 5, 25 and 500 nM rapamycin for 24 h, followed by treatment with either 0, 2 or 8 Gy of IR and incubation for an additional 24 h. Cells were fixed and proliferation rate was determined as outlined in (A). Results of 3–4 independent experiments (mean±s.e.) are shown. Statistically significant differences of MET or rapamycin treatments from no-drug treatment control in the same radiation dose group are shown: *P<0.05; **P<0.001 for 0 Gy group; #P<0.05; ##P<0.001 for 2 Gy group; xP<0.05; xxP<0.001 for 8 Gy treatment group.
A549, H1299 and SK-MES cells were subjected to treatments with MET alone and in combination with IR (2 or 8 Gy; Figure 1B). At 5 μ M, MET reduced significantly proliferation in all cell lines (11±4.2%, 22±3.74% and 26±5.12% inhibition for A549, H1299 and SK-MES1 cells, respectively). Dose-dependent inhibition of proliferation was observed with 5 μ M–5 mM MET (up to 65–70% or 30–35% of control). Metformin inhibited proliferation further when combined with 2 or 8 Gy IR (Figure1B). Exact proliferation values with each MET and IR dose, the combinations and the incremental inhibition of proliferation by MET in irradiated cells is shown in Supplementary Data (Supplementary Table S1). To analyse the type of interaction between MET and IR, we used the Chou and Talalay (1984) method as described in Materials and Methods (Chou and Talay, 1984). We detected CI<1 (0.74–0.1) for all MET and IR combinations, indicating synergy between the two treatments in inhibiting NSCLC cell proliferation (see Supplementary Table S1).
As MET showed similar antiproliferative action in SK-MES and H1299 cells, we pursued the majority of subsequent experiments with A549 or H1299 cells. In clonogenic assays, MET (5 μ M or higher) inhibited colony formation in A549 and H1299 cells (Figure 1C). H1299 cells showed greater sensitivity to MET with 32.0±2.6% inhibition of colony formation at 2.5 μ M MET and 54.4±1.7% inhibition at 5 μ M, compared with 8.7±5.3 and 38.2±8.4% for A549 cells. Next, we assessed the ability of MET (5 and 25 μ M) to radiosensitise. Results were fitted into the linear quadratic model. Metformin (5 and 25 μ M) radiosensitised cells indicated by the steeper slopes of the curves of MET-treated cells. This reached statistical significance (P<0.05) at all IR doses at 25 μ M in both A549 and H1299 cells, and at 5 μ M in A549 cells treated with 8 Gy and H1299 cells treated with either 6 or 8 Gy, compared with control cells treated with the same doses of IR but without MET (Figure 1C).
Comparison of MET with rapamycin
Metformin (5–100 μ M) inhibited proliferation to levels comparable to those achieved by widely used concentrations of rapamycin (5–25 nM; Figure 1D). Although rapamycin inhibited proliferation of non-irradiated cells further than MET, in irradiated cells the inhibition was similar with that of MET (5–100 μ M). A complete dose-response analysis of rapamycin in combination with IR is shown in Supplementary Data (Supplementary Figure S3). Both drugs exhibited parallel dose-dependent inhibition of proliferation in non-irradiated and irradiated cells.
Modulation of AMPK–p53 and mTOR pathways
Ionising radiation stimulated AMPKT172 phosphorylation and activity, marked by phosphorylation of its direct target acetylCoA carboxylase (ACC), in response to 8 Gy IR (Figure 2A and B). Similarly, IR increased total p53 and p21cip1 levels and enhanced p53Ser15 phosphorylation. No significant alterations in total and phosphorylated (T308 and S473) Akt levels or the total levels of mTOR were observed 24 h after IR, but we detected a trend for inhibition of 4EBP1 phosphorylation (by 34.3±14.3%), indicating reduced mTOR activity (Figure 2A–C).

Metformin and IR mediate sustained modulation of molecular tumour growth and suppression pathways. A549 cells were treated with either MET (0, 5 or 100 μmol) for 48 h, IR (0, 8 Gy) for 24 h or combined MET+IR treatments. Cells were washed and lysed. Lysates were analysed with immunoblotting using indicated antibodies. (A) Representative immunoblots are shown. (B and C) Mean±s.e. densitometric quantification values from three independent immunoblotting experiments are shown for markers of the AMPK–p53–p21cip1 and the Akt–mTOR–4EBP1 pathways, respectively. (xP<0.05 between 0 μ M MET treatment groups (0 Gy vs 8 Gy); #P<0.05, ##P<0.001 between 5 μ M MET group (0 Gy vs 8 Gy). *P<0.05 compared to cells not treated with MET in the same IR group, respectively).
Treatment with MET (48 h) and IR (24 h after initiation of MET) phosphorylated AMPKαT172. Metformin stimulated AMPK activity detected as enhanced ACC phosphorylation (at 100 μ M MET). The MET pretreatment potentiated IR induction of total and phosphorylated p53, but did not induce further p21cip1 expression in these conditions (Figure 2A and B). Metformin (5 μ M) did not affect the levels of phosphorylated and total Akt, and showed minor trends for reduced total mTOR and phosphorylated 4EBP1. At 100 μ M MET, we did observe small but significant reductions of Akt-S473 phosphorylation, total mTOR and phosphorylated 4EBP1 levels. However, MET both at 5 μ M and 100 μ M mediated a robust inhibition of 4EPB1 phosphorylation in irradiated cells (Figure 2A and C). We postulate that the enhanced inhibition of 4EBP1 phosphorylation by MET in irradiated cells is the result of a potentiation of signalling events downstream of AMPK as a result of the combined IR and MET treatments.
Regulation of the cell cycle
Untreated A549 cells distributed mainly at the G1 phase (G1: 86.3%, S phase: 10.5%, G2-M: 3.5%; Figure 3A). Metformin shifted cells into S phase (5 μ M: G1: 76.5%, S: 19.7%, G2-M: 3.8% and 100 μ M: G1:77.4%, S: 16.7% and G2-M: 5.83%, respectively). Ionising radiation (8 Gy) induced the expected G2-M checkpoint arrest G2-M: 28.9% vs 3.5% in non-irradiated cells. Combined MET+IR treatment caused G1 shift, and reduced S and G2-M distribution, compared with IR-treated cells (G1: IR: 50.8% vs 5 μ M MET/8 Gy: 65.8% and 100 μ M MET/8 Gy: 66.9% and S: IR: 20.2% vs 5 μ M MET/8 Gy: 12.2% and 100 μ M MET/8 Gy: 14.7%).

Modulation of cell cycle, apoptosis and DDR by MET and IR. (A) Cell cycle regulation by MET and IR. A549 cells were treated with 0, 5 or 100 μ M MET for 24 h before being subjected to either 0 or 8 Gy IR for additional 48 h. Cells were fixed with ethanol and analysed by flow cytometry, as described in Materials and Methods. Representative images are shown. Graph shows results from three independent experiments. (B) Induction of apoptosis by MET and IR. Cells were treated with the indicated concentrations of MET for 48 h and exposure to 0 or 8 Gy IR. Twenty-four hours later, cells were fixed and labelled with an anti-Annexin-V antibody, and visualised under a fluorescent microscope. Representative images from two independent experiments are shown. (C) Response of the DDR pathway to MET treatment. A549 cells were treated with 5 μ M MET for a period of 1–72 h, after which cells were lysed and lysates were probed with indicated antibodies. Representative immunoblots of four independent experiments are shown. (D) Induction of γH2AX foci by MET and IR. Cells were treated with the indicated concentrations of MET 24 h before 4 Gy IR. Following the indicated times after IR, cells were fixed and stained with DAPI (blue) and an antibody against γH2Ax (green) and visualised at × 40. Representative images of multiple fields are shown for each treatment group.
Induction of apoptosis
Forty-eight hours after MET treatment, A549 cells exhibited detectable levels of Annexin-V signal. However, IR (8 Gy) elevated Annexin-V signal, and this increased further in cells treated with combined 5 μ M or 25 μ M of MET and IR, respectively (Figure 3B).
Modulation of DDR signals by IR and MET
Similar to IR (Sanli et al, 2012b), MET treatment alone induced time-dependent increase in ATM levels and its activity detected as phosphorylated histone H2AX (γH2AX) on immunoblotting (Figure 3C). To evaluate the subcellular distribution of this event, we examined γH2AX foci by immunofluorescence microscopy also. Ionising radiation (4 Gy) caused transiently γH2AX foci, an established marker of IR-induced DNA double-strand breaks (DSBs). Foci were reversed within 24 h, indicating DNA repair (Figure 3D). In agreement with immunoblotting, MET alone also induced nuclear γH2AX foci, and combined MET and IR treatment led to sustained γH2AX foci at 24 h, when IR effects were reversed.
Involvement of ATM and AMPK in the mechanism of action of MET and IR
To examine whether the ATM–AMPK pathway is involved in the mechanism of action of MET, we utilised first biochemical and molecular approaches to inhibit ATM. Figure 4A shows that MET (5 μ M) induces, within 48 h, ATM activity (increased γH2Ax), AMPK phosphorylation and expression of p21cip1. However, knocking down of ATM with specific siRNA eliminated cellular ATM levels and inhibited MET-induced activation of ATM, seen as blockade of γH2AX phosphorylation and AMPKα phosphorylation. We inhibited ATM with siRNA and the specific ATM inhibitor KU60019 (Golding et al, 2009) in irradiated cells also. Metformin induced ATM phosphorylation and activity (detected as γH2AX phosphorylation) in control cells, and enhanced further IR-induced ATM phosphorylation and activation in A549 cells (Figure 4B and C). Ataxia telengiectasia-mutated knockdown eliminated total and phosphorylated ATM levels and blocked AMPK phosphorylation by both MET and IR (Figure 4B). Similarly, ATM inhibition with KU60019 blocked ATM activation (lack of γH2Ax) by MET and IR, and inhibited AMPKα phosphorylation by MET in irradiated cells (Figure 4C).

Role of ATM and AMPK in the signalling and antiproliferative effects of MET and IR. (A–C) Ataxia telengiectasia-mutated regulates AMPK in response to MET and IR. A549 cells were either transfected with ATM-specific siRNA or control vector and incubated for 72 h (B) or incubated with the ATM-specific inhibitor KU60019 or vehicle for a period of 24 h (C), before treatment with 5 μ M MET for 48 h (A–C) and/or IR of 0, 2 or 8 Gy 24 h (C) after initiation of MET treatment. After treatments, cells were washed, lysed and probed with indicated antibodies. Representative immunoblots of three independent experiments are shown. AMP-activated kinase mediates the signalling and antiproliferative effects of MET and IR. A549 cells were pretreated with siRNAs against (D) AMPKα1 and AMPKα2 catalytic subunits or control vector for a period of 72 h before treatment with 5 μ M MET for a 48-h period and/or IR dose of 0, 2 or 8 Gy for a 24-h period. After treatment, cells were washed, lysed and probed with indicated antibodies. Representative immunoblots of three independent experiments are shown. (E) A549 cells were pretreated with control vector (vehicle) or siRNA sequences against AMPKα1 and AMPKα2 catalytic subunits for a period of 72 h before a 48-h treatment with MET (0 μ M–1 mM) and a 24-h treatment with 0, 2 or 8 Gy dose of IR. Proliferation results (mean±s.e.) of three independent experiments (six replicates per condition in each experiment) are shown. (F) WT and AMPKα1/2−/−-MEFs were treated with MET (0 μ M–100 μ M) for a period of 48 h. After treatment, cells were washed, lysed and probed with indicated antibodies. Representative immunoblots of three independent experiments are shown. (G) WT and AMPKα1/2−/−-MEFs were treated with MET (0 μ M–5 mM) for a period of 48 h and/or the indicated doses of IR for 24 h. Proliferation results (mean±s.e.) of three independent experiments are shown.
To examine the role of AMPK in the mechanism of action of MET and IR, first A549 cells were pretreated with either vehicle alone or anti-AMPKα1 and -α2-specific siRNAs, and subjected to immunoblotting or proliferation assays. AMPKα1/2-siRNAs diminished significantly the expression of the enzyme and its activation by MET and IR, indicated by absence of phosphorylated AMPKα, inhibition of ACC phosphorylation and blockage of p21cip1 induction (Figure 4D). Furthermore, knockdown of AMPK induced a dramatic stimulation of the activity of the Akt–mTOR pathway, similar to our earlier observations in AMPKα1/2−/−-MEFs (Sanli et al, 2012b). Lack of AMPKα1/2 induced AktT-308 phosphorylation, enhanced mTOR expression and phosphorylation as well as 4EBP1 phosphorylation. Importantly, AMPK inhibition eliminated the antiproliferative effects of both MET, IR and their combined effects (Figure 4E). To ensure the specificity of the effects of AMPKα1/2-siRNA, we subjected also WT- and AMPKα1/2−/−-MEFs to MET (5–100 μ M). AMPKα1/2−/−-MEFs lacked AMPKα expression and phosphorylation, as well as the robust induction of p21cip1 expression in response to MET seen in WT-MEFs. AMPKα1/2−/−-MEFs had higher basal levels of phosphorylated 4EBP1, which did not respond to MET with reduction of this marker (Figure 4F). Further, AMPKα1/2−/−-MEFs showed severe resistance to the antiproliferative effects of both MET and IR (Figure 4G).
Metformin and IR effects on tumour growth
Balb/c-nude mice grafted with A549 or H1299 cells were subjected to treatments: MET, IR, both or neither. Animals tolerated tumour grafting and MET treatment with no noticeable changes in behaviour, eating habits or weight. Within 15 days after treatment initiation, A549 tumours of combined MET and IR treatment and H1299 tumours of the MET, IR or combined treatment began to show significant inhibition of growth rate compared with untreated tumours (P<0.01; Figure 5A). At 60 days, xenografts treated with MET or IR alone were on average 31.0±5.8 and 49.0±8.2% smaller than controls for A549, and 42.4±4.02 and 61.9±5.62% for H1299 tumours. Combined treatment (MET+IR) showed over 62±8.4% growth reduction for A549 and 73.2±6.19% for H1299 tumours. As shown (Figure 5A), animals grafted with H1299 reached end point size earlier than the treated animals, and were euthanised at 50 days. Overall, in both animal models, MET and IR (10 Gy) treatments inhibited significantly tumour growth kinetics, the combined MET+IR treatment inhibited tumour growth further, but the combined effects were not additive.

Inhibition of growth and molecular effects of MET and IR in human LC xenografts. (A) Effects on xenograft growth. Twenty-four and sixteen male Balb/c-nude mice were grafted into the right flank with 1 × 106 A549 or H1299 cells respectively. Treatment with MET, IR or combination was initiated once tumour volume reached 100 mm3. Tumour volume was measured every 5 days for a period of 8 weeks, as described in Materials and Methods. Average tumour volume data of four or six animals per treatment group are shown (mean±s.e.). Final tumour volumes were: A549: control: 679.5±50.6 mm3, MET: 470.53±35.2 mm3, IR: 342.9±21.8 mm3 and MET+IR: 257.9±15.2 mm3 and H1299: control: 1129.39±39.17 mm3, MET: 646.02±40.43 mm3, IR: 428.02±18.89 mm3 and MET+IR: 300.25±24.25 mm3. Statistically significant differences between the indicated treatment groups (*P<0.05 and **P<0.001). (B) Effects of MET and IR treatments on expression and activation of molecular markers. Tumours collected from control (CON), MET, IR and MET+IR treatment groups of A549 cohort were subjected to lysis and immunoblotting with antibodies against the indicated markers. Representative immunoblots of three independent experiments are shown. (C) Immunohistochemical analysis of P-AMPK expression. Analysis of A549 tumours from four different treatment groups using an anti-P-AMPKα (Thr172) antibody. Representative images from three independent experiments are shown. (D) Densitometric analysis of immunoblotting experiments (n=6 in each group). Data are presented as mean±s.e. of normalised densitometry values. *P⩽0.05; **P⩽0.001 statistically significant difference between treatment groups and control for each marker.
Chronic regulation of expression and activity of ATM–AMPK/Akt–mTOR pathways
Excised tumours, 8 weeks after a single dose of IR (10 Gy), continued MET or combined treatment, showed sustained enhancement of total ATM protein levels and its activity in irradiated tumours, evident by increased γH2AX (Figure 5B–D). However, ATM levels and activity (γH2AX levels) were also increased by MET treatment alone, which induced further enhancement of the IR-induced levels of ATM expression and activity in tumours treated with MET and IR.
Ionising radiation and MET treatment also caused sustained enhancement of total AMPKα, P-AMPKαThr172 and phospho-(P)-ACC levels, indicating stimulation of AMPK expression and activity (Figure 5B and D). Combined IR+MET treatment enhanced those markers further. AMPKα was increased by over two-fold by MET+IR compared to control; P-AMPKαThr172 was increased three-fold and four-fold by MET and IR vs five-fold by MET+IR (Figure 5B and D) whereas P-ACC followed similar trends (Figure 5B and D). To confirm the immunoblotting results and examine the tumour gross cellular and subcellular distribution of AMPK activation, P-AMPKαThr172 levels were examined with IHC (Figure 5C). Activated P-AMPKαThr172 distributed in the cytoplasm and nucleus of tumour cells with MET- and IR-treated tumours, showing increased levels of AMPKαThr172 phosphorylation and further increase by the combined treatment.
Similar regulation to ATM and AMPK was shown for p53 and p21cip1, respectively. Metformin and IR enhanced total p53, P-p53S15and p21cip1 levels but the combined MET+IR treatments produced significant further enhancement of those markers that approached additive effects on p21cip1 expression (Figure 5B and D).
We detected no significant differences in the total AKT levels between individual MET or IR treatments and untreated tumours (Figure 5B and D). Combined MET and IR treatment induced a trend for reduced total AKT expression. Ionising radiation- and MET-treated tumours showed reduced Akt-T308 and S473 phosphorylation, and so did the ones treated with combined MET+IR, although this was lower than MET alone (Figure 5B and D). We observed no consistent modulation of total mTOR levels by either treatment; however, IR and MET led to a significant additive inhibition of mTOR activity indicated by reduced 4EBP1 phosphorylation (MET: 26.8±4.8%; IR: 29.5±5.2% vs MET+IR: 51.5±8.3% reduction, Figure 5B and D), consistent with in vitro results (Figure 2).
Modulation of tumour vasculature by MET and IR
Irradiated tumours showed reduced endothelial angiogenesis marker CD31 expression using immunoblotting (by 29.3±3.9%, Figure 6A). However, tumours of animals treated with MET or MET+IR showed higher reductions of CD31 levels (by 51±4.1% and 47±4.9%, respectively, Figure 6C). In agreement with these results, IHC experiments showed reduced microvessel density in tumours treated with MET, IR or the combined treatment (Figure 6B).

Metformin and IR reduce microvessels and enhance apoptosis markers in A549 LC tumours. (A) Effects on apoptosis markers. Lysates from control, MET-, IR- and MET+IR-treated tumours were analysed with immunoblotting using anti-CD31, -Puma, -Bax and -actin antibodies. Representative immunoblots are shown. (B) Representative images of anti-CD31 IHC analysis of A549 control (CON), MET-, IR- and MET+IR-treated tumours. (C) Densitometric analysis of immunoblotting results from lysates of tumours in all experimental groups (n=6 in each group). Data are mean±s.e. of normalised densitometry values (*, P⩽0.05; **, P⩽0.001 statistically significant difference between treatment groups and control for each marker). (D) Effects MET and IR on expression of CC3. Representative images of IHC analysis of control (CON), MET-, IR- and MET+IR-treated tumours with an anti-CC3 antibody.
Regulation of pro-apoptotic markers
Individual MET and IR treatments increased the apoptotic markers BAX and PUMA in tumours. This was statistically significant only for irradiated tumours (Figure 6A and B). Metformin increased further the IR induction of the two markers. To verify further the activation of the apoptotic pathway, we examined the levels of CC3 with IHC. Metformin increased CC3 levels in non-irradiated tumours but more significantly so in irradiated tumours (Figure 6D).
Discussion
The potential of MET to offer true systemic antiproliferative effects in humans has been debated since, to date, studies used clinically intolerable (mM) concentrations of the drug to achieve significant inhibition of cancer cell growth. Ben Sahra et al (2008) and Buzzai et al (2007) showed inhibition of prostate, breast and colon cancer cell survival with 1–5 mM MET. In early studies, we also observed resistance of breast (MCF7 and MDAMB231) and prostate cancer (PC3) cells to low μ M dose MET, compared with NSCLC A549, H1299 and SK-MES1 cells (Supplementary Figure S1). Those results formed the basis of the present study, which shows significant antiproliferative effects of 5–100 μ M MET in NSCLC models (Figure 1). Most human studies report plasma Cmax of 1–2 μg ml−1 (∼10 μ M). Steady state concentrations of 7.8 μ M were described with daily oral MET of 850–1700 mg, whereas maximum concentrations up to 38 μ M or higher were shown in controlled studies (Lipska et al, 2011). Diabetic patients are treated with up to 2.5–3.0 g MET daily without significant toxicity. Lactic acidosis is the most significant, but rare, complication of MET therapy but no threshold plasma concentration of MET for lactic acidosis has been defined (Lipska et al, 2011). To date, there are no reports of increased normal human tissue toxicity with radiotherapy in patients treated with MET. Consistently, we observed no inhibition of proliferation of normal human embryonic lung fibroblasts (MRC5) with low MET doses (1–25 μ M; Supplementary Figure S2), indicating that MET has the potential to increase the therapeutic ratio of radiotherapy in LC.
Effects of MET on NSCLC genotypes
To explore the antitumour effect of MET in different NSCLC genotypes, we used lines that represent common histologies and mutation profile of NSCLC (A549 adenocarcinoma: LKB1-deficient (nonsense mutation of codon 37 leading to stop codon), K-Ras G12S-activating mutant, p53-WT; H1299 adenocarcinoma: LKB1-WT, p53-deficient (TP53 partial deletion) and SK-MES squamous cell carcinoma: LKB1-WT, p53-deficient (exon 8 mutation); Carretero et al, 2004). The efficacy of MET to inhibit proliferation or clonogenic survival in all these models of NSCLC indicates potential for clinical development in most NSCLC histologies.
Radiosensitisation by MET
For relevance to clinical radiotherapy, we treated cells and tumours with standard radiation doses of 2–10 Gy, using clinical linear accelerators. Both low and higher doses of MET augmented the antiproliferative effects of both 2 and 8 Gy IR, and statistical analysis of the interaction indicated synergy between the two treatments (see Results and Supplementary Table S1). Further, we observe global antitumour and radiosensitising action of MET (5–25 μ M) in clonogenic assays (Figure 1C). We show radiosensitising effects in LC at clinically achievable doses of MET (5–25 μ M). Recently, Song et al (2012) showed radiosensitisation of breast cancer and sarcoma cells by MET, but this was achieved at mM doses of MET. However, Skinner et al (2012) showed that 25 μ M MET could decrease the surviving fraction of head+neck cancer cells after 2 Gy IR (SF2). Overall, MET inhibited significantly NSCLC cell and tumour growth, and enhanced the IR-mediated cytotoxicity (Figures 1 and 5).
Comparison with other targeted therapeutics
As MET modulates mTOR activity, we compared it with the macrocyclic lactone mTOR inhibitor rapamycin (Sirolimus). In human pharmacokinetic studies, 2–3 mg oral rapamycin led to Cmax plasma levels of 5–29 nM, with limited grade 1–2 toxicity, but higher doses produced grade 3–4 dose-limiting toxicity including mucositis, thrombocytopenia, diarrhoea and hyperglycaemia (Jimeno et al, 2008). In irradiated cells, MET (5–100 μ M) caused similar antiproliferative effects as 5–500 nM rapamycin (Figure 1D). One of the reasons specific mTOR inhibitors failed to show clinical benefit in some tumours is their property to induce feedback activation of the PI3k-Akt axis, promoting survival of tumours and resistance to cytotoxics (Vazquez-Martin et al, 2009). Similar to others (Zakikhani et al, 2010), we observed inhibition of 4EBP1 phosphorylation by rapamycin but enhancement of Akt-T308 and Akt-S473 phosphorylation at 24 and 48 h of treatment (Supplementary Figure S4). This suggests that MET offers benefits over mTOR inhibitors as it inhibits not only mTOR but also Akt phosphorylation in tumours (Figures 2, 4, 5). We have also compared MET with EGFR inhibitor gefitinib (1–4 μ M used earlier; Tanaka et al, 2008). Gefitinib (1 μ M or higher) inhibited proliferation of non-irradiated NSCLC cells more effectively than 5 μ M MET, showed comparable antiproliferative action with 5–100 μ M MET in cells irradiated with 2 Gy but weaker radio-enhancing action compared with 5–100 μ M MET when combined with 8 Gy IR (Supplementary Figure S5).
Mechanism of action of IR and MET
Role of LKB1 and p53
Metformin is an inhibitor of complex I of the mitochondria respiratory chain and is proposed to function as an energy restriction mimetic, which leads to increased AMP levels and activation of AMPK through binding to γ-subunit (Steinberg and Kemp, 2009). It was suggested that MET requires LKB1 to activate AMPK, inhibit mTOR and suppresses proliferation and oncogenesis (Dowling et al, 2007; Shaw et al, 2005). Others argued that MET inhibits growth (Buzzai et al, 2007) or induces radiosensitisation (Skinner et al, 2012) selectively in tumours and cancer cells lacking functional p53. It is possible that specific p53-null tumours may be more sensitive to MET. We did observe greater sensitivity to low-dose MET in non-irradiated and irradiated p53-null NSCLC cells H1299 and SK-MES compared with A549 (p53-WT; Figure1 and Supplementary Figure S5). However, MET caused significant dose-dependent inhibition of proliferation and radiosensitisation in both LKB1- and p53-WT or -deficient cells, and inhibited growth of LKB1-deficient p53-WT (A549) xenografts (Figures 1 and 5). Further, p53-null MDAMB231 breast cancer and PC3 prostate cancer cells demonstrated higher resistance to MET compared with NSCLC cells. (Supplementary Figure S1).
Role of ATM
Earlier, we suggested that activation of the AMPK–p53–p21cip1 pathway in response to IR was ATM-dependent (Sanli et al, 2010 and Sanli et al, 2012b). Other studies showed that ATM regulates AMPK in response to cytotoxics such as etoposide (47), and recent reports suggest that MET mediates an ATM-mediated DDR-like response (Vazquez-Martin et al, 2011). Here, we showed that MET increased expression and activation of ATM in cells and tumours in association with activation of the AMPK-p53-p21cip1 axis. Metformin induced nuclear γH2AX foci, resembling those assembled at sites of DNA DSBs (Figure 3), indicating that the drug or its effectors are active within the nucleus. Presently, there is no evidence that MET induces genomic stress or DSBs. γH2AX foci sustained long after irradiation are likely the result of enhanced ATM activity related to potential replication stress (Menendez et al, 2011) or oxidative stress developing in cells, as MET suppresses mitochondria respiration. Such results are consistent with a developing notion that MET mediates a pseudo DDR that could protect against oncogenesis (Menendez et al, 2011). Future studies should investigate in depth the mechanism of regulation of ATM by MET. Here, we show that inhibition of ATM with either KU60019 or siRNA blocks MET- and IR-induced phosphorylation of AMPKα (Figure 4A–C) indicated that ATM functions upstream of AMPK in the pathways of action of MET and IR.
Role of AMPK
To date, our work suggested that (i) AMPK is a key sensor of IR signals (Sanli et al, 2010, ii) AMPK subunit expression is tightly regulated by IR (Sanli et al, 2012b) and (iii) lack of AMPK destabilises the Akt–mTOR and DDR pathways, leading to inhibition of radiation responsiveness (Sanli et al, 2012b). Consistently, here we observed enhanced expression and activation of AMPK in NSCLC cells and tumours in response to IR (Figures 2 and 5). Metformin induced a robust phosphorylation of AMPK in non-irradiated and irradiated cells and tumours, and increased total AMPKα levels in tumours (Figures 1, 2, 5). Metformin may indeed regulate AMPKα gene expression in addition to activation. Earlier, we suggested that sestrin 2, member of a family of p53-dependent stress-induced genes, facilitates AMPK subunit gene expression and activation in response to IR (Sanli et al, 2012a) and others (Rocha et al, 2011) proposed that sestrins mediate the AMPK response to combined MET and chemotherapy treatments.
Individual and combined treatments with MET and IR produced a sustained enhancement of signalling events downstream of AMPK, such as increased total and phosphorylated p53 and p21cip1 levels, as well as inhibition of mTOR activity, indicated by inhibition of 4EBP1 phosphorylation in cells and tumours (Figures 2 and 5). With AMPKα1/2 knockdown in NSCLC cells (siRNA) and knock-out models (AMPKα1/2−/−-MEFs), we demonstrate the dependency of those signalling events on AMPK and the key role of this enzyme in mediation of the antiproliferative effects of both MET and IR (Figure 4D–G). Knockdown of AMPKα1/2 produced in cancer cells the radio-resistant molecular expression phenotype we observed earlier in AMPKα1/2−/−-MEFs (Sanli et al, 2012b) with induction of mTOR expression and activity (P-4EBP1) and phosphorylation of Akt-T308 and mTORS2448 (Rosner et al, 2010; Figure 4D), both of which are markers of increased activity of the PI3k–Akt survival pathway. These results verify and strengthen our earlier observations on the role of AMPK in radiation responses of cancer cells and support the notion that ATM and AMPK are points of convergence of molecular signals activated by both IR and MET.
Inhibition of 4EBP1 phosphorylation and induction of p21cip1, which were observed in response to MET, are expected to inhibit protein synthesis and growth of cellular biomass, and to induce G1/S checkpoints. We observed an increase of cells at S phase with MET treatment (Figure 3). More effective modulation of p53/p21cip1 and mTOR by MET in irradiated cells is expected to inhibit cycling further and promote G1 arrest, which we did observe (Figure 3) and has been described by others (Ben Sahra et al, 2008; Rattan et al, 2011).
Metformin effects on angiogenesis
Reports on the effects of MET on tumour microvasculature remain controversial. AMP-activated kinase is suggested to participate in angiogenesis (Nagata et al, 2003) and MET was suggested to normalise tumour vasculature in breast cancer and inhibit angiogenesis in ovarian cancer xenografts (Rattan et al, 2011). Consistent with the latter, MET and IR reduced microvascular density and expression of the endothelial marker CD31 in NSCLC tumours (Figure 5A–C). These events may participate in the enhancement of IR-induced cytotoxicity seen with MET and contribute to sustained activation of the ATM-AMPK axis.
Metformin effects on apoptosis
The significant induction of expression and phosphorylation of p53 we detected with MET in vitro and in vivo (Figures 2 and 5) suggested that apoptotic cell death is activated. Combined IR+MET induced the greatest enhancement in apoptosis markers in cultured NSCLC cells, detected by the Annexin-V assay (Figure 3B), and in tumours, detected as enhanced expression of the apoptotic markers Bax, Puma and CC3 (Figure 6). They suggest activation of the intrinsic (mitochondria-dependent) and the common apoptotic pathways, which lead to apoptotic death. AMP-activated kinase activation, MET and IR were described to stimulate autophagy (Chaachouay et al, 2011; Mihaylova and Shaw, 2011; Shi et al, 2012), which is suggested to facilitate survival under metabolic stress. However, it remains highly controversial whether autophagy is a reliable marker of cell survival or cell death in cells subjected to cytotoxic therapy (Galluzzi et al, 2007). Our results are consistent with those obtained when MET was combined with paclitaxel (Rocha et al, 2011).
Conclusions
In conclusion, this study demonstrates that MET doses that are achieved safely in human plasma inhibit proliferation and radiosensitise NSCLC. Metformin inhibits tumour growth and enhances tumour response and the pro-apoptotic and antiangiogenic effects of IR. In cells and tumours, MET mediates activation of tumour suppressor and inhibition of survival pathways. The ATM–AMPK pathway is a point of convergence of IR and MET signals and the mediator of the antitumour action of the two agents. The work presented here provides a preclinical basis to begin investigation of MET in combination with radiotherapy in early-phase clinical trials in NSCLC.
Change history
28 May 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F (2008) The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 27: 3576–3586
Bowker SL, Majumdar SR, Veugelers P, Johnson JA (2006) Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin: Response to Farooki and Schneider. Diabetes Care 29: 1990–1991
Brachmann S, Fritsch C, Maira SM, Garcia-Echeverria C (2009) PI3K and mTOR inhibitors: a new generation of targeted anticancer agents. Curr Opin Cell Biol 21: 194–198
Bussink J, van der Kogel AJ, Kaanders JH (2008) Activation of the PI3-K/AKT pathway and implications for radioresistance mechanisms in head and neck cancer. Lancet Oncol 9: 288–296
Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB (2007) Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res 67: 6745–6752
Carretero J, Medina PP, Pio R, Montuenga LM, Sanchez-Cespedes M (2004) Novel and natural knockout lung cancer cell lines for the LKB1/STK11 tumor suppressor gene. Oncogene 23: 4037–4040
Chaachouay H, Ohneseit P, Toulany M, Kehlbach R, Multhoff G, Rodemann HP (2011) Autophagy contributes to resistance of tumor cells to ionizing radiation. Radiother Oncol 99: 287–292
Chou TC (2010) Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 70: 440–446
Chou TC, Talalay P (1984) Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 22: 27–55
Curran WJ Jr (2003) Evolving chemoradiation treatment strategies for locally advanced non-small-cell lung cancer. Oncology 17: 7–14
Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N (2007) Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res 67: 10804–10812
Dowling RO, Niraula S, Stambolic V, Goodwin PJ (2012) Metformin in cancer: translational challenges. J Mol Endocrinol 48 (3): R31–R43
Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD (2005) Metformin and reduced risk of cancer in diabetic patients. BMJ 330: 1304–1305
Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, Kroemer G (2007) Cell death modalities: classification and pathophysiological implications. Cell Death Differ 14: 1237–1243
Golding SE, Rosenberg E, Valerie N, Hussaini I, Frigerio M, Cockcroft XF, Chong WY, Hummersone M, Rigoreau L, Menear KA, O'Connor MJ, Povirk LF, van Meter T, Valerie K (2009) Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol Cancer Ther 8: 2894–2902
Goodwin PJ, Ligibel JA, Stambolic V (2009) Metformin in breast cancer: time for action. J Clin Oncol 27: 3271–3273
Goodwin PJ, Stambolic V, Lemieux J, Chen BE, Parulekar WR, Gelmon KA, Hershman DL, Hobday TJ, Ligibel JA, Mayer IA, Pritchard KI, Whelan TJ, Rastogi P, Shepherd LE (2011) Evaluation of metformin in early breast cancer: a modification of the traditional paradigm for clinical testing of anti-cancer agents. Breast Cancer Res Treat 126: 215–220
Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30: 214–226
Huang X, Wullschleger S, Shpiro N, McGuire VA, Sakamoto K, Woods YL, McBurnie W, Fleming S, Alessi DR (2008) Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem J 412: 211–221
Iliopoulos D, Hirsch HA, Struhl K (2011) Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer Res 71: 3196–3201
Jimeno A, Rudek MA, Kulesza P, Ma WW, Wheelhouse J, Howard A, Khan Y, Zhao M, Jacene H, Messersmith WA, Laheru D, Donehower RC, Garrett-Mayer E, Baker SD, Hidalgo M (2008) Pharmacodynamic-guided modified continuous reassessment method-based, dose-finding study of rapamycin in adult patients with solid tumors. J Clin Oncol 26: 4172–4179
Jiralerspong S, Palla SL, Giordano SH, Meric-Bernstam F, Liedtke C, Barnett CM, Hsu L, Hung MC, Hortobagyi GN, Gonzalez-Angulo AM (2009) Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol 27: 3297–3302
Laplante M, Sabatini DM (2009) mTOR signaling at a glance. J Cell Sci 122: 3589–3594
Lipska KJ, Bailey CJ, Inzucchi SE (2011) Use of metformin in the setting of mild-to-moderate renal insufficiency. Diabetes Care 34: 1431–1437
Lord JM, Flight IH, Norman RJ (2003) Metformin in polycystic ovary syndrome: systematic review and meta-analysis. BMJ 327: 951–953
Mak IWY, Turcotte RE, Popovic S, Singh G, Ghert M (2011) AP-1 as a Regulator of MMP-13 in the Stromal Cell of Giant Cell Tumor of Bone. Biochem Res Int 2011: 164197
Menendez JA, Cufi S, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Vellon L, Vazquez-Martin A (2011) Metformin and the ATM DNA damage response (DDR): accelerating the onset of stress-induced senescence to boost protection against cancer. Aging 3: 1063–1077
Mihaylova MM, Shaw RJ (2011) The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 13: 1016–1023
Nagata D, Mogi M, Walsh K (2003) AMP-activated protein kinase (AMPK) signaling in endothelial cells is essential for angiogenesis in response to hypoxic stress. J Biol Chem 278: 31000–31006
Petroulakis E, Mamane Y, Le Bacquer O, Shahbazian D, Sonenberg N (2006) mTOR signaling: implications for cancer and anticancer therapy. Br J Cancer 94: 195–199
Rattan R, Graham RP, Maguire JL, Giri S, Shridhar V (2011) Metformin suppresses ovarian cancer growth and metastasis with enhancement of cisplatin cytotoxicity in vivo. Neoplasia 13: 483–491
Rocha GZ, Dias MM, Ropelle ER, Osorio-Costa F, Rossato FA, Vercesi AE, Saad MJ, Carvalheira JB (2011) Metformin amplifies chemotherapy-induced AMPK activation and antitumoral growth. Clin Cancer Res 17: 3993–4005
Rosner M, Siegel N, Valli A, Fuchs C, Hengstschlager M (2010) mTOR phosphorylated at S2448 binds to raptor and rictor. Amino Acids 38: 223–228
Sanli T, Linher-Melville K, Tsakiridis T, Singh G (2012a) Sestrin2 modulates AMPK subunit expression and its response to ionizing radiation in breast cancer cells. PLoS One 7: e32035
Sanli T, Liu C, Rashid A, Hopmans S, Tsiani E, Schultz C, Farrell T, Singh G, Wright J, Tsakiridis T (2011) Lovastatin sensitizes lung cancer cells to ionizing radiation. Modulation of growth and tumour suppressor signalling pathways and induction of apoptosis. J Thorac Oncol 6 (3): 439–450
Sanli T, Rashid A, Liu C, Harding S, Bristow RG, Cutz JC, Singh G, Wright J, Tsakiridis T (2010) Ionizing radiation activates AMP-activated kinase (AMPK): a target for radiosensitization of human cancer cells. Int J Radiat Oncol Biol Phys 78: 221–229
Sanli T, Storozhuk Y, Linher-Melville K, Bristow RG, Laderout K, Viollet B, Wright J, Singh G, Tsakiridis T (2012b) Ionizing radiation regulates the expression of AMP-activated protein kinase (AMPK) in epithelial cancer cells: modulation of cellular signals regulating cell cycle and survival. Radiother Oncol 102: 459–465
Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC (2005) The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 310: 1642–1646
Shi WY, Xiao D, Wang L, Dong LH, Yan ZX, Shen ZX, Chen SJ, Chen Y, Zhao WL (2012) Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell Death Dis 3: e275
Skinner HD, Sandulache VC, Ow TJ, Meyn RE, Yordy JS, Beadle BM, Fitzgerald AL, Giri U, Ang KK, Myers JN (2012) TP53 disruptive mutations lead to head and neck cancer treatment failure through inhibition of radiation-induced senescence. Clin Cancer Res 18: 290–300
Song CW, Lee H, Dings RP, Williams B, Powers J, Santos TD, Choi BH, Park HJ (2012) Metformin kills and radiosensitizes cancer cells and preferentially kills cancer stem cells. Sci Rep 2: 362
Steinberg GR, Kemp BE (2009) AMPK in health and disease. Physiol Rev 89: 1025–1078
Tanaka T, Munshi A, Brooks C, Liu J, Hobbs ML, Meyn RE (2008) Gefitinib radiosensitizes non-small cell lung cancer cells by suppressing cellular DNA repair capacity. Clin Cancer Res 14: 1266–1273
Taubes G (2012) Cancer research. Cancer prevention with a diabetes pill? Science 335: 29
Tsakiridis T, Cutz JC, Singh G, Hirte H, Okawara G, Corbett T, Sur R, Cai W, Whelan T, Wright JR (2008) Association of phosphorylated epidermal growth factor receptor with survival in patients with locally advanced non-small cell lung cancer treated with radiotherapy. J Thorac Oncol 3: 716–722
Vazquez-Martin A, Oliveras-Ferraros C, Cufi S, Martin-Castillo B, Menendez JA (2011) Metformin activates an ataxia telangiectasia mutated (ATM)/Chk2-regulated DNA damage-like response. Cell Cycle 10: 1499–1501
Vazquez-Martin A, Oliveras-Ferraros C, Del Barco S, Martin-Castillo B, Menendez JA (2009) If mammalian target of metformin indirectly is mammalian target of rapamycin, then the insulin-like growth factor-1 receptor axis will audit the efficacy of metformin in cancer clinical trials. J Clin Oncol 27 (33): e207–e209
Zakikhani M, Blouin MJ, Piura E, Pollak MN (2010) Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast Cancer Res Treat 123: 271–279
Acknowledgements
We thank Carrie Schultz and Dr Thomas Farrell, Radiation Therapy and Physics, Juravinski Cancer Center, for their work to facilitate the planning and delivery of radiotherapy to tissue cultures and tumours of grafted animals. We thank Dr Jacek M Kwiecien, McMaster University, for his kind assistance with animal autopsies and Dr Robert Bristow, Department of Radiation Oncology, University of Toronto for scientific advice. We greatly appreciate the donation of WT- and AMPKα1/2−/−-MEFs by Dr Benoit Viollet (INSERM, France). This work was supported by grants by the Canadian Association of Radiation Oncologists –RAZCER program to TT and the Canadian Institutes of Health Research to GS.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Storozhuk, Y., Hopmans, S., Sanli, T. et al. Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (NSCLC) through ATM and AMPK. Br J Cancer 108, 2021–2032 (2013). https://doi.org/10.1038/bjc.2013.187
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.187
Keywords
This article is cited by
-
Radioproteomics modeling of metformin-enhanced radiosensitivity: an animal study
Japanese Journal of Radiology (2023)
-
Metformin in therapeutic applications in human diseases: its mechanism of action and clinical study
Molecular Biomedicine (2022)
-
The role of AMPK-dependent pathways in cellular and molecular mechanisms of metformin: a new perspective for treatment and prevention of diseases
Inflammopharmacology (2022)
-
AMPK activation by ASP4132 inhibits non-small cell lung cancer cell growth
Cell Death & Disease (2021)
-
Molecular mechanisms underlining the role of metformin as a therapeutic agent in lung cancer
Cellular Oncology (2021)
