
Abstract
Matricellular proteins have been classified as a family of non-structural matrix proteins capable of modulating a variety of biological processes within the extracellular matrix (ECM). These proteins are expressed dynamically and their cellular functions are highly dependent upon cues from the local environment. Recent studies have shown an increasing appreciation of the key roles these ECM proteins play within the tumour microenvironment. Induced by either tumour cells or tumour stromal components, matricellular proteins initiate downstream signalling events that lead to proliferation, invasion, matrix remodelling and dissemination to pre-metastatic niches in other organs. In this review, we summarise and discuss the current knowledge of the diverse roles these proteins play within the microenvironment that influences tumour progression and potential for future therapies targeting the tumour microenvironment.
Similar content being viewed by others

Matricellular proteins and the tumour microenvironment
To understand the roles matricellular proteins play within the tumour microenvironment, it is important to recognise first the complexity of interactions occurring between malignant tumour cells and the different components of the surrounding tumour stroma such as the extracellular matrix (ECM), fibroblasts, inflammatory/immune and endothelial cells. The ECM is critical during development as it not only maintains tissue homoeostasis and structure, but it also performs a diverse set of regulatory functions that orchestrates downstream signalling pathways for proliferation and survival, and serving as a scaffold for mediating physical interactions between different cell types (Lu et al, 2012). Mounting evidence has revealed that the ECM serves as a crucial nexus for the establishment of ECM protein-growth factor receptor interactions, ECM-growth factor/cytokine interactions and other ECM protein–protein associations (Lu et al, 2012). During tumour progression, alterations in tumour suppressor genes and oncogenes cause cancer cells to activate adjacent stromal components and induce the release of cytokines, growth factors and ECM proteins into the tumour stroma to create a microenvironment conducive for growth and dissemination (Mueller and Fusenig, 2004; Kalluri and Zeisberg, 2006). This array of stromal modulating factors that includes, but not limited to, transforming growth factor β (TGF-β), interleukins, colony stimulating factor 1, tumour necrosis factor α and epidermal growth factor receptor (EGFR) ligands, thereby leading to a perturbation in tissue homoeostasis and creating a condition not dissimilar to wound healing (Christofori, 2006). Additionally, deposition of ECM proteins such as collagen I and IV, fibronectin, laminin and matricellular proteins leads to matrix remodelling and subsequent release of proteases such as metalloproteinases (MMPs) and cathepsins, which degrade the basement membrane and initiate a pro-invasion programme for tumour cells (Joyce and Pollard, 2009). Tumour angiogenesis also plays an important role within the tumour microenvironment toward supporting a permissive environment that fuels tumour growth by increased production of vascular endothelial growth factor (VEGF) (Goel et al, 2011). Aberrant pro-angiogenic signalling driven by VEGF leads to the formation of abnormal tumour vasculature that contributes to tumours’ abilities for growth, evasion from immune surveillance as well as facilitating metastasis (Goel et al, 2011).
Matricellular proteins have been defined as a subset of non-structural ECM proteins that modulate cell–matrix interactions and cell regulatory functions by different mechanisms and can be highly context dependent (Roberts, 2011). These proteins, which are highly responsive to cues received by their local environment, exert their functions by either binding to cell surface receptors or binding directly to other ECM proteins to form a scaffold that can trigger extracellular signalling pathways for upregulation of inflammatory cytokines and other growth factors (Chiodoni et al, 2010). Members of this family of proteins include osteopontin (OPN), periostin (POSTN), secreted protein acidic and rich in cysteine, (SPARC), tenascin C (TNC) and others. Although the matricellular family of proteins has been growing in recent years, we focus on the aforementioned matricellular proteins in this review as they have been well-studied in terms of their clinical relevance in tumorigenesis. In particular, their roles in cancer development, metastasis and potential strategies toward augmenting current cancer therapeutics will be discussed.
Knockout mouse models of these specific matricellular proteins are often viable but characteristically display impaired responses to mechanical stress and wound healing (Bradshaw et al, 2003; Rios et al, 2005; Vetrone et al, 2009; Okamura et al, 2010). This group of proteins is dynamically expressed and is highly elevated during embryonic development but show diminished activity in normal adult tissues. Wound injury, tissue remodelling, inflammation, cancer and other chronic diseases induce the re-expression of these proteins (Chiodoni et al, 2010). In cancer, overexpression of matricellular proteins by activated fibroblasts or malignant cells can potentiate pro-tumorigenic interactions in the tumour microenvironment (Chiodoni et al, 2010). Given the range of their binding capabilities due to multiple domains that contain many binding sites to different cell adhesion receptors, matricellular proteins are able to direct a multitude of biological processes essential for tumorigenesis such as proliferation, cell adhesion, migration, invasion, angiogenesis and survival (Bornstein and Sage, 2002).
Clinical implications of matricellular proteins in cancer
The deregulation of matricellular proteins’ expression has been reported in many solid tumours. Immunohistochemical analyses of human tumour samples have revealed high SPARC levels in astrocytomas, breast and pancreatic cancers (Huang et al, 2000; Barth et al, 2005; Prenzel et al, 2006). Similarly, quantitative gene expression and proteomic data have identified OPN, POSTN and TNC to be upregulated in diverse tumour types (Wikman et al, 2002; Kwon et al, 2009; Rocco et al, 2011). Aberrant upregulation of these matricellular proteins have been found to correlate with poorer prognosis in cancer patients (Chiodoni et al, 2010) and may have important clinical implications in the field of cancer molecular diagnostics.
For example, TNC has been proposed also as a potential diagnostic marker for certain cancers such as gliomas and bladder cancer because of its correlation with poor patient survival (Herold-Mende et al, 2002; Brunner et al, 2004). POSTN has also been found to be associated with lower survival in non-small cell lung cancer (Sasaki et al, 2001) and our group has also observed similar findings in a cohort of oesophageal squamous cell carcinoma patients (Wong et al, 2012). Moreover, a prospective clinical study charting serial plasma OPN levels in women with metastatic breast cancer over time revealed a strong correlation of high OPN plasma levels with poorer survival (Bramwell et al, 2006). Thus, the evidence from these data suggests matricellular proteins may have some efficacy as potential biomarkers in cancer.
Concomitant with the premise of using these matricellular proteins as tumour biomarkers, they have also been considered as candidates for adjuvant therapy. Experimental work has revealed that neutralising antibodies targeted against POSTN lead to decreased formation of metastases in human ovarian cancer xenografts and in a murine breast cancer model (Kyutoku et al, 2011; Zhu et al, 2011). Additionally, human breast cancer cells that constitutively express high levels of TNC showed decreased primary xenograft tumour growth as well as decreased number of metastatic foci in the lungs with genetic abalation of TNC (Calvo et al, 2008). Promising results have emerged from clinical trials using TNC-specific antibodies conjugated with cytotoxic agents both in malignant glioma patients (Reardon et al, 2002) and in patients with non-Hodgkin lymphoma (Rizzieri et al, 2004) with potential applicability for a broader range of cancers.
Induction of migration, EMT and invasion by matricellular proteins
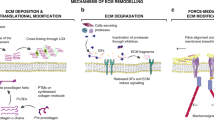
Tumour initiation and subsequent invasion of tumour cells into the surrounding stroma marks a vital step towards metastases. This process, known as epithelial–mesenchymal transition (EMT), which is a distinctive morphological change whereby the tumour cells switch from a well-differentiated epithelial phenotype to a more invasive mesenchymal phenotype (Kalluri and Weinberg, 2009). Detachment of tumour cells from the primary tumour requires a progressive loss of cell–cell contact at adherens junctions, which is facilitated by downregulation of epithelial proteins such as E-cadherin (E-cad) and cytokeratins and increase in cell motility through increase of mesenchymal proteins such as vimentin and N-cadherin (N-cad) (Kalluri and Weinberg, 2009). Induction of EMT can occur via growth factors such as hepatocyte growth factor, TGF-β, epidermal growth factor (EGF) and fibroblast growth factor (FGF) as well as matricellular proteins (Kalluri and Zeisberg, 2006). Matricellular proteins can support and induce EMT by triggering several receptor-mediated signalling pathways (Figure 1A) (Chiodoni et al, 2010) and this family of proteins has been shown to have important functions in promoting tumour cell migration and invasion.

Roles of matricellular proteins within the tumour microenvironment and metastatic niche. (A) After induction by soluble factors such as EGF and TGF-β as well as cytokines in the tumour microenvironment (red arrow), matricellular proteins act as ligands to activate integrin signalling or receptor tyrosine kinases such as EGFR or FAK receptors, which in turn induces downstream biological processes such as EMT, cell motility and invasion and ultimately metastasis. (B) Matricellular proteins induce ECM remodelling by activating intracellular ADAMTs or undergoing proteolytic cleavage by extracellular MMPs. These interactions promote downstream tumour cell signalling, invasion and metastasis. Matricellular proteins can also act as scaffolds for facilitating interactions with other matricellular/ECM proteins that promotes upregulation of LOXP activity and fibrillar collagen crosslinking. The resulting matrix remodelling and stiffness cluster receptor tyrosine kinases or integrins, which then initiate downstream cell signalling pathways for mediating invasion. (C) Matricellular proteins, derived from growth factors or cytokines secreted from primary tumours or stromal cells (red arrow), aid in maintenance of cancer stem cell (CSC) signalling such as Notch1 or Wnt signalling to maintain the metastatic niche. Matricellular proteins may also form interactions with other matricellular proteins to augment CSC survival in metastatic niche.
POSTN is a secreted TGF-β inducible, N-glycoprotein that comprises multiple domains such as an N-terminal signal peptide sequence, a cysteine-rich domain, a hydrophilic C-terminus with heparin binding domains and Fasciclin 1 adhesion domains that has been found to bind to several ECM proteins such as fibronectin, collagen I/V, TNC and integrins (Skonier et al, 1992; Takeshita et al, 1993; Maruhashi et al, 2010). POSTN has been shown to increase the metastatic potential of tumorigenic 293T cells in vivo as well as stimulating EMT (Yan and Shao, 2006). Moreover, our group has demonstrated the functional importance of POSTN in initiating tumour cell invasion when secreted at the invasive front through the convergence of mutant p53 and EGFR overexpression (Michaylira et al, 2010). Although it does not possess an Arginine–Glutamate–Aspartate (RGD) domain, a classical structural element for binding to integrins, POSTN also been reported to bind to αVβ5 and αVβ3 integrins to coactivate EGFR signalling that modulates cell motility and proliferation (Yan and Shao, 2006). Similar to these observations, POSTN has been reported to promote cell survival in colon cancer cells via integrin signalling and consequent downstream activation of the PKB/Akt pathway (Bao et al, 2004).
TNC has been also implicated in mediating cell motility and invasion in a variety of epithelial cancers. TNC is a secreted glycoprotein with a hexabranchion structure containing EGF and fibronectin-like repeats as well as a fibrinogen-like domain that binds to a variety of proteins in the ECM (Chung et al, 1995). Experimental evidence has established that the role of TNC in colorectal cancer and EMT is modulated by β-catenin (Beiter et al, 2005). In addition, it has also been proposed that there is a link between TNC and Notch-mediated invasion in glioma cells as Notch1 was found to be capable of regulating the expression of TNC, although its effect in tumour invasion remains to be elucidated (Sivasankaran et al, 2009). TNC knockdown in metastatic breast cancer lines (LM2) not only decreased cell migration in vitro but also showed a significant reduction in the formation of lung metastases (Tavazoie et al, 2008). Intriguingly, miRNA profiling of MDA-MB-231 breast cancer derivatives identified miR-335 as an important regulator of a set of metastatic genes, one of which was shown to be TNC (Tavazoie et al, 2008).
Similar to both TNC and POSTN, OPN can induce cell motility and invasion and was first characterised in an immune response against bacterial infection (Nau et al, 1997). OPN is a secreted phosphoglycoprotein with multiple domains that are highly conserved and allow it to bind to a variety of cellular receptors such as integrins and hyaluronan receptor, CD44 (Shevde et al, 2010). OPN is found also to be upregulated in epithelial cancers and can be produced by either the tumour cells or surrounding stromal cells, such as endothelial cells or macrophages (Shevde et al, 2010). Secreted OPN is then deposited in the ECM and mediates cell–matrix interactions to initiate cell signalling, such as binding to growth receptors, for example EGFR or c-Met, to induce cell proliferation and migration (Tuck et al, 2003). Like POSTN, OPN undergoes alternative splicing and produces three isoforms, OPN-a, OPN-b and OPN-c. These isoforms are expressed differentially during cancer progression and can promote tumorigenesis to a varying degree. For instance, a recent study reports that only the OPN-c isoform is expressed highly in ovarian tumours compared with normal tissue samples (Tilli et al, 2011). In addition, OPN-c is capable of augmenting cell proliferation and invasion of the ovarian cancer cell line OvCar-3 and this was found to be mediated through PI3K signalling (Tilli et al, 2011). However, there was no change in function with OPN-a and OPN-b. Similar observations have been reported for prostate cancer cells except OPN-b also plays a pro-tumorigenic role by inducing invasion (Tilli et al, 2012).
SPARC, also known as osteonectin, is a secreted glycoprotein with multidomains such as N- and C-termini Ca2+-binding domains and a disulphide, copper binding follistatin domain (Clark and Sage, 2008). SPARC has been shown also to bind directly to components within the ECM compartment such as collagen I and III, which promotes collagen fibrillogenesis and integrin receptors αVβ4, β1 and β5 (Rentz et al, 2007). SPARC also binds directly to growth factors such as VEGF and affects regulation of FGF by affecting the phosphorylation of the FGF receptor (Clark and Sage, 2008). The functions of SPARC have been characterised broadly as anti-proliferative, deadhesive and promoting cell–ECM interactions which have important pro-tumorigenic functions when SPARC protein expression is dysregulated in tumours. SPARC overexpression in normal melanocytes and melanoma cells reduces E-cadherin expression and induces EMT through upregulation of mesenchymal markers such as Snail as well as promoting cell motility and invasion (Robert et al, 2006). Acquisition of an invasive phenotype through SPARC overexpression has been reported in glioma cells and was shown to be attenuated when SPARC was knocked down with accompanying inhibition of the focal adhesion kinase (FAK) signalling pathway (Shi et al, 2007). However, the pro-tumorigenic roles of SPARC appear to be dependent upon cellular context as the paracrine and autocrine effects of SPARC towards tumour invasion remain to be elucidated. Experimental findings from several groups in melanoma and ovarian cancer have demonstrated the importance of tumour-derived SPARC on tumour invasion compared with stromal-derived SPARC. In their studies, tumour xenografts in SPARC-null mice led to larger tumours compared with tumour xenografts in control wild-type mice (Brekken et al, 2003; Prada et al, 2007). A possible mechanism mediating this finding could be due to aberrant TGF-β1 activation by tumour-derived SPARC. In pancreatic adenocarcinoma, it has been recently demonstrated that tumour-derived SPARC enhances tumour progression by controlling the activation of TGF-β1 in SPARC-null mice (Arnold and Rivera, 2012).
Matricellular proteins and remodelling of the ECM
Given the importance of ECM dynamics during development and maintenance of tissue homoeostasis, there has been a burgeoning appreciation about how the deregulation of the ECM leads to or promotes tumour initiation and progression. Extracellular matrix remodelling forms an integral part in this process by changing the structural architecture of the tumour cell matrix and effecting the physical changes that facilitate tumour cell signalling and behaviour (Lu et al, 2012). Excess ECM deposition due to increased production of fibrillar type I collagens, fibronectin, proteoglycans, cytokines and secretion of matricellular proteins can be subject to enzymatic proteolysis which can lead to increased matrix crosslinking, thereby creating a fibrotic matrix environment that has been associated with accelerated neoplastic transformation (Cox and Erler, 2011). Investigating the links between mechanotransduction due to increased tissue stiffness and enhanced tumour cell signalling has been the subject of recent intense interest (Cox and Erler, 2011).
Increased tissue stiffness in breast tumour lesions compared with normal breast tissues has been demonstrated to be induced by upregulation of the enzymatic activity of lysyl oxidase (LOX), an enzyme that is responsible for collagen I and IV fibre crosslinking together with other ECM proteins (Levental et al, 2009). It is this collagen crosslinking via LOX that has been shown to be one of the contributing factors that lead to increased tissue stiffness in cancers. In addition, it has also been proposed that increased tissue stiffness is due to increased collagen content (Netti et al, 2000) as well as collagen remodelling by tumour fibroblasts (Gaggioli et al, 2007), all of which provide a network of tracks in the tumour microenvironment that supports the migration of cancer cells and fibroblasts as well as clustering receptors such as integrins, FAK and other tyrosine kinases to transduce downstream signalling for cell motility and invasion (Friedl and Alexander, 2011). Release of pro-fibrotic growth factors such as TGF-β, FGF2 and platelet-derived growth factor by cancer-associated fibroblasts also augment the induction of matricellular protein secretion (Kalluri and Zeisberg, 2006). Overexpression of matricellular proteins within the tumour microenvironment aids these processes by not only serving as ligands to receptor-mediating signalling but also acting as scaffolds for the formation of a dense ECM meshwork (Figure 1B). Expression of endogenous SPARC has been shown to affect ECM organisation by influencing collagen deposition (Brekken et al, 2003). POSTN has been shown to interact with bone morphogenetic protein 1 (BMP-1) and fibronectin that consequently leads to the activation of LOX in the ECM and enhanced collagen crosslinking (Maruhashi et al, 2010). Interestingly, biochemical studies have revealed that POSTN supports the incorporation of TNC onto ECM fibrils within the ECM architecture, suggesting that the interactions of these matricellular proteins in the ECM may form the basis of a niche that might be conducive for metastasis (Kii et al, 2010).
During tumorigenesis, another key characteristic of ECM remodelling is the deregulation in the activities of extracellular proteinases such as metalloproteinases (MMPs), cathepsins and membrane-bound proteinases such as a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTs) (Kessenbrock et al, 2010). These proteinases serve as effectors of ECM remodelling and function as enzymes responsible for degradation of the ECM and other structural ECM molecules such as laminins and collagens (Kessenbrock et al, 2010). Enzymatic proteolysis of these structural ECM proteins also effectively remodels the ECM to create tracks for tumour cells to invade into the underlying mesechymal stroma as well as releasing short peptide fragments that provide cues for the recruitment of myeloid cells to metastatic sites (Kessenbrock et al, 2010). Matricellular proteins can both undergo proteolysis by these proteinases as well as induce their activities. Upregulation of MMP-2 by SPARC has been observed in breast cancer and glioma cell lines (Chlenski and Cohn, 2010). TNC has been demonstrated to increase the invasiveness of glioma cells through interaction with MMP-12 (Sarkar et al, 2006). Release of OPN fragments through cleavage by MMP-9 in hepatic cell carcinoma (HCC) cells has been shown to be essential for tumour cell invasion and correlates with increased propensity for hepatocellular cell carcinoma (HCC) metastasis (Takafuji et al, 2007). Overall, these data suggest that matricellular proteins have the capacity to mediate tumour cell invasion and dissemination through ECM remodelling either by serving as scaffolds for bridging ECM protein interactions and helping to increase tissue stiffness through upregulation of collagen fibrillogenesis or by upregulation of ECM proteinase activity.
Since the tumour ECM has been reported to be a barrier to efficient drug delivery, several groups have demonstrated that targeting specific populations within the stroma leads to improved drug delivery and therapeutic efficacy. For instance, Olive et al (2009) established that pharmacologic disruption of the Hedgehog (Hh) signalling pathway depleted the desmoplastic stroma in a mouse model of pancreatic cancer. Hence, the removal of this physical barrier allowed more effective exposure of the tumour cells to chemotherapeutic agents. When hyaluronic acid was specifically ablated from the stroma in the same mouse model of pancreatic cancer, this restored tumour interstitial fluid pressure and subsequently increased sensitivity for the tumour cells to chemotherapy (Provenzano et al, 2012). Additionally, it has been shown that the mechanism of increased chemotherapy delivery is facilitated through increased perfusion by virtue of vessel decompression (Stylianopoulos et al, 2012). In aggregate, it is conceivable that targeting of matricellular proteins might augment the abovementioned strategies to ablate the tumour stroma, although this is conceptual in nature at the present time.
Priming of the pre-metastatic niche by matricellular proteins
Metastasis represents the final step in the cascade initiated by tumour cells after egress from the primary tumour site, invasion and intravasation into the blood circulation. In order for the disseminated tumour cells to colonise and thrive at distant, metastatic sites, it would need an environment or ‘niche’ that is permissive for successful tumour cell colonisation and growth (Chaffer and Weinberg, 2011). However, the low number of metastasis initiating cells suggests that microenvironments of distant sites are generally inhospitable for tumour metastases. In order to overcome this obstacle, it has been shown that soluble factors, ECM proteins and growth factors, are secreted from the primary tumour or stromal cells, in turn priming the microenvironment at distant sites to prepare for metastatic cell seeding (Psaila and Lyden, 2009). Recent studies have proposed that one source of these stromal factors are matricellular proteins derived from the ECM of primary tumours (Figure 1C). McAllister et al (2008) highlighted the capacity of tumour-derived OPN in instigating tumour growth in indolent tumours at distant sites and enhancing lung metastases via recruitment of bone marrow derived cells. In an orthotopic model of 4T1 breast cancer, S100A4+ stromal cells were found to promote metastatic colonisation to the lung by producing elevated levels of TNC (O’Connell et al, 2011). Furthermore, metastatic assays done with 4T1 breast cancer cells implanted in mice with a TNC-deletion led to smaller metastatic lesions compared with wild-type littermate control mice, confirming the importance of TNC in the metastatic microenvironment (O’Connell et al, 2011).
Another facet in the study of tumour metastasis has been the putative association between cancer stem cells (CSC) and subsequent growth in their metastatic niches. In a polyoma virus middle T antigen (PyMT) mouse model of breast cancer, elevated POSTN expression was observed both in mammary tumours and in lung metastases. However, the number of lung metastases was reduced significantly when this model of cancer was studied in the context of POSTN-null mice compared with wild-type control mice, suggesting that POSTN mediates initiation of metastatic lesions in the lung (Malanchi et al, 2012). Moreover, the tumour initiating capacity of CSC was diminished when inhibited by POSTN blocking antibodies and stromal POSTN was shown to be responsible for maintaining CSC signalling in the metastatic niche by interacting with Wnt ligands (Malanchi et al, 2012). Strikingly, parallel observations were made in another study where TNC was found to be important for breast cancer cell metastatic colonisation to the lungs as well as maintaining Wnt and Notch signalling in CSCs (Oskarsson et al, 2011). It has been speculated that since POSTN anchors TNC to the ECM, it is possible that in the context of the metastatic niche, these two matricellular proteins collaborate to promote important signalling pathways for CSC survival.
Conclusions and future perspectives
Matricellular proteins have been demonstrated to be key components in several aspects of cancer progression especially in the context of the tumour microenvironment. Since the ECM forms a dynamic component towards mediating tumorigenesis, the abilities of matricellular proteins to modulate protein interactions for various essential receptor-mediated signalling pathways prove critical for facilitating cell survival, proliferation, EMT, invasion and metastasis. In addition, matricullar proteins have been established as directly influential toward coordinating aberrant ECM remodelling, thereby leading to the acquisition of an invasive and subsequently metastatic phenotype. In terms of the metastatic niche, it is becoming increasingly apparent that stromal factors form a supportive foundation for metastatic colonisation and matricellular proteins may play a pivotal role in maintaining signalling cascades for tumour growth. We have summarised the roles of several members of the matricellular family at different stages of tumour progression and present how the functional diversity of these proteins allows them to serve dynamically in advancing tumour progression. Other matricellular proteins such as thrombospondins, CCN family of proteins and galectins may also play similar roles in tumour development. Future opportunities are to gain a better understanding of the underlying molecular mechanisms that direct matricellular protein function in the tumour ECM and metastatic niche in order to implement targeted therapeutic approaches that may prove beneficial in combination with conventional therapies.
References
Arnold SA, Rivera LB, Carbon JG, Toombs JE, Chang CL, Bradshaw AD, Brekken RA (2012) Losartan slows pancreatic tumor progression and extends survival of SPARC-null mice by abrogating aberrant TGFbeta activation. PLoS One 7: e31384
Bao S, Ouyang G, Bai X, Huang Z, Ma C, Liu M, Shao R, Anderson RM, Rich JN, Wang XF (2004) Periostin potently promotes metastatic growth of colon cancer by augmenting cell survival via the Akt/PKB pathway. Cancer Cell 5: 329–339
Barth PJ, Moll R, Ramaswamy A (2005) Stromal remodeling and SPARC (secreted protein acid rich in cysteine) expression in invasive ductal carcinomas of the breast. Virchows Arch 446: 532–536
Beiter K, Hiendlmeyer E, Brabletz T, Hlubek F, Haynl A, Knoll C, Kirchner T, Jung A (2005) beta-Catenin regulates the expression of tenascin-C in human colorectal tumors. Oncogene 24: 8200–8204
Bornstein P, Sage EH (2002) Matricellular proteins: extracellular modulators of cell function. Curr Opin Cell Biol 14: 608–616
Bradshaw AD, Puolakkainen P, Dasgupta J, Davidson JM, Wight TN, Helene Sage E (2003) SPARC-null mice display abnormalities in the dermis characterized by decreased collagen fibril diameter and reduced tensile strength. J Invest Dermatol 120: 949–955
Bramwell VH, Doig GS, Tuck AB, Wilson SM, Tonkin KS, Tomiak A, Perera F, Vandenberg TA, Chambers AF (2006) Serial plasma osteopontin levels have prognostic value in metastatic breast cancer. Clin Cancer Res 12: 3337–3343
Brekken RA, Puolakkainen P, Graves DC, Workman G, Lubkin SR, Sage EH (2003) Enhanced growth of tumors in SPARC null mice is associated with changes in the ECM. J Clin Invest 111: 487–495
Brunner A, Mayerl C, Tzankov A, Verdorfer I, Tschorner I, Rogatsch H, Mikuz G (2004) Prognostic significance of tenascin-C expression in superficial and invasive bladder cancer. J Clin Pathol 57: 927–931
Calvo A, Catena R, Noble MS, Carbott D, Gil-Bazo I, Gonzalez-Moreno O, Huh JI, Sharp R, Qiu TH, Anver MR, Merlino G, Dickson RB, Johnson MD, Green JE (2008) Identification of VEGF-regulated genes associated with increased lung metastatic potential: functional involvement of tenascin-C in tumor growth and lung metastasis. Oncogene 27: 5373–5384
Chaffer CL, Weinberg RA (2011) A perspective on cancer cell metastasis. Science 331: 1559–1564
Chiodoni C, Colombo MP, Sangaletti S (2010) Matricellular proteins: from homeostasis to inflammation, cancer, and metastasis. Cancer Metastasis Rev 29: 295–307
Chlenski A, Cohn SL (2010) Modulation of matrix remodeling by SPARC in neoplastic progression. Semin Cell Dev Biol 21: 55–65
Christofori G (2006) New signals from the invasive front. Nature 441: 444–450
Chung CY, Zardi L, Erickson HP (1995) Binding of tenascin-C to soluble fibronectin and matrix fibrils. J Biol Chem 270: 29012–29017
Clark CJ, Sage EH (2008) A prototypic matricellular protein in the tumor microenvironment--where there's SPARC, there's fire. J Cell Biochem 104: 721–732
Cox TR, Erler JT (2011) Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis Model Mech 4: 165–178
Friedl P, Alexander S (2011) Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 147: 992–1009
Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, Sahai E (2007) Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol 9: 1392–1400
Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK (2011) Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev 91: 1071–1121
Herold-Mende C, Mueller MM, Bonsanto MM, Schmitt HP, Kunze S, Steiner HH (2002) Clinical impact and functional aspects of tenascin-C expression during glioma progression. Int J Cancer 98: 362–369
Huang H, Colella S, Kurrer M, Yonekawa Y, Kleihues P, Ohgaki H (2000) Gene expression profiling of low-grade diffuse astrocytomas by cDNA arrays. Cancer Res 60: 6868–6874
Joyce JA, Pollard JW (2009) Microenvironmental regulation of metastasis. Nat Rev Cancer 9: 239–252
Kalluri R, Weinberg RA (2009) The basics of epithelial-mesenchymal transition. J Clin Invest 119: 1420–1428
Kalluri R, Zeisberg M (2006) Fibroblasts in cancer. Nat Rev Cancer 6: 392–401
Kessenbrock K, Plaks V, Werb Z (2010) Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141: 52–67
Kii I, Nishiyama T, Li M, Matsumoto K, Saito M, Amizuka N, Kudo A (2010) Incorporation of tenascin-C into the extracellular matrix by periostin underlies an extracellular meshwork architecture. J Biol Chem 285: 2028–2039
Kwon YJ, Lee SJ, Koh JS, Kim SH, Kim YJ, Park JH (2009) Expression patterns of aurora kinase B, heat shock protein 47, and periostin in esophageal squamous cell carcinoma. Oncol Res 18: 141–151
Kyutoku M, Taniyama Y, Katsuragi N, Shimizu H, Kunugiza Y, Iekushi K, Koibuchi N, Sanada F, Oshita Y, Morishita R (2011) Role of periostin in cancer progression and metastasis: inhibition of breast cancer progression and metastasis by anti-periostin antibody in a murine model. Int J Mol Med 28: 181–186
Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM (2009) Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139: 891–906
Lu P, Weaver VM, Werb Z (2012) The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol 196: 395–406
Malanchi I, Santamaria-Martinez A, Susanto E, Peng H, Lehr HA, Delaloye JF, Huelsken J (2012) Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 481: 85–89
Maruhashi T, Kii I, Saito M, Kudo A (2010) Interaction between periostin and BMP-1 promotes proteolytic activation of lysyl oxidase. J Biol Chem 285: 13294–13303
McAllister SS, Gifford AM, Greiner AL, Kelleher SP, Saelzler MP, Ince TA, Reinhardt F, Harris LN, Hylander BL, Repasky EA, Weinberg RA (2008) Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell 133: 994–1005
Michaylira CZ, Wong GS, Miller CG, Gutierrez CM, Nakagawa H, Hammond R, Klein-Szanto AJ, Lee JS, Kim SB, Herlyn M, Diehl JA, Gimotty P, Rustgi AK (2010) Periostin, a cell adhesion molecule, facilitates invasion in the tumor microenvironment and annotates a novel tumor-invasive signature in esophageal cancer. Cancer Res 70: 5281–5292
Mueller MM, Fusenig NE (2004) Friends or foes—bipolar effects of the tumour stroma in cancer. Nat Rev Cancer 4: 839–849
Nau GJ, Guilfoile P, Chupp GL, Berman JS, Kim SJ, Kornfeld H, Young RA (1997) A chemoattractant cytokine associated with granulomas in tuberculosis and silicosis. Proc Natl Acad Sci USA 94: 6414–6419
Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK (2000) Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res 60: 2497–2503
Okamura N, Hasegawa M, Nakoshi Y, Iino T, Sudo A, Imanaka-Yoshida K, Yoshida T, Uchida A (2010) Deficiency of tenascin-C delays articular cartilage repair in mice. Osteoarthritis Cartilage 18: 839–848
Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, Madhu B, Goldgraben MA, Caldwell ME, Allard D, Frese KK, Denicola G, Feig C, Combs C, Winter SP, Ireland-Zecchini H, Reichelt S, Howat WJ, Chang A, Dhara M, Wang L, Ruckert F, Grutzmann R, Pilarsky C, Izeradjene K, Hingorani SR, Huang P, Davies SE, Plunkett W, Egorin M, Hruban RH, Whitebread N, McGovern K, Adams J, Iacobuzio-Donahue C, Griffiths J, Tuveson DA (2009) Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324: 1457–1461
Oskarsson T, Acharyya S, Zhang XH, Vanharanta S, Tavazoie SF, Morris PG, Downey RJ, Manova-Todorova K, Brogi E, Massague J (2011) Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med 17: 867–874
O’Connell JT, Sugimoto H, Cooke VG, MacDonald BA, Mehta AI, LeBleu VS, Dewar R, Rocha RM, Brentani RR, Resnick MB, Neilson EG, Zeisberg M, Kalluri R (2011) VEGF-A and Tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proc Natl Acad Sci USA 108: 16002–16007
Prada F, Benedetti LG, Bravo AI, Alvarez MJ, Carbone C, Podhajcer OL (2007) SPARC endogenous level, rather than fibroblast-produced SPARC or stroma reorganization induced by SPARC, is responsible for melanoma cell growth. J Invest Dermatol 127: 2618–2628
Prenzel KL, Warnecke-Eberz U, Xi H, Brabender J, Baldus SE, Bollschweiler E, Gutschow CA, Holscher AH, Schneider PM (2006) Significant overexpression of SPARC/osteonectin mRNA in pancreatic cancer compared to cancer of the papilla of Vater. Oncol Rep 15: 1397–1401
Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR (2012) Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 21: 418–429
Psaila B, Lyden D (2009) The metastatic niche: adapting the foreign soil. Nat Rev Cancer 9: 285–293
Reardon DA, Akabani G, Coleman RE, Friedman AH, Friedman HS, Herndon JE, Cokgor I, McLendon RE, Pegram CN, Provenzale JM, Quinn JA, Rich JN, Regalado LV, Sampson JH, Shafman TD, Wikstrand CJ, Wong TZ, Zhao XG, Zalutsky MR, Bigner DD (2002) Phase II trial of murine (131)I-labeled antitenascin monoclonal antibody 81C6 administered into surgically created resection cavities of patients with newly diagnosed malignant gliomas. J Clin Oncol 20: 1389–1397
Rentz TJ, Poobalarahi F, Bornstein P, Sage EH, Bradshaw AD (2007) SPARC regulates processing of procollagen I and collagen fibrillogenesis in dermal fibroblasts. J Biol Chem 282: 22062–22071
Rios H, Koushik SV, Wang H, Wang J, Zhou HM, Lindsley A, Rogers R, Chen Z, Maeda M, Kruzynska-Frejtag A, Feng JQ, Conway SJ (2005) periostin null mice exhibit dwarfism, incisor enamel defects, and an early-onset periodontal disease-like phenotype. Mol Cell Biol 25: 11131–11144
Rizzieri DA, Akabani G, Zalutsky MR, Coleman RE, Metzler SD, Bowsher JE, Toaso B, Anderson E, Lagoo A, Clayton S, Pegram CN, Moore JO, Gockerman JP, DeCastro C, Gasparetto C, Chao NJ, Bigner DD (2004) Phase 1 trial study of 131I-labeled chimeric 81C6 monoclonal antibody for the treatment of patients with non-Hodgkin lymphoma. Blood 104: 642–648
Robert G, Gaggioli C, Bailet O, Chavey C, Abbe P, Aberdam E, Sabatie E, Cano A, Garcia de Herreros A, Ballotti R, Tartare-Deckert S (2006) SPARC represses E-cadherin and induces mesenchymal transition during melanoma development. Cancer Res 66: 7516–7523
Roberts DD (2011) Emerging functions of matricellular proteins. Cell Mol Life Sci 68: 3133–3136
Rocco M, Malorni L, Cozzolino R, Palmieri G, Rozzo C, Manca A, Parente A, Chambery A (2011) Proteomic profiling of human melanoma metastatic cell line secretomes. J Proteome Res 10: 4703–4714
Sarkar S, Nuttall RK, Liu S, Edwards DR, Yong VW (2006) Tenascin-C stimulates glioma cell invasion through matrix metalloproteinase-12. Cancer Res 66: 11771–11780
Sasaki H, Dai M, Auclair D, Fukai I, Kiriyama M, Yamakawa Y, Fujii Y, Chen LB (2001) Serum level of the periostin, a homologue of an insect cell adhesion molecule, as a prognostic marker in nonsmall cell lung carcinomas. Cancer 92: 843–848
Shevde LA, Das S, Clark DW, Samant RS (2010) Osteopontin: an effector and an effect of tumor metastasis. Curr Mol Med 10: 71–81
Shi Q, Bao S, Song L, Wu Q, Bigner DD, Hjelmeland AB, Rich JN (2007) Targeting SPARC expression decreases glioma cellular survival and invasion associated with reduced activities of FAK and ILK kinases. Oncogene 26: 4084–4094
Sivasankaran B, Degen M, Ghaffari A, Hegi ME, Hamou MF, Ionescu MC, Zweifel C, Tolnay M, Wasner M, Mergenthaler S, Miserez AR, Kiss R, Lino MM, Merlo A, Chiquet-Ehrismann R, Boulay JL (2009) Tenascin-C is a novel RBPJkappa-induced target gene for Notch signaling in gliomas. Cancer Res 69: 458–465
Skonier J, Neubauer M, Madisen L, Bennett K, Plowman GD, Purchio AF (1992) cDNA cloning and sequence analysis of beta ig-h3, a novel gene induced in a human adenocarcinoma cell line after treatment with transforming growth factor-beta. DNA Cell Biol 11: 511–522
Stylianopoulos T, Martin JD, Chauhan VP, Jain SR, Diop-Frimpong B, Bardeesy N, Smith BL, Ferrone CR, Hornicek FJ, Boucher Y, Munn LL, Jain RK (2012) Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc Natl Acad Sci USA 109: 15101–15108
Takafuji V, Forgues M, Unsworth E, Goldsmith P, Wang XW (2007) An osteopontin fragment is essential for tumor cell invasion in hepatocellular carcinoma. Oncogene 26: 6361–6371
Takeshita S, Kikuno R, Tezuka K, Amann E (1993) Osteoblast-specific factor 2: cloning of a putative bone adhesion protein with homology with the insect protein fasciclin I. Biochem J 294 (Part 1): 271–278
Tavazoie SF, Alarcon C, Oskarsson T, Padua D, Wang Q, Bos PD, Gerald WL, Massague J (2008) Endogenous human microRNAs that suppress breast cancer metastasis. Nature 451: 147–152
Tilli TM, Franco VF, Robbs BK, Wanderley JL, da Silva FR, de Mello KD, Viola JP, Weber GF, Gimba ER (2011) Osteopontin-c splicing isoform contributes to ovarian cancer progression. Mol Cancer Res 9: 280–293
Tilli TM, Mello KD, Ferreira LB, Matos AR, Accioly MT, Faria PA, Bellahcene A, Castronovo V, Gimba ER (2012) Both osteopontin-c and osteopontin-b splicing isoforms exert pro-tumorigenic roles in prostate cancer cells. Prostate 72 (15): 1688–1699
Tuck AB, Hota C, Wilson SM, Chambers AF (2003) Osteopontin-induced migration of human mammary epithelial cells involves activation of EGF receptor and multiple signal transduction pathways. Oncogene 22: 1198–1205
Vetrone SA, Montecino-Rodriguez E, Kudryashova E, Kramerova I, Hoffman EP, Liu SD, Miceli MC, Spencer MJ (2009) Osteopontin promotes fibrosis in dystrophic mouse muscle by modulating immune cell subsets and intramuscular TGF-beta. J Clin Invest 119: 1583–1594
Wikman H, Kettunen E, Seppanen JK, Karjalainen A, Hollmen J, Anttila S, Knuutila S (2002) Identification of differentially expressed genes in pulmonary adenocarcinoma by using cDNA array. Oncogene 21: 5804–5813
Wong GS, Habibollahi P, Heidari P, Lee JS, Klein-Szanto AJ, Waldron TJ, Gimotty P, Nakagawa H, Taylor PR, Wang TC, Mahmood U, Rustgi AK (2012) Optical imaging of periostin enables early endoscopic detection and characterization of esophageal cancer in mice. Gastroenterology pii: S0016-5085 (12): 01546–01546
Yan W, Shao R (2006) Transduction of a mesenchyme-specific gene Periostin into 293 T cells induces cell invasive activity through epithelial-mesenchymal transformation. J Biol Chem 28: 19700–19708
Zhu M, Saxton RE, Ramos L, Chang DD, Karlan BY, Gasson JC, Slamon DJ (2011) Neutralising monoclonal antibody to periostin inhibits ovarian tumour growth and metastasis. Mol Cancer Ther 10: 1500–1508
Acknowledgements
This work was supported by NIH/NCI P01-CA098101 (GSW, AKR), NIH T32-CA115299 (GSW), NIH/NCI U01-CA14305603 (GSW, AKR) and NIH/NIDDK Center for Molecular Studies in Digestive and Liver Diseases and its Core facilities-Molecular Pathology, Molecular Biology/Gene Expression, Cell Culture and Mouse (P30-DK050306) and American Cancer Society (RP-10-033-01-CCE). We are grateful to other members of the Rustgi lab for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Wong, G., Rustgi, A. Matricellular proteins: priming the tumour microenvironment for cancer development and metastasis. Br J Cancer 108, 755–761 (2013). https://doi.org/10.1038/bjc.2012.592
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2012.592
Keywords
This article is cited by
-
A concise review on miRNAs as regulators of colon cancer stem cells and associated signalling pathways
Clinical and Translational Oncology (2023)
-
SMOC2 promotes an epithelial-mesenchymal transition and a pro-metastatic phenotype in epithelial cells of renal cell carcinoma origin
Cell Death & Disease (2022)
-
Time-series biological responses toward decellularized bovine tendon graft and autograft for 52 consecutive weeks after rat anterior cruciate ligament reconstruction
Scientific Reports (2022)
-
Transcription factor Runx2 changes the expression of some matricellular proteins in metastatic breast cancer cells
Molecular Biology Reports (2022)
-
Antitumor effect of 4MU on glioblastoma cells is mediated by senescence induction and CD44, RHAMM and p-ERK modulation
Cell Death Discovery (2021)
