
Abstract
Radioactive iodine (RAI) is a key therapeutic modality for thyroid cancer. Loss of RAI uptake in thyroid cancer inversely correlates with patient’s survival. In this review, we focus on the challenges encountered in delivering sufficient doses of I-131 to eradicate metastatic lesions without increasing the risk of unwanted side effects. Sodium iodide symporter (NIS) mediates iodide influx, and NIS expression and function can be selectively enhanced in thyroid cells by thyroid-stimulating hormone. We summarize our current knowledge of NIS modulation in normal and cancer thyroid cells, and we propose that several reagents evaluated in clinical trials for other diseases can be used to restore or further increase RAI accumulation in thyroid cancer. Once validated in preclinical mouse models and clinical trials, these reagents, mostly small-molecule inhibitors, can be readily translated into clinical practice. We review available genetically engineered mouse models of thyroid cancer in terms of their tumor development and progression as well as their thyroid function. These mice will not only provide important insights into the mechanisms underlying the loss of RAI uptake in thyroid tumors but will also serve as preclinical animal models to evaluate the efficacy of candidate reagents to selectively increase RAI uptake in thyroid cancers. Taken together, we anticipate that the optimal use of RAI in the clinical management of thyroid cancer is yet to come in the near future.
Similar content being viewed by others


Introduction
The ability of thyroid follicular cells to concentrate iodine allows the use of radioactive iodine (RAI) to ablate post-surgical thyroid remnants and to eradicate residual, recurrent, and metastatic thyroid cancer cells. Thyroidal RAI accumulation is mainly contributed by Na+/I− symporter (NIS)-mediated iodide influx [1, 2]. Since NIS expression is often reduced in malignant thyroid tissues [3], much effort has been focused on studying NIS modulation in thyroid cells with the hope that NIS expression and function can be restored and further enhanced in thyroid cancer cells. Accordingly, most RAI administered would be delivered to targeted thyroid cancers to ensure the efficacy of RAI therapy with minimal RAI-induced toxicity in non-targeted tissues.
Recently, several excellent reviews were published to summarize advances made in NIS molecular characterization and regulation in detail [4–6]. In addition, Spitzweg et al. [7] wrote an excellent review focusing on NIS deregulation in thyroid cancer and therapeutic potential of NIS restoration in advanced thyroid cancer patients. In this mini-review, we list clinical issues that remain to be addressed for current I-131 therapy, in particular, the challenge of delivering sufficient I-131 dose to targeted metastatic lesions without increasing the risk of unwanted side effects. Based on current knowledge of NIS modulation in normal and cancer thyroid cells, we list several reagents in clinical trials for other diseases may selectively increase thyroidal RAI accumulation. We summarize genetically engineered mouse models that lead to various types of thyroid cancer. These mice will serve to reveal the mechanisms underlying the loss of RAI uptake in thyroid tumors and will also serve to evaluate the efficacy of candidate reagents to selectively increase RAI uptake in thyroid cancers.
Radioiodine Ablation and Therapy for Differentiated Thyroid Cancer
For patients with differentiated thyroid cancer, the benefit of administering I-131 to ablate remnants of normal thyroid tissue and/or to target residual or metastatic lesions must also take into consideration the risk of I-131-induced damages in non-targeted tissues.
RAI Ablation for Thyroid Remnants
For patients who had complete surgical resection without distant metastatic disease, RAI ablation for thyroid remnants can ensure accuracy of tumor staging and facilitate follow-up [8]. Post-ablation whole-body I-131 scintigraphy may identify undiagnosed lesions resulting in a change in tumor staging that may have an impact on clinical management of the disease. The absence of thyroid remnants allows the use of serum thyroglobulin (Tg) measurement for early detection of recurrent disease. For patients who are cured by surgery and are at low risk for recurrence, the clinical benefit of RAI remnant ablation is limited and is not recommended. For patients who have gross extra-thyroidal extension, incomplete tumor resection, or distant metastasis, RAI ablation for a thyroid remnant is routinely recommended as these patients are likely to have undiagnosed lesions and are at high risk for recurrence. However, one cannot always be certain of risk assessment based on the initial presentation of the disease, and the prognosis of disease may change over time depending on their responsiveness to ongoing therapy. Thus, risk reassessment should be conducted periodically for all patients.
RAI Therapy for Suspected or Known Metastatic Thyroid Cancer Lesions
I-131 has been proven to be effective in decreasing recurrence rate and in improving overall survival for thyroid cancer patients who had gross extra-thyroidal extension or distant metastasis [9, 10]. Patients of young age, who have small metastatic lesions with significant I-131 uptake, can be cured with a few doses of I-131 after thyroidectomy. However, patients of older age who have large metastatic lesions with absent or insufficient I-131 uptake do not benefit from I-131 therapy. Some of these patients may benefit from I-131 therapy if I-131 uptake can be restored and enhanced in their metastatic lesions. However, metastatic lesions with evident I-131 uptake are not always responsive to I-131 therapy. The efficacy of I-131 therapy is inversely related to the size of metastatic lesions, and the underlying mechanisms are not well elucidated except that hypoxia in bulky tumors may account for RAI resistance. Without conducting lesion dosimetry to determine how much radiation each metastatic lesion will receive (NCT00673010) [11], it is difficult to distinguish lesions that are not responsive to I-131 from lesions that do not receive sufficient radioactivity of I-131. For metastatic lesions not responsive to I-131, co-treatment with radiosensitizers may be beneficial.
RAI-Induced Damage in Non-thyroidal Tissues
NIS is expressed not only in thyroid follicular cells but also in salivary striated ducts, lactating breast, gastric mucosa, lacrimal ducts, etc. Accordingly, these NIS-expressing tissues as well as I-131-handling organs are subjected to I-131-induced damage. The side effects of I-131 therapy include temporary or permanent salivary gland dysfunction, temporary GI upset, lacrimal duct obstruction, gonadal dysfunction, and possible secondary malignancy. Among these side effects, many I-131-treated thyroid cancer survivors suffer from lifelong morbidity of I-131-induced salivary gland dysfunction, including recurrent sialadenitis, persistent xerostomia, and progressive susceptibility to dental caries and periodontal diseases. More than half-million people are living with thyroid cancer in the USA, and many of these patients are at risk to suffer from newly developed or worsening I-131-induced salivary gland dysfunction. Accordingly, prevention strategies for I-131-induced salivary gland dysfunction are warranted. Finally, continued I-131 therapy is not recommended for patients who have received a cumulative dose of I-131 greater than 600 mCi.
Clinical Questions Regarding I-131 Therapy
In the 2009 revised American Thyroid Association guidelines for patients with thyroid nodules and differentiated thyroid cancer [8], a series of clinically relevant questions regarding the use of RAI for patients with differentiated thyroid cancer were identified and evidence-based recommendations were made. One of these major issues is patient selection, i.e., who would benefit from I-131 ablation and therapy. This issue involves multiple factors that are beyond the scope of this review. The other major issue is to deliver sufficient doses of I-131 to eradicate targeted lesions without increasing the risk of unwanted side effects. This issue can be addressed if selective enhancement of RAI uptake in targeted lesions can be achieved.
Modulation of NIS-Mediated Iodide Influx, Iodide Efflux, and Iodide Organification in Thyroid Cells
The extent of RAI accumulation in thyroid follicular cells is determined by NIS-mediated iodide influx, iodide efflux, and iodide organification (reviewed in [12]). NIS expression and function are mainly modulated at transcriptional and post-translational levels. Mutations in the NIS gene do not appear to be a major cause for reduced NIS expression/function in thyroid cancers. A mutation in NIS gene (A581G) was found only in one patient, and a homozygous deletion was found in another patient from the TCGA database of 399 papillary thyroid cancer (PTC) samples [13, 14]. Currently, not much is known whether NIS modulation occurs at messenger RNA (mRNA) stability or translational level in thyroid cells. Studies on transcription factors that bind to NIS promoter and/or enhancer are summarized in Table 1. Various reagents that selectively increase thyroidal RAI uptake and the underlying mechanistic actions are summarized in Table 2. Note that NIS regulation studies were mostly conducted with normal thyroid cells and sometimes verified by restoration in thyroid cancer cells.
Transcription Factors That Bind to NIS Promoter and/or Enhancer
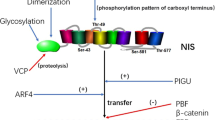
Thyroid-stimulating hormone (TSH), secreted by the pituitary gland, is the main regulator of NIS transcription in normal thyroid cells. TSH not only stimulates NIS proximal promoter (NIS_PP) activity [15] but also stimulates the NIS upstream enhancer (NUE) activity [16, 17]. Based on consensus motifs in NIS_PP and/or NUE, along with electrophoretic mobility shift assays (EMSA) using nuclear extracts of thyroid cells and chromatin immunoprecipitation (ChIP) assays, several transcription factors binding to NIS_PP or NUE were identified. Thyroid transcription factor-1 (TTF-1) was shown [15] and hairy and enhancer of split-1 (Hes-1) was predicted [18] to bind to NIS_PP, and both the transcription factors increased NIS_PP activity by luciferase reporter assay. The paired domain transcription factor-8 (Pax-8) [16], cAMP-response element binding protein (CREB) [17], β-catenin [19], and forkhead transcription factor (FoxE1) [20] were shown to bind to NUE and to enhance NUE activity by luciferase reporter assay. Sterol regulatory element binding proteins (SREBPs) [21] were shown to bind to NIS 5′UTR and to enhance NIS promoter activity by luciferase reporter assay. The pituitary tumor transforming gene (PTTG) binds to NUE and PTTG binding factor (PBF) binds to both NUE and NIS_PP, yet both repress NUE/NIS_PP activity by luciferase reporter assay [22]. TTF-1, CREB, Pax-8, β-catenin, Hes-1, SREBPs, and FoxE1 modulate NIS_PP or NUE activity in a TSH-dependent manner, yet PTTG and PBF modulate NIS_PP or NUE activity in a TSH-independent manner.
Among these transcription factors, forced expression of TTF-1 and Pax-8 by adenoviral vector results in an increased NIS mRNA/protein levels as well as NIS-mediated RAI uptake in K-1 and F133 thyroid cancer cells [23]. Forced expression of Hes-1 increased NIS mRNA in FRTL-5 thyroid cells and WRO thyroid cancer cells [18], and NIS protein level was found to be decreased in Hes-1−/− mouse thyroid cells [24]. SREBP small interfering RNA (siRNA) or SREBP maturation inhibitor, 25-HC, decreased NIS protein levels, and 25-HC decreased RAI uptake in FRTL-5 thyroid cells [21]. FoxE1 siRNA decreased NIS mRNA and protein in PCCl3 thyroid cells [20]. The effects of CREB and β-catenin on endogenous NIS mRNA/protein levels have not been evaluated.
Reagents Known to Increase Thyroidal NIS mRNA/Protein Levels
In addition to recombinant human TSH (rhTSH) [25–28], several reagents have been shown to increase NIS mRNA/protein levels. Many of them target signaling nodes known to participate in the development and progression of thyroid cancers, most of which have reduced NIS expression. Specifically, inhibitors for MEK [29–31], PI3K [32, 33], BRAF [31], HDAC [34–38], and TGF-β [39] are shown to increase thyroid NIS expression, and these reagents are in clinical trials for various diseases. Accordingly, the use of these reagents to further enhance TSH-stimulated thyroidal RAI uptake to facilitate I-131 therapy for thyroid cancer patients could be imminent, if their effects are validated in preclinical animal models and clinical trials. Ferretti et al. reported that thyroidal NIS expression was increased by Notch activation [18]. Indeed, Notch-1 activators, resveratrol and hesperetin, increased NIS expression in HTh7 and 8505C thyroid cancer cell lines [40, 41], and resveratrol increases RAI uptake in FRTL-5 thyroid cells [42].
It is interesting to note that signaling nodes targeted by these reagents modulate many transcription factors listed in Table 1. Indeed, Pax-8 is decreased via the TGF-β-SMAD3 pathway in PCCl3 thyroid cells carrying BRAF(V600E) mutation [43, 39] and is also decreased in thyroid cells carrying RET/PTC rearrangement [44]. NIS expression induced by Notch-1 activators in anaplastic thyroid cancer cells is likely in part mediated by increased expression of TTF-1, Pax-8, and Hes-1 [18, 40, 41]. PI3K inhibitors [32] and BRAF inhibitors [43] increase NIS expression likely by increasing Pax-8 levels. HDAC inhibitors increase NIS expression likely by increasing TTF-1 levels [34]. MEK inhibitors increase NIS expression likely by increasing Pax-8 levels [29, 30] and TTF-1 levels [30]. Finally, almost all transcription factors that bind to NIS promoter/enhancer are modulated by rhTSH.
NIS Post-translational Modifications and NIS-Associated Proteins Known to Modulate NIS Protein Stability, Cell Surface Trafficking, and Iodide Influx Velocity
NIS expression and function can be modulated by NIS post-translational modifications and NIS-associated proteins. TSH increases NIS phosphorylation, protein stability, and cell surface trafficking in FRTL-5 thyroid cells [28]. In addition to decreasing NIS expression, TGF-β most likely also decreases NIS protein stability. NIS protein level was decreased by 43 % upon 12-h treatment of TGF-β in PCCl3 thyroid cells under chronic TSH stimulation, where the half-life of NIS protein is 5 days in the absence of TGF-β (Lakshmanan and Jhiang, unpublished data). We reported that Akt inhibitor alone [33] or in combination with apigenin [45] increased iodide influx rate without increasing cell surface NIS protein levels.
NIS phosphorylation sites and NIS-associated proteins were identified by exogenous NIS expression in non-thyroid cells. Five in vivo phosphorylation sites have been identified in exogenously expressed rat NIS in HEK-293 cells [46]. The phosphorylation status of Ser-227 did not alter NIS expression or function. Thr-49 appears to be critical for proper NIS conformation as both phospho-mimic and phospho-defective mutants decreased RAI uptake. Phosphorylation of Ser-43 or Ser-581 is essential for NIS-mediated RAI uptake without affecting total or cell surface NIS protein levels. Kinetic studies indicated that Ser-43 and Ser-581 phospho-defective NIS mutants had decreased iodide influx rate. Ser-43 was conserved in human NIS, and its phosphorylation is also critical for hNIS activity. Interestingly, we noted that NIS mobility was shifted upwards in SDS-denatured polyacrylamide gel electrophoresis upon combination treatment of Akt inhibitor and apigenin in PCCl3 rat thyroid cells [45]. The nature of this mobility shift in NIS remains unclear and it may be due to a post-translational modification. Thr-577 site has been implicated in NIS protein stability, as phospho-defective NIS mutant protein was completely degraded, yet phospho-mimic NIS mutant protein level was comparable to wild-type NIS protein. Indeed, we found that exogenously expressed NIS is associated with ubiquitin in HEK-293 cells [47]. In addition, MEK inhibitors lead to lysosome-mediated NIS protein degradation in trans-retinoic acid/hydrocortisone-treated MCF-7 breast cancer cells [48].
In addition to acting as a repressor for NIS transcription [22], PBF may also play a role in NIS cell surface trafficking [49]. NIS and PBF complex formation was demonstrated by a pull-down assay as well as co-immunoprecipitation of NIS and PBF exogenously expressed in COS-7 cells. Cell surface localization of exogenously expressed NIS in COS-7 cells was decreased by co-expression of exogenous PBF. However, the role of PBF on NIS cell surface trafficking in physiological or pathological conditions is yet to be demonstrated in thyroid cells expressing endogenous NIS and PBF. Strong intracellular staining with anti-NIS antibodies has been reported in thyroid cancers [50–52]. It was first proposed that this staining reveals intracellular NIS, but later, it has been demonstrated that the intracellular staining was probably due to non-specific binding of the anti-NIS antibody [53, 54]. Thus, the contribution of impairment in NIS cell surface trafficking in both thyroid cancer and breast cancer remains circumstantial.
Reagents Known to Modulate Iodide Efflux and Iodine Organification
In normal thyroid follicular cells, NIS-mediated iodide influx occurs at the basolateral membrane and iodide efflux occurs at the apical membrane where iodide oxidation and organification occur. These processes are stimulated by TSH and together contribute to prolonged iodine accumulation in the thyroid gland. However, the follicle structure is not maintained in most thyroid cancers; thus, an increase in iodide efflux may not result in an increase but rather a decrease in iodine accumulation in thyroid cancer cells. Indeed, iodide efflux decreased in the presence of an HSP90 inhibitor, 17-AAG, which resulted in an increased RAI accumulation in monolayer-cultured PCCl3 rat thyroid cells as well as PCCl3 cells expressing RET/PTC1 oncoprotein [55]. The possible molecules that mediate iodide efflux in thyroid cells are pendrin, apical iodide transporter, CFTR (reviewed in [56]), and ClC-5 [57], yet the mechanisms underlying the actions of HSP90 inhibitor in decreasing iodide efflux in thyroid cells are yet to be elucidated.
Iodide organification is a unique property of the thyroid gland. Iodine effluxed into the follicular lumen at the apical membrane is oxidized by thyroperoxidase (TPO) using H2O2 produced by the dual oxidase-2 (DUOX-2) and its essential partner dual oxidase-2A (DUOX-2A). Oxidized iodine is then incorporated into tyrosyl residues of Tg, and the iodinated Tg are stored in the follicular lumen as colloid (reviewed in [12]). For monolayer-cultured thyroid cells, and likely most thyroid cancer cells in patients, iodide organification may occur randomly at intracellular locations as the polarity of iodide influx and iodide efflux/organification may no longer exist. Pax-8 is the main transcription factor for Tg and TPO genes, and forced expression of Pax-8 by adenoviral vector in human anaplastic thyroid cancer cell lines, K-1 and F133, resulted in an increase in mRNA and protein levels of Tg and TPO, thereby resulting in an increase in iodide organification and retention [58]. Forced expression of both TTF-1 and Pax-8 further increased RAI uptake in K-1 and F133 cells by increasing NIS, Tg, and TPO expression levels [23]. Depsipeptide, an HDAC inhibitor, also induced iodide organification by increasing NIS, Tg, and TPO expression levels in BHP-7 cells that express high levels of endogenous Pax-8 [34].
NIS Expression and Modulation in Salivary Glands
In human salivary glands, NIS protein was abundantly expressed in striated ducts, expressed at lower levels in excretory ducts, but not in acinar cells [59, 60]. No immortalized salivary ductal cells maintain endogenous NIS expression; thus, NIS modulation in salivary ductal cells is not well studied. Based on NIS immunohistochemical staining, NIS protein levels are decreased in inflamed or malignant salivary glands. The mechanisms underlying the transition of NIS expression from intercalated ducts (no NIS expression) to striated ducts (high NIS expression) and to excretory ducts (low NIS expression) remain to be elucidated. While the parotid gland is the largest salivary gland in humans, the submandibular gland is the largest in mice. In addition, the submandibular gland in mouse contains granular convoluted ducts that do not exist in the human salivary duct system. The convoluted ducts are located between intercalated ducts and striated ducts. NIS expression level in convoluted ducts is lower than that in striated ducts but is higher than excretory ducts (La Perle and Jhiang, unpublished observation). Male mouse salivary glands have significantly higher NIS-mediated radioisotope accumulation than females [61], as NIS-expressing convoluted duct is larger and more prominent in male mice. Finally, TSH does not modulate NIS expression and iodide is not organified in the salivary gland.
Genetically Engineered Mouse Models of Thyroid Cancer
Several lines of genetically modified mice have been created that resemble human papillary, follicular, and anaplastic thyroid cancers. The development of these in vivo models has provided valuable insights into the effects of different mutations that lead to various types of thyroid cancer and has allowed us to examine radioiodine uptake and retention in various tumor stages. Available genetically engineered mouse models of thyroid cancer and their thyroid function status are summarized in Table 3.
Genetically Engineered Mouse Models of Papillary Thyroid Cancer
RTK rearrangements, such as RET or TRK, and the BRAF(V600E) mutation account for the driver mutations for most PTCs in humans. Mice with thyroid-targeted expression of RET/PTC1 [62, 63], RET/PTC3 [64], TRK-T1 [65], or BRAF(V600E) [31, 66, 67] developed PTCs, yet lymph node metastasis was rare and distant metastasis was not found. Since BRAF(V600E) mutation was detected in 40–50 % of human PTCs and was associated with progressive disease [68], three different BRAF(V600E) mouse models were established. In bTg-BRAF(V600E) mice, BRAF(V600E) was overexpressed in the thyroid gland at embryonic stages when the bTg promoter becomes active [67]. In bTgCreER:BRAF(V600E) mice, BRAF(V600E) knock-in allele was induced by tamoxifen in thyroid glands when mice were at 1 month of age [66]. In bTg-rtTA:tetO-BRAF(V600E) mice, BRAF(V600E) overexpression in thyroid gland is induced by doxycycline [31]. PTCs were progressed to anaplastic thyroid cancer (ATC) when the bTgCreER:BRAF(V600E) mouse model was crossed with thyroid-targeted PTEN−/− or PI3KCAH1047R mouse models [69], in which both Raf/MEK and PI3K signaling are overactivated in their thyroid gland. PTCs were progressed to ATC with distant metastasis when bTg-RET/PTC1 [70] or hTPOCreER:BRAF(V600E) [71] mouse models were crossed with a p53−/− mouse model, indicating that p53 loss is needed for metastatic spread of PTC. Finally, the latency of PTC development in the bTg-TRK-T1 mouse model was shortened by crossing it with a p27−/− mouse model [72].
Similar to human PTCs, thyroid tumors that developed in mouse models of PTC had decreased expression of thyroid differentiation genes. It is of interest to note that all PTC mouse models that were examined for serum TSH levels had increased serum TSH levels [31, 63, 66, 67, 71]. This is in contrast to PTC patients, who are euthyroid. The difference can be attributed to the fact that almost all thyroid follicular cells are expressing the oncogene, leading to a tissue-wide de-differentiation effect in the thyroids of these mouse models. In patients with PTC, oncogenes are expressed only in tumor foci, but not in the surrounding normal thyroid follicular cells. Furthermore, oncogene expression in thyroid follicular cells of mouse models can be dynamic in nature. For example, in bTg-BRAF(V600E) mice, BRAF(V600E) de-differentiation effects can diminish the activity of the bTg promoter such that BRAF(V600E) expression itself is reduced [67]. Concomitantly, increased serum TSH levels, due to thyroid de-differentiation, can further enhance the bTg promoter if the thyroid follicular cells have intact TSHR-mediated signaling pathways. Taken together, the level of BRAF(V600E) driven by the bTg promoter in a given thyroid follicular cell is determined by the equilibrium between the effects of BRAF(V600E)-driven de-differentiation and the cell’s responsiveness to increased serum TSH levels. However, in bTgCre:BRAF(V600E) mice, once BRAF(V600E) knock-in allele was established in thyroid follicular cells by bTg-driven Cre expression; bTg promoter activity became irrelevant [66]. Regardless of the various BRAF(V600E) mouse models, the differentiation status in any given thyroid follicular cell is dictated by the equilibrium between BRAF(V600E) de-differentiation effects and TSH differentiation effects. The fact that all BRAF(V600E) mouse models have elevated TSH level indicates that BRAF(V600E) de-differentiation effects are dominant over TSH differentiation effects.
Genetically Engineered Mouse Models of Follicular Thyroid Cancer
TRβPV/PV mice [73], as well as thyroid-targeted PRKAR1A−/− [74], PTEN−/− [75], or NRAS(Q61K) [76] mice, developed follicular thyroid cancer (FTC) with varying penetrance. Except PRKAR1A−/− mice, all other mouse models had several mice develop lung metastasis. TRβPV/PV mice had elevated T4 and TSH levels due to thyroid hormone resistance. In contrast to mouse models of PTC characterized with increased serum TSH levels, all FTC mouse models except bTg-NRAS(Q61K) mouse model had decreased serum TSH levels due to an increase of T4 levels. Accordingly, signaling deregulation (mainly cAMP and PI3K) that leads to FTC development does not seem to greatly interfere with thyroid differentiation such that increased proliferation of thyroid follicular cells can still lead to sufficient or increased T4 production. Furthermore, signaling deregulation leading to FTC development seems to be permissive for distant metastasis to occur. The latency of FTC development in TRβPV/PV mice was shortened by crossing to PPARγ+/− mice [77] or PTEN+/− mice [78]. The thyroid-targeted PRKAR1A−/−:PTEN−/− mouse model had a shorter latency for FTC development with 100 % penetrance of FTC, and 27 % of the mice developed lung metastasis [79]. Remarkably, 100 % of the thyroid-targeted PTEN−/−:KRASG12D mice developed FTC and lung metastasis by 3 months of age [80]. FTCs were progressed to ATC with lung or liver metastasis when the thyroid-targeted PTEN−/− mouse model was crossed with the thyroid-targeted p53−/− mouse model [81].
In Vivo Imaging of Thyroidal RAI Uptake and Retention in Mice
Thyroidal RAI accumulation is contributed by RAI uptake and RAI retention. Micro-SPECT [61, 82, 83] or micro-PET imaging allows non-invasive quantification of thyroidal RAI uptake as well as RAI retention. Ultrasound imaging allows non-invasive measurement of thyroid volume such that thyroidal RAI uptake can be normalized by anatomic volume. At 1 h post-RAI injection (t1), when blood circulating level of RAI remains high, thyroidal RAI uptake is mostly contributed by the equilibrium between NIS-mediated RAI influx and RAI efflux. At 24 h post-RAI injection (t24), when most blood circulating level of RAI is eliminated by urinary excretion, thyroidal RAI accumulation is contributed by both RAI uptake and subsequent retention by RAI organification. Accordingly, RAI retention rate can be defined as % injected dose (ID) at t24 divided by %ID at t1 [83]. With ultrasound and SPECT or PET imaging, thyroid tumor progression can be monitored non-invasively and can be defined by sudden increase in tumor size and/or abrupt decrease in RAI uptake and retention. Different from isolated thyroid cultured cells, RAI uptake and retention in thyroid tumor of live animals are most likely also influenced by local factors in surrounding microenvironments as well as by dynamic interactions with systemic cytokines, hormones, etc. Thus, the extent of increase in thyroidal RAI uptake per anatomic volume and RAI retention rate upon treatment of selected reagents could be investigated and compared at distinct tumor stages.
Reduced Thyroidal RAI Accumulation in Thyroid Cancer Mouse Models
In a doxycycline-induced BRAF(V600E) mouse model, the mice became hypothyroid within 2 days of doxycycline administration. NIS, Tg, and TPO expression levels were almost completely abolished upon a 1-week induction of BRAF(V600E). The expression levels of TSHR, TTF-2, and Pax-8 were also greatly reduced. I-124 accumulation in the thyroid was minimal in these mice, despite several hundredfold increases in serum TSH levels. This finding indicates that BRAF(V600E)-expressing thyroid tumors were not responsive to elevated TSH levels. However, I-124 accumulation in the thyroid, as well as expression of thyroid-differentiated genes, was extensively recovered after doxycycline withdrawal for 1 week, suggesting that BRAF or MEK inhibitors may restore thyroidal iodine accumulation in BRAF(V600E)-expressing tumors. Indeed, thyroidal I-124 accumulation was considerably enhanced after 1 week of administration of BRAF or MEK inhibitors in the continued presence of doxycycline. The dosing schedule of the MEK inhibitor was critical as 2 weeks treatment with 25 mg/kg once a day did not restore thyroid function, yet 6 days treatment with 12.5 mg/kg twice a day did. This suggests that a sustained pERK inhibition is more important than the extent of pERK inhibition [31]. All PTC mouse models had elevated TSH levels, indicating that differentiation status of the thyroid, and thus thyroidal RAI accumulation, is extremely sensitive to RTK/BRAF/MEK-activated pathway. Consequently, MEK inhibitors could be applied to further enhance TSH-stimulated RAI accumulation in PTC mouse models.
All FTC mouse models had normal or elevated T4 levels, indicating that thyroidal RAI accumulation was much less compromised by the signaling that leads to FTC development. For hTPOCre:PTEN−/− mice [75], expression levels of NIS, TPO, and Tg were only decreased by about 50 % in the thyroids of young mice compared to those of wild-type mice. NIS and Tg levels were slightly changed or decreased to varying degrees among FTCs examined. In comparison, TPO expression in FTCs was comparable to the thyroids of wild-type mice. For mice with FTCs that had decreased thyroidal RAI accumulation, PI3K inhibitors may be effective in restoring or further enhancing TSH-stimulated thyroidal RAI accumulation.
Mouse models of ATC that progress from PTC or FTC are available [69–71, 81]. It would be of great interest to investigate whether or not selected reagents could restore the loss of thyroidal RAI accumulation. With state-of-the-art mouse imaging modalities, the extent of increase in thyroidal RAI uptake per anatomic volume and RAI retention rate upon treatment of selected reagents could be investigated and compared at distinct tumor stages.
Conclusion Remarks and Future Research Direction
RAI is a key therapeutic modality in thyroid cancer. Loss of RAI uptake inversely correlates with survival. For patients with RAI refractory disease, there are few treatment options, as these tumors are generally resistant to external radiation and conventional chemotherapy [84]. To this date, no novel treatment has been shown to improve overall survival despite improved progression-free survival in some patients with RAI refractory disease [85]. The side effects of I-131 therapy are much more tolerable than external radiation, conventional chemotherapy, or small-molecule inhibitors. Accordingly, strategies to restore and enhance thyroidal RAI accumulation for patients with RAI refractory disease are of great clinical importance. Indeed, a recent success of using MEK inhibitors to enhance RAI uptake in advanced thyroid cancer is most encouraging [86].
We have summarized transcription factors reported to modulate NIS expression as co-activators or co-repressors. In addition, we have listed several reagents evaluated in clinical trials for other diseases as possible candidates to enhance thyroidal RAI accumulation by increasing NIS expression/function, decreasing iodide efflux rate, or increasing iodine organification. For transcription factors acting as co-activators for NIS expression, thyroid-targeted forced expression by viral or non-viral vectors remains challenging. For transcription factors acting as co-repressors, it is considered undruggable by conventional drug discovery methods. However, Liu and Altman recently describe a novel computational algorithm, DrugFEATURE, to precisely calculate target druggability and predict candidate drug or fragment leads [87]. Small interfering RNAs (siRNAs) could be used to knockdown molecular targets repressing NIS expression or function. Furthermore, microRNAs (miRs) or anti-miRs may also serve as possible candidates to further enhance TSH-stimulated RAI accumulation in thyroid cancer cells if critical miRs that modulate NIS expression or function are identified. However, thyroid-targeted delivery of siRNAs or miRs continues to be a major obstacle.
The fact that several reagents that are being evaluated in clinical trials for other types of cancer may restore or further enhance TSH-stimulated RAI accumulation in thyroid cancer is most exciting. If validated, these reagents could be readily translated to clinical practice, as their pharmacokinetics and toxicity profiles are favorable in humans. Various genetically engineered mouse models of thyroid cancer predisposed by mutations found in patients with PTC or FTC may provide insights into the selection of appropriate reagents based on their driver mutations. Since signaling context in normal thyroid tissues is quite different from that in malignant thyroid tumors, strategies to increase efficacy of RAI ablation for thyroid remnants may be different from those of RAI therapy for metastatic lesions. Finally, I-131-induced salivary gland dysfunction could be prevented if salivary NIS expression could be temporarily shut down during 24–48 h post-I-131 administration when blood-circulating I-131 is high. Taken together, we anticipate that the optimal use of RAI in the clinical management of thyroid cancer is yet to come in the near future.
References
Dai G, Levy O, Carrasco N (1996) Cloning and characterization of the thyroid iodide transporter. Nature 379(6564):458–460
Smanik PA, Liu Q, Furminger TL, Ryu K, Xing S, Mazzaferri EL, Jhiang SM (1996) Cloning of the human sodium iodide symporter. Biochem Biophys Res Commun 226(2):339–345
Lazar V, Bidart JM, Caillou B, Mahe C, Lacroix L, Filetti S, Schlumberger M (1999) Expression of the Na+/I- symporter gene in human thyroid tumors: a comparison study with other thyroid-specific genes. J Clin Endocrinol Metab 84(9):3228–3234
Darrouzet E, Lindenthal S, Marcellin D, Pellequer JL, Pourcher T (2014) The sodium/iodide symporter: state of the art of its molecular characterization. Biochim Biophys Acta 1838(1 Pt B):244–253
Kogai T, Brent GA (2012) The sodium iodide symporter (NIS): regulation and approaches to targeting for cancer therapeutics. Pharmacol Ther 135(3):355–370
Portulano C, Paroder-Belenitsky M, Carrasco N (2014) The Na+/I- symporter (NIS): mechanism and medical impact. Endocr Rev 35(1):106–149
Spitzweg C, Bible KC, Hofbauer LC, Morris JC (2014) Advanced radioiodine-refractory differentiated thyroid cancer: the sodium iodide symporter and other emerging therapeutic targets. Lancet Diabetes Endocrinol. doi:10.1016/S2213-8587(14)70051-8
American Thyroid Association Guidelines Taskforce on Thyroid N, Differentiated Thyroid C, Cooper DS, Doherty GM, Haugen BR, Kloos RT, Lee SL, Mandel SJ, Mazzaferri EL, Mciver B, Pacini F, Schlumberger M, Sherman SI, Steward DL, Tuttle RM (2009) Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid 19(11):1167–1214
Mazzaferri EL, Jhiang SM (1994) Long-term impact of initial surgical and medical therapy on papillary and follicular thyroid cancer. Am J Med 97(5):418–428
Reiners C, Hanscheid H, Luster M, Lassmann M, Verburg FA (2011) Radioiodine for remnant ablation and therapy of metastatic disease. Nat Rev Endocrinol 7(10):589–595
Memorial Sloan-Kettering Cancer Center; Gustave Roussy Cancer Center Grand Paris (2008)- [cited 2014 Jul 29]. Lesion dosimetry with 124-iodine in metastatic thyroid carcinoma. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). Available from: http://clinicaltrials.gov/ct2/show/NCT00673010 NLM Identifier: NCT00673010
Kopp PA (2008) Reduce, recycle, reuse—iodotyrosine deiodinase in thyroid iodide metabolism. N Engl J Med 358(17):1856–1859
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2(5):401–404
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6(269):pl1
Endo T, Kaneshige M, Nakazato M, Ohmori M, Harii N, Onaya T (1997) Thyroid transcription factor-1 activates the promoter activity of rat thyroid Na+/I- symporter gene. Mol Endocrinol 11(11):1747–1755
Ohno M, Zannini M, Levy O, Carrasco N, Di Lauro R (1999) The paired-domain transcription factor Pax8 binds to the upstream enhancer of the rat sodium/iodide symporter gene and participates in both thyroid-specific and cyclic-AMP-dependent transcription. Mol Cell Biol 19(3):2051–2060
Taki K, Kogai T, Kanamoto Y, Hershman JM, Brent GA (2002) A thyroid-specific far-upstream enhancer in the human sodium/iodide symporter gene requires Pax-8 binding and cyclic adenosine 3′,5′-monophosphate response element-like sequence binding proteins for full activity and is differentially regulated in normal and thyroid cancer cells. Mol Endocrinol 16(10):2266–2282
Ferretti E, Tosi E, Po A, Scipioni A, Morisi R, Espinola MS, Russo D, Durante C, Schlumberger M, Screpanti I, Filetti S, Gulino A (2008) Notch signaling is involved in expression of thyrocyte differentiation markers and is down-regulated in thyroid tumors. J Clin Endocrinol Metab 93(10):4080–4087
Sastre-Perona A, Santisteban P (2014) Wnt-independent role of beta-catenin in thyroid cell proliferation and differentiation. Mol Endocrinol 28(5):681–695
Fernandez LP, Lopez-Marquez A, Martinez AM, Gomez-Lopez G, Santisteban P (2013) New insights into FoxE1 functions: identification of direct FoxE1 targets in thyroid cells. PLoS ONE 8(5):e62849
Ringseis R, Rauer C, Rothe S, Gessner DK, Schutz LM, Luci S, Wen G, Eder K (2013) Sterol regulatory element-binding proteins are regulators of the NIS gene in thyroid cells. Mol Endocrinol 27(5):781–800
Boelaert K, Smith VE, Stratford AL, Kogai T, Tannahill LA, Watkinson JC, Eggo MC, Franklyn JA, Mccabe CJ (2007) PTTG and PBF repress the human sodium iodide symporter. Oncogene 26(30):4344–4356
Mu D, Huang R, Li S, Ma X, Lou C, Kuang A (2012) Combining transfer of TTF-1 and Pax-8 gene: a potential strategy to promote radioiodine therapy of thyroid carcinoma. Cancer Gene Ther 19(6):402–411
Carre A, Rachdi L, Tron E, Richard B, Castanet M, Schlumberger M, Bidart JM, Szinnai G, Polak M (2011) Hes1 is required for appropriate morphogenesis and differentiation during mouse thyroid gland development. PLoS ONE 6(2):e16752
Saito T, Endo T, Kawaguchi A, Ikeda M, Nakazato M, Kogai T, Onaya T (1997) Increased expression of the Na+/I- symporter in cultured human thyroid cells exposed to thyrotropin and in Graves’ thyroid tissue. J Clin Endocrinol Metab 82(10):3331–3336
Kogai T, Curcio F, Hyman S, Cornford EM, Brent GA, Hershman JM (2000) Induction of follicle formation in long-term cultured normal human thyroid cells treated with thyrotropin stimulates iodide uptake but not sodium/iodide symporter messenger RNA and protein expression. J Endocrinol 167(1):125–135
Kogai T, Endo T, Saito T, Miyazaki A, Kawaguchi A, Onaya T (1997) Regulation by thyroid-stimulating hormone of sodium/iodide symporter gene expression and protein levels in FRTL-5 cells. Endocrinology 138(6):2227–2232
Riedel C, Levy O, Carrasco N (2001) Post-transcriptional regulation of the sodium/iodide symporter by thyrotropin. J Biol Chem 276(24):21458–21463
Knauf JA, Kuroda H, Basu S, Fagin JA (2003) RET/PTC-induced dedifferentiation of thyroid cells is mediated through Y1062 signaling through SHC-RAS-MAP kinase. Oncogene 22(28):4406–4412
Liu D, Hu S, Hou P, Jiang D, Condouris S, Xing M (2007) Suppression of BRAF/MEK/MAP kinase pathway restores expression of iodide-metabolizing genes in thyroid cells expressing the V600E BRAF mutant. Clin Cancer Res 13(4):1341–1349
Chakravarty D, Santos E, Ryder M, Knauf JA, Liao XH, West BL, Bollag G, Kolesnick R, Thin TH, Rosen N, Zanzonico P, Larson SM, Refetoff S, Ghossein R, Fagin JA (2011) Small-molecule MAPK inhibitors restore radioiodine incorporation in mouse thyroid cancers with conditional BRAF activation. J Clin Invest 121(12):4700–4711
Kogai T, Sajid-Crockett S, Newmarch LS, Liu YY, Brent GA (2008) Phosphoinositide-3-kinase inhibition induces sodium/iodide symporter expression in rat thyroid cells and human papillary thyroid cancer cells. J Endocrinol 199(2):243–252
Liu YY, Zhang X, Ringel MD, Jhiang SM (2012) Modulation of sodium iodide symporter expression and function by LY294002, Akti-1/2 and rapamycin in thyroid cells. Endocr Relat Cancer 19(3):291–304
Furuya F, Shimura H, Suzuki H, Taki K, Ohta K, Haraguchi K, Onaya T, Endo T, Kobayashi T (2004) Histone deacetylase inhibitors restore radioiodide uptake and retention in poorly differentiated and anaplastic thyroid cancer cells by expression of the sodium/iodide symporter thyroperoxidase and thyroglobulin. Endocrinology 145(6):2865–2875
Hou P, Bojdani E, Xing M (2010) Induction of thyroid gene expression and radioiodine uptake in thyroid cancer cells by targeting major signaling pathways. J Clin Endocrinol Metab 95(2):820–828
Kitazono M, Robey R, Zhan Z, Sarlis NJ, Skarulis MC, Aikou T, Bates S, Fojo T (2001) Low concentrations of the histone deacetylase inhibitor, depsipeptide (FR901228), increase expression of the Na(+)/I(−) symporter and iodine accumulation in poorly differentiated thyroid carcinoma cells. J Clin Endocrinol Metab 86(7):3430–3435
Pugliese M, Fortunati N, Germano A, Asioli S, Marano F, Palestini N, Frairia R, Boccuzzi G, Catalano MG (2013) Histone deacetylase inhibition affects sodium iodide symporter expression and induces 131I cytotoxicity in anaplastic thyroid cancer cells. Thyroid 23(7):838–846
Sherman EJ, Su YB, Lyall A, Schoder H, Fury MG, Ghossein RA, Haque S, Lisa D, Shaha AR, Tuttle RM, Pfister DG (2013) Evaluation of romidepsin for clinical activity and radioactive iodine reuptake in radioactive iodine-refractory thyroid carcinoma. Thyroid 23(5):593–599
Riesco-Eizaguirre G, Rodriguez I, De La Vieja A, Costamagna E, Carrasco N, Nistal M, Santisteban P (2009) The BRAFV600E oncogene induces transforming growth factor beta secretion leading to sodium iodide symporter repression and increased malignancy in thyroid cancer. Cancer Res 69(21):8317–8325
Patel PN, Yu XM, Jaskula-Sztul R, Chen H (2014) Hesperetin activates the Notch1 signaling cascade, causes apoptosis, and induces cellular differentiation in anaplastic thyroid cancer. Ann Surg Oncol. doi:10.1245/s10434-013-3459-7
Yu XM, Jaskula-Sztul R, Ahmed K, Harrison AD, Kunnimalaiyaan M, Chen H (2013) Resveratrol induces differentiation markers expression in anaplastic thyroid carcinoma via activation of Notch1 signaling and suppresses cell growth. Mol Cancer Ther 12(7):1276–1287
Sebai H, Hovsepian S, Ristorcelli E, Aouani E, Lombardo D, Fayet G (2010) Resveratrol increases iodide trapping in the rat thyroid cell line FRTL-5. Thyroid 20(2):195–203
Costamagna E, Garcia B, Santisteban P (2004) The functional interaction between the paired domain transcription factor Pax8 and Smad3 is involved in transforming growth factor-beta repression of the sodium/iodide symporter gene. J Biol Chem 279(5):3439–3446
De Vita G, Zannini M, Cirafici AM, Melillo RM, Di Lauro R, Fusco A, Santoro M (1998) Expression of the RET/PTC1 oncogene impairs the activity of TTF-1 and Pax-8 thyroid transcription factors. Cell Growth Differ 9(1):97–103
Lakshmanan A, Doseff AI, Ringel MD, Saji M, Rousset B, Zhang X, Jhiang SM (2014) Apigenin in combination with Akt inhibition significantly enhances thyrotropin-stimulated radioiodide accumulation in thyroid cells. Thyroid 24(5):878–887
Vadysirisack DD, Chen ES, Zhang Z, Tsai MD, Chang GD, Jhiang SM (2007) Identification of in vivo phosphorylation sites and their functional significance in the sodium iodide symporter. J Biol Chem 282(51):36820–36828
Vadysirisack DD (2007) Modulation of sodium iodide symporter expression and activity at post-translational levels. Doctoral dissertation, The Ohio State University, Columbus, OH. Retrieved from http://www.ohiolink.edu/etd/
Zhang Z, Beyer S, Jhiang SM (2013) MEK inhibition leads to lysosome-mediated Na+/I- symporter protein degradation in human breast cancer cells. Endocr Relat Cancer 20(2):241–250
Smith VE, Read ML, Turnell AS, Watkins RJ, Watkinson JC, Lewy GD, Fong JC, James SR, Eggo MC, Boelaert K, Franklyn JA, Mccabe CJ (2009) A novel mechanism of sodium iodide symporter repression in differentiated thyroid cancer. J Cell Sci 122(Pt 18):3393–3402
Wapnir IL, Van De Rijn M, Nowels K, Amenta PS, Walton K, Montgomery K, Greco RS, Dohan O, Carrasco N (2003) Immunohistochemical profile of the sodium/iodide symporter in thyroid, breast, and other carcinomas using high density tissue microarrays and conventional sections. J Clin Endocrinol Metab 88(4):1880–1888
Dohan O, Baloch Z, Banrevi Z, Livolsi V, Carrasco N (2001) Rapid communication: predominant intracellular overexpression of the Na(+)/I(−) symporter (NIS) in a large sampling of thyroid cancer cases. J Clin Endocrinol Metab 86(6):2697–2700
Riesco-Eizaguirre G, Santisteban P (2006) A perspective view of sodium iodide symporter research and its clinical implications. Eur J Endocrinol 155(4):495–512
Peyrottes I, Navarro V, Ondo-Mendez A, Marcellin D, Bellanger L, Marsault R, Lindenthal S, Ettore F, Darcourt J, Pourcher T (2009) Immunoanalysis indicates that the sodium iodide symporter is not overexpressed in intracellular compartments in thyroid and breast cancers. Eur J Endocrinol 160(2):215–225
Beyer SJ, Jimenez RE, Shapiro CL, Cho JY, Jhiang SM (2009) Do cell surface trafficking impairments account for variable cell surface sodium iodide symporter levels in breast cancer? Breast Cancer Res Treat 115(1):205–212
Marsee DK, Venkateswaran A, Tao H, Vadysirisack D, Zhang Z, Vandre DD, Jhiang SM (2004) Inhibition of heat shock protein 90, a novel RET/PTC1-associated protein, increases radioiodide accumulation in thyroid cells. J Biol Chem 279(42):43990–43997
Fong P (2011) Thyroid iodide efflux: a team effort? J Physiol 589(Pt 24):5929–5939
Van Den Hove MF, Croizet-Berger K, Jouret F, Guggino SE, Guggino WB, Devuyst O, Courtoy PJ (2006) The loss of the chloride channel, ClC-5, delays apical iodide efflux and induces a euthyroid goiter in the mouse thyroid gland. Endocrinology 147(3):1287–1296
Mu D, Huang R, Ma X, Li S, Kuang A (2012) Radioiodine therapy of thyroid carcinoma following Pax-8 gene transfer. Gene Ther 19(4):435–442
Jhiang SM, Cho JY, Ryu KY, Deyoung BR, Smanik PA, Mcgaughy VR, Fischer AH, Mazzaferri EL (1998) An immunohistochemical study of Na+/I- symporter in human thyroid tissues and salivary gland tissues. Endocrinology 139(10):4416–4419
La Perle KM, Kim DC, Hall NC, Bobbey A, Shen DH, Nagy RS, Wakely PE Jr, Lehman A, Jarjoura D, Jhiang SM (2013) Modulation of sodium/iodide symporter expression in the salivary gland. Thyroid 23(8):1029–1036
Brandt MP, Kloos RT, Shen DH, Zhang X, Liu YY, Jhiang SM (2012) Micro-single-photon emission computed tomography image acquisition and quantification of sodium-iodide symporter-mediated radionuclide accumulation in mouse thyroid and salivary glands. Thyroid 22(6):617–624
Cho JY, Sagartz JE, Capen CC, Mazzaferri EL, Jhiang SM (1999) Early cellular abnormalities induced by RET/PTC1 oncogene in thyroid-targeted transgenic mice. Oncogene 18(24):3659–3665
Jhiang SM, Sagartz JE, Tong Q, Parker-Thornburg J, Capen CC, Cho JY, Xing S, Ledent C (1996) Targeted expression of the ret/PTC1 oncogene induces papillary thyroid carcinomas. Endocrinology 137(1):375–378
Powell DJ, Russell J, Nibu K, Li G, Rhee E, Liao M, Goldstein M, Keane WM, Santoro M, Fusco A, Rothstein JL (1998) The RET/PTC3 oncogene: metastatic solid-type papillary carcinomas in murine thyroids. Cancer Res 58(23):5523–5528
Russell JP, Powell DJ, Cunnane M, Greco A, Portella G, Santoro M, Fusco A, Rothstein JL (2000) The TRK-T1 fusion protein induces neoplastic transformation of thyroid epithelium. Oncogene 19(50):5729–5735
Charles RP, Iezza G, Amendola E, Dankort D, Mcmahon M (2011) Mutationally activated BRAF(V600E) elicits papillary thyroid cancer in the adult mouse. Cancer Res 71(11):3863–3871
Knauf JA, Ma X, Smith EP, Zhang L, Mitsutake N, Liao XH, Refetoff S, Nikiforov YE, Fagin JA (2005) Targeted expression of BRAFV600E in thyroid cells of transgenic mice results in papillary thyroid cancers that undergo dedifferentiation. Cancer Res 65(10):4238–4245
Xing M (2005) BRAF mutation in thyroid cancer. Endocr Relat Cancer 12(2):245–262
Charles RP, Silva J, Iezza G, Phillips WA, Mcmahon M (2014) Activating BRAF and PIK3CA mutations cooperate to promote anaplastic thyroid carcinogenesis. Mol Cancer Res 12(7):979–986
La Perle KM, Jhiang SM, Capen CC (2000) Loss of p53 promotes anaplasia and local invasion in ret/PTC1-induced thyroid carcinomas. Am J Pathol 157(2):671–677
Mcfadden DG, Vernon A, Santiago PM, Martinez-Mcfaline R, Bhutkar A, Crowley DM, Mcmahon M, Sadow PM, Jacks T (2014) p53 constrains progression to anaplastic thyroid carcinoma in a Braf-mutant mouse model of papillary thyroid cancer. Proc Natl Acad Sci U S A 111(16):E1600–E1609
Fedele M, Palmieri D, Chiappetta G, Pasquinelli R, De Martino I, Arra C, Palma G, Valentino T, Pierantoni GM, Viglietto G, Rothstein JL, Santoro M, Fusco A (2009) Impairment of the p27kip1 function enhances thyroid carcinogenesis in TRK-T1 transgenic mice. Endocr Relat Cancer 16(2):483–490
Suzuki H, Willingham MC, Cheng SY (2002) Mice with a mutation in the thyroid hormone receptor beta gene spontaneously develop thyroid carcinoma: a mouse model of thyroid carcinogenesis. Thyroid 12(11):963–969
Pringle DR, Yin Z, Lee AA, Manchanda PK, Yu L, Parlow AF, Jarjoura D, La Perle KM, Kirschner LS (2012) Thyroid-specific ablation of the Carney complex gene, PRKAR1A, results in hyperthyroidism and follicular thyroid cancer. Endocr Relat Cancer 19(3):435–446
Antico-Arciuch VG, Dima M, Liao XH, Refetoff S, Di Cristofano A (2010) Cross-talk between PI3K and estrogen in the mouse thyroid predisposes to the development of follicular carcinomas with a higher incidence in females. Oncogene 29(42):5678–5686
Vitagliano D, Portella G, Troncone G, Francione A, Rossi C, Bruno A, Giorgini A, Coluzzi S, Nappi TC, Rothstein JL, Pasquinelli R, Chiappetta G, Terracciano D, Macchia V, Melillo RM, Fusco A, Santoro M (2006) Thyroid targeting of the N-ras(Gln61Lys) oncogene in transgenic mice results in follicular tumors that progress to poorly differentiated carcinomas. Oncogene 25(39):5467–5474
Kato Y, Ying H, Zhao L, Furuya F, Araki O, Willingham MC, Cheng SY (2006) PPARgamma insufficiency promotes follicular thyroid carcinogenesis via activation of the nuclear factor-kappaB signaling pathway. Oncogene 25(19):2736–2747
Guigon CJ, Zhao L, Willingham MC, Cheng SY (2009) PTEN deficiency accelerates tumour progression in a mouse model of thyroid cancer. Oncogene 28(4):509–517
Pringle DR, Vasko VV, Yu L, Manchanda PK, Lee AA, Zhang X, Kirschner JM, Parlow AF, Saji M, Jarjoura D, Ringel MD, La Perle KM, Kirschner LS (2014) Follicular thyroid cancers demonstrate dual activation of PKA and mTOR as modeled by thyroid-specific deletion of Prkar1a and Pten in mice. J Clin Endocrinol Metab 99(5):E804–E812
Miller KA, Yeager N, Baker K, Liao XH, Refetoff S, Di Cristofano A (2009) Oncogenic Kras requires simultaneous PI3K signaling to induce ERK activation and transform thyroid epithelial cells in vivo. Cancer Res 69(8):3689–3694
Antico Arciuch VG, Russo MA, Dima M, Kang KS, Dasrath F, Liao XH, Refetoff S, Montagna C, Di Cristofano A (2011) Thyrocyte-specific inactivation of p53 and Pten results in anaplastic thyroid carcinomas faithfully recapitulating human tumors. Oncotarget 2(12):1109–1126
Franken PR, Guglielmi J, Vanhove C, Koulibaly M, Defrise M, Darcourt J, Pourcher T (2010) Distribution and dynamics of (99 m)Tc-pertechnetate uptake in the thyroid and other organs assessed by single-photon emission computed tomography in living mice. Thyroid 20(5):519–526
Liu YY, Brandt MP, Shen DH, Kloos RT, Zhang X, Jhiang SM (2011) Single photon emission computed tomography imaging for temporal dynamics of thyroidal and salivary radionuclide accumulation in 17-allyamino-17-demothoxygeldanamycin-treated thyroid cancer mouse model. Endocr Relat Cancer 18(1):27–37
Schlumberger M, Sherman SI (2012) Approach to the patient with advanced differentiated thyroid cancer. Eur J Endocrinol 166(1):5–11
Sherman SI (2011) Targeted therapies for thyroid tumors. Mod Pathol 24(Suppl 2):S44–S52
Ho AL, Grewal RK, Leboeuf R, Sherman EJ, Pfister DG, Deandreis D, Pentlow KS, Zanzonico PB, Haque S, Gavane S, Ghossein RA, Ricarte-Filho JC, Dominguez JM, Shen R, Tuttle RM, Larson SM, Fagin JA (2013) Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N Engl J Med 368(7):623–632
Liu T, Altman RB (2014) Identifying druggable targets by protein microenvironments matching: application to transcription factors. CPT Pharmacometrics Syst Pharmacol 3:e93
Heaney AP, Nelson V, Fernando M, Horwitz G (2001) Transforming events in thyroid tumorigenesis and their association with follicular lesions. J Clin Endocrinol Metab 86(10):5025–5032
Nilsson M, Bjorkman U, Ekholm R, Ericson LE (1990) Iodide transport in primary cultured thyroid follicle cells: evidence of a TSH-regulated channel mediating iodide efflux selectively across the apical domain of the plasma membrane. Eur J Cell Biol 52(2):270–281
Yu KH, Youn H, Song MG, Lee DS, Chung JK (2012) The effect of tanespimycin (17-AAG) on radioiodine accumulation in sodium-iodide symporter expressing cells. Nucl Med Mol Imaging 46(4):239–246
Rothenberg Sm MD, Palmer E, Daniels G, Wirth L (2013) Re-differentiation of radioiodine-refractory BRAFV600E-mutant thyroid carcinoma with dabrafenib: a pilot study. J Clin Oncol 31(suppl):abstr 6025
Acknowledgments
This work was supported by the National Institutes of Health P01 CA124570 (PI: Ringel, M; Project 3 leader: Jhiang, SM) and P50 CA168505 (PI: Ringel, M; Project 2 leader: Jhiang, SM).
Conflict of Interest
No competing financial interests exist.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Lakshmanan, A., Scarberry, D., Shen, D.H. et al. Modulation of Sodium Iodide Symporter in Thyroid Cancer. HORM CANC 5, 363–373 (2014). https://doi.org/10.1007/s12672-014-0203-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-014-0203-0