
Abstract
Aggregated tau protein is a major neuropathological substrate central to the pathophysiology of neurodegenerative diseases such as Alzheimer’s disease (AD), frontotemporal dementia, progressive supranuclear palsy, corticobasal degeneration and chronic traumatic encephalopathy. In AD, it has been shown that the density of hyperphosphorylated tau tangles correlates closely with neuronal dysfunction and cell death, unlike β-amyloid. Until now, diagnostic and pathologic information about tau deposition has only been available from invasive techniques such as brain biopsy or autopsy. The recent development of selective in-vivo tau PET imaging ligands including [18F]THK523, [18F]THK5117, [18F]THK5105 and [18F]THK5351, [18F]AV1451(T807) and [11C]PBB3 has provided information about the role of tau in the early phases of neurodegenerative diseases, and provided support for diagnosis, prognosis, and imaging biomarkers to track disease progression. Moreover, the spatial and longitudinal relationship of tau distribution compared with β - amyloid and other pathologies in these diseases can be mapped. In this review, we discuss the role of aggregated tau in tauopathies, the challenges posed in developing selective tau ligands as biomarkers, the state of development in tau tracers, and the new clinical information that has been uncovered, as well as the opportunities for improving diagnosis and designing clinical trials in the future.
Similar content being viewed by others

Introduction
Alzheimer’s disease (AD), Parkinson’s disease without (PD) and with later dementia (PDD), Lewy body dementia (LBD), frontotemporal dementia (FTD), and corticobasal degeneration (CBD) are common neurodegenerative disorders. Tau is a microtubule-associated protein which is essential for neuronal stability and transport of axonal nutrients. Aggregated tau, due to hyperphosphorylation, is a pathological characteristic of a group of neurodegenerative conditions known as the tauopathies [1]. The neuropathological substrates of AD are tau neurofibrillary tangles (NFT) and β-amyloid (Aβ) plaques, while activated microglia, astrocytes, and neuropil threads also play a significant role in disease pathogenesis. It has been shown that Aβ plaque deposition can begin decades before symptom onset, while tau deposition is more closely associated with symptom onset due to neuronal dysfunction, its levels at autopsy correlating well pre-morbid cognitive status [2, 3].
Recent advances in selective tau tracer development for positron emission tomography (PET) imaging have, for the first time, allowed in-vivo exploration of the presence and extent of tau pathology in patients suspected of having tauopathies. Clinically, tau PET imaging can provide valuable support in the early differential diagnosis of neurodegenerative disorders by revealing whether a characteristic pattern of aggregated tau is present. It also provides a potential biomarker of disease progression. Over the next decade, tau imaging is likely to dominate the field of dementia research and, in this review, we will discuss current developments in novel tau tracers, future applications, and how this could extend our knowledge of dementia.
Tau protein and its role in the pathophysiology of the tauopathies
Tau is a natively unfolded phosphorylated protein that is present mainly in axons, and binds to microtubules, stabilizing them. Microtubules comprise the cell cytoskeleton, and are critical for maintaining the structural integrity of the cell and for transporting nutrients from the soma down the axons to synaptic terminals [1, 4, 5]. Tau protein exists as six distinct isoforms that result from alternate mRNA splicing of the MAPT (microtubule associated protein tau) gene on chromosome 17 (cytogenetic location 17q21.1). The isoforms differ in the number of microtubule binding repeats (which are encoded by exon 10) that are present, and tau can exist in 3-repeat (3R) or 4-repeat (4R) forms. The healthy adult human cortex has equal numbers of 3R and 4R isoforms, and its tau expression is roughly double that seen in the white matter and cerebellum [1]. Through the tandem repeats, tau assembles into filaments which have a cross β structure, similar to that of Aβ. The ability of tau to bind to microtubules is also regulated by post-translational modification of the protein by phosphorylation, glycosylation, glycation, ubiquitination, sumoylation, and nitration [1, 6]. The functions of tau are regulated in part by its phosphorylation state, and the protein has multiple kinase phosphorylation sites [7, 8].
In all neurodegenerative diseases in which tau is implicated, it is in a hyperphosphorylated form, and this is responsible for its aggregation, leading to neuronal dysfunction and death. Hyperphosphorylation prevents tau binding to microtubules, reducing their stability, which in turn leads to impaired axon transport. Aggregated tau in AD exists as paired helical filaments which further coalesce into neurofibrillary tangles (NFTs) [5, 9, 10]. These then lead to impaired synaptic and neuronal dysfunction [11].
In AD, the distribution of extracellular Aβ plaques can be variable between individuals, but amyloid deposition is thought to start in the inferior frontal cingulate areas and then spread to association cortex [12]. According to Braak staging of AD, the deposition of NFTs follows a more predictable and stereotyped course than Aβ as the disease progresses [12]. NFTs are first detected in the transentorhinal cortex (stages 1 and 2) in the presymptomatic stage of AD, and then spread to the limbic areas, by which time symptoms become evident. NFTs finally involve association cortical areas by stages 5–6, when symptoms become severe. Braak et al. have also reported that at post mortem occasional NFTs can be found in the brains of apparently healthy 30-year-olds [13].
Multiple studies have reported that the density of NFTs correlates more closely with cell dysfunction and symptoms than does Aβ plaque density. In-vivo Aβ PET imaging studies have demonstrated that Aβ deposition can occur 1–2 decades before the symptoms appear, levels approaching a plateau by the onset of cognitive symptoms. The Aβ cascade hypothesis posits that Aβ deposition is central to the pathology of disease, leading to a series of downstream events which cause tau hyperphosphorylation and aggregation, which results in neuronal dysfunction. Recent Aβ imaging studies support the Aβ cascade hypothesis to some extent [14], but it is now recognised that Aβ deposition alone cannot explain AD progression and pathogenesis. Thirty percent of elderly healthy subjects have significant levels of cortical amyloid deposition when imaged with 11C-PIB PET but manifest no overt symptoms [15]. Additionally, several high-profile anti-Aβ treatments have failed to halt or reverse the symptoms of AD [5].
Although the cascade hypothesis may be over simplistic, cognitively normal subjects who have brain Aβ deposition show cortical thinning [16] and have a higher risk of progressing to dementia [17, 18] so clearly amyloid aggregates are toxic to the brain. Aβ deposition, however, occurs at a slow rate over time, preceding neurodegenerative changes and cognitive deterioration [19], and levels correlate poorly with severity of cognitive symptoms. This suggests that strategies to remove amyloid could be most effective if used to protect early asymptomatic cases. In contrast, histopathological studies have shown that the presence of NFTs and neuronal loss increase in parallel with the duration and severity of symptoms [2]. While Aβ deposition plateaus early in the disease, NFT deposition and cell dysfunction continue to progress throughout the course of disease, correlating with symptom severity [3].
The emerging evidence, therefore, supports a complex pathological relationship between Aβ and tau aggregation that may also involve neuroinflammation in the form of microglial activation. This has been described as a ‘toxic pas de deux’ [20]. Histopathological comparisons of brains of non-demented, mildly demented and severely demented patients have shown that NFTs increase in all individuals with increasing age, but have a different distribution from that of Aβ plaques, indicating that the formation of tau and Aβ occurs independently. While NFT formation can occur early, the pathology progresses only slowly in isolation; however, if Aβ plaques are also present then their density increases rapidly. While tau and Aβ aggregation occur independently of one another, neither are sufficient alone to cause AD, and their pathologies may be interdependent [21].
This view is supported by animal studies. Injection of Aβ42 into the brains of transgenic mice expressing P301L pathological tau caused a 5-fold increase in NFT deposition compared with controls, indicating that the introduction of Aβ drove the tau pathology [22]. Furthermore, the offspring of transgenic mice expressing mutant tau crossed with mutant APP mice developed Aβ plaques at the same age, but had a significantly higher density of NFTs in the limbic system and cortex [23]. When brain extracts from APP transgenic mice were introduced into P301L tau transgenic mice, tau pathology was later identified not only at injection sites, but also in distant brain regions [24], indicating that introduction of Aβ triggered tau pathology and that tau aggregation was capable of ‘spreading’ across vulnerable neuronal networks in the brain. This may explain the predictable pattern of tau deposition throughout the cortex described by Braak staging. Aggregated tau is capable of leaving cells and causing normal tau to undergo aggregation and fibrillation [25]. For this reason, the transmission of tau aggregation has been likened to that of prions [26].
The presence of aggregated tau is a defining feature of several other neurodegenerative diseases, which include progressive supranuclear palsy [27], frontotemporal dementia related to chromosome 17 [28], argyrophilic grain disease (an age-related disease, caused by degeneration of argyrophilic grains, correlating with NFT deposition and cognitive impairment [29]), senile dementia of the NFT type [30], corticobasal degeneration and Pick’s disease [31]. Different ultrastructural forms of tau can cause different disease phenotypes. While normal human and Alzheimer brains contain equal amounts of 3R and 4R isoforms, Pick’s disease is characterised by aggregation of 3R isoforms into Pick bodies, while CBD, PSP, and argyrophilic grain disorders contain aggregated 4R isoforms as globose tangles in the case of PSP [1].
Chronic traumatic encephalopathy (CTE) is a progressive dementing neuropsychological illness in people who have suffered serial mild concussive brain injuries, which result in axonal injury. The condition has received significant attention in players of contact sports such as American football, rugby, and boxing, and also in horse riders who have frequent falls. It is characterised histologically by the deposition of tau in areas of axonal injury [32]. In one autopsy study of 85 people with mild and repetitive traumatic brain injury, compared with 18 controls, a clear and predictable range of NFT pathology across multiple regions was found, allowing for a grading system of tau pathology [33].
Table 1 shows the characteristic topographic features and distribution of tau aggregates in the common tauopathies. [31, 33–36]
Tau PET imaging as a biomarker in AD
The National Institute of Ageing–Alzheimer’s Association (NIA–AA) Working Group Guidelines have emphasised the concept of AD as a spectrum or continuum of disease, consistent with the idea that pathophysiological changes of AD occur long before the onset of cognitive symptoms and ultimate dementia. Stages of disease can therefore be considered as an ‘AD Preclinical Stage’, reflecting the asymptomatic stage during which underlying pathology develops, and an ‘AD Clinical’ stage including mild cognitive impairment, when symptoms occur secondary to synaptic dysfunction and neuronal loss. The long prodrome has been identified as a key target time for disease-modifying therapy [37]. During the asymptomatic prodrome, imaging biomarkers can potentially be used to stage disease and follow its progression. Biomarkers in current use for detecting AD pathology are those reflecting Aβ deposition (reduced CSF Aβ levels and raised brain Aβ load) and markers of neurodegeneration (MRI atrophy, reduced glucose metabolism on FDG-PET, and raised CSF p-tau) [38]. These biomarkers have clinical utility in that they can predict risk of progression of mild cognitive impairment (MCI) to AD and health to MCI [39, 40] and give an indication of the sequence of pathological processes that occur in AD [39]. However, CSF measurement of tau requires a painful invasive spinal tap requiring a skilled operator, and does not provide critical information about the spatial distribution of tau. Aβ imaging provides useful information about the spatial and temporal distribution of Aβ deposition, but is not a marker of disease progression, due to the plateauing of plaque deposition.
There has been a global effort to identify a selective tau tracer to enable in-vivo PET imaging of NFT load as a method of identifying the spatial and temporal progression of tau pathology. As NFT density correlates with neuronal dysfunction and symptom onset, tau imaging should provide a valuable marker of disease progression. Simultaneous imaging of tau and Aβ aggregant load will promote a deeper understanding of the complex synergistic relationship between the two, helping to prove or refute much of the current speculation.
Tau PET imaging could be useful in clinical trials assessing the efficacy of anti-tau strategies. It will aid recruitment of subjects with a significant tau load (no longer relying on clinical assessment, which can be unhelpful, and markers of neuronal degeneration, which occur late in the course of disease), will provide proof of mechanism, and monitor tau clearance as an end-point. In the clinical setting, it can be used in the differential diagnosis of early dementias (differentiating between AD and non-AD pathologies), and also in differentiating between MCI and normal ageing [41–44].
The search for a suitable tau tracer
While molecular imaging in dementia has been stimulated by the success of Aβ imaging, particularly using the Aβ tracer [11C]PIB, the identification of selective tau tracers has proved more difficult until the last few years. There are certain idiosyncrasies of the tau protein that need to be taken in consideration during tracer design. Tau is an inherently more complex and unpredictable protein, with multiple isoforms and many post-translational modifications. Therefore, tracers may bind specifically to a particular isoform or to multiple isoforms. Tau is an intracellular protein, so any ligand must cross the plasma cell membrane as well as the blood–brain barrier, which confers requirements about the molecular size and lipophilicity of the ligand. Furthermore, tau is present in the brain at much lower concentrations than Aβ, so a selective ligand will need to have a high binding affinity for tau over Aβ. This problem is confounded by the fact that Aβ and tau often co-exist in the same cortical areas, and both manifest β-sheet structure, which is where planar polyaromatic ligands tend to bind [41–45].
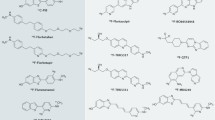
Therefore, requirements of an ideal tau tracer are: (1) high sensitivity and selectivity for its target (20–50-fold selectivity is required for tau over Aβ), (2) low toxicity, (3) rapid uptake and clearance from the brain, and (4) no active brain metabolism. From a practical view, the radioactive half-life of the isotope used to label the ligand should also be taken into consideration. Use of 18F (half-life of 110 minutes) rather than 11C (half-life 20 minutes) can preclude the need for onsite production and allows longer imaging times, but increases the dosimetry. An 11C tracer may be preferred if multiple PET scans are to be performed, due to its lower dosimetry [41, 42, 44, 45]. Figure 1 shows the chemical structure of different tau tracers.

Chemical structures of current tau tracers. The chemical structures of: a [18F] THK-523, b [18F]THK-5105, c [18F] THK-5117, d [18F] THK-5351, e [11C] PBB3, f [18F]T808, and g [18F]-T807
[18F]FDDNP
2-(1-{6-[2-[18F]fluoroethyl)(methyl)amino]-2-naphthylethylidene)malononitrile (FDDNP) was developed as an amyloid marker by Barrio et al. in 2008 [46]. It is extracted well by the brain and shows moderate affinity for both amyloid plaques and neurofibrillary tangles, though the specific signal is low. FDDNP PET will separate groups of normal, MCI, and AD subjects by their levels of cortical uptake. The pattern of FDDNP uptake in AD reflects both amyloid and tau deposition, as signal is seen in both association cortex and hippocampus [47]. The uptake of FDDNP increases over time in AD and MCI due to increasing tau accumulation, so the tracer can be used to track disease progression. FDDNP PET has also been used to detect brain amyloid in Down syndrome [48] and dementia with Lewy bodies [49]. Drawbacks of FDDNP PET as a biomarker are its low sensitivity and selectivity. While it has been reported that [18F]FDDNP PET can predict progressive cognitive impairment in MCI [50], it is less sensitive than [18F]FDG PET for detecting disease progression. As a consequence, further searches for selective tau ligands were performed. A further consideration of FDDNP PET is the rapid metabolism and clearance of the tracer [51], and it does not reach a steady state for a long time. The optimal analysis method is therefore Logan graphical analysis using the cerebellum as a reference region, as used by Small et al. [52]. This requires long scanning periods (up to 125 minutes), making it a difficult tracer to use in clinical practice.
THK compounds
The first tau-selective ligand, [18F]THK523, was identified by Okamura and colleagues at Tohuku University, after screening a series of quinolone and benzimidazole derivatives [59].
Pharmacokinetic studies showed excellent brain uptake and rapid clearance in mice, with no lipophilic metabolites and higher binding to tau over Aβ [59].[60] In-vitro studies of this ligand demonstrated binding to NFTs in AD brain sections and a higher affinity for tau fibrils than Aβ fibrils [61]. MicroPET studies in tau transgenic mice showed a correlation between in-vivo binding with subsequent histofluorescence [62]. However, while in-vivo testing in humans demonstrated selective tau binding, which correlated with the known tau distribution in AD, and a correlation of tracer uptake with impaired cognition, there was significant white matter retention which prevented accurate visual interpretation of signals, thus precluding its widespread use as a PET tracer [60]. Furthermore, it does not bind tau aggregates in non-AD tauopathies, further limiting its diagnostic utility [63].
Subsequently, the same researchers identified two further 2-arylquinoline derivatives, [18F]THK5105 and [18F]THK5117 which have superior binding affinity (Ki = 59.3nM for THK523, 7.8nM for THK5105, and 10.9nM for THK5117) and selectivity for tau in AD brains than [18F]THK523, as well as higher brain uptake and more rapid clearance. The tracer also has good penetration of the blood–brain barrier and no toxic effects [64]. In-vivo studies showed that there was higher cortical retention in AD patients compared with healthy controls, and retention correlated well with impaired performance on cognitive testing, and loss of brain volume [54].
[18F]THK5105 has a binding affinity to tau 25 times greater than that of amyloid, with peak brain entry higher than that for [18F}THK523, [18F]AV1451, [18F]T808, and [11C]PBB3 after 6 minutes. There is no obvious accumulation in the skull reported, but there is non-specific tracer retention in the brainstem, thalamus, subcortical white matter, probably due to binding to β-sheet structures in the myelin basic protein. This is not reported to be visually noticeable [54]. Compared to [18F]THK5117, [18F]THK5105 has a relatively slower clearance from the brain and higher lipophilicity, resulting in a lower signal to noise ratio [54]. Figure 2 shows [18F]THK523, [18F]THK5105, and [18F]THK5117 PET in different stages and types of dementia.

PET images using the [18F] THK family of tracers. a The first tau tracer, [18F] THK 523 in a healthy control, a subject with semantic dementia and an AD subject. There is increased tracer retention in the AD subject, but no difference between the control and SD Reproduced from Villemagne 2014 [53]. b [18F] THK5105 PET images in a 72-year-old healthy control (MMSE 29) and a 68-year-old AD subject (MMSE 20). Reproduced from Okamura 2014 [54]. c Tau tracer [18F]THK 5117 in a subject with mild, moderate, and severe AD, showing increasing retention of tracer as disease progresses from the medial, anterior, and inferior temporal cortex in mild AD, spreading to association areas in moderate AD, and throughout the neocortex in severe AD. Reproduced from Okamura 2014 [44]
Brain uptake of [18F]THK5117 has been shown to have high affinity for tau in saturation binding assays, with nanomolar binding affinity [65]. It has been compared with that of [11C]PIB and [18F]FDG in subjects with MCI and AD. The authors noted a significant correlation between tracer retention of [18F]THK5117 and cognitive performance. In addition, they noted a different regional pattern of retention compared with [11C]PIB. The investigators reported lower [18F]THK5117 uptake in MCI compared with AD subjects, though both were raised compared to healthy controls, thus demonstrating the ability of the tau tracer to distinguish the spectrum of the Alzheimer disease process [66].
One report concerned three AD patients who had had [18F]FDG and [11C]PIB PET scans in life and donated their brains for subsequent post-mortem analysis, allowing in-vitro binding of [18F]THK5117 to be investigated by autoradiography. Binding of the tracer was highest in the mesial temporal region in all subjects, consistent with known tau pathology, but levels showed poor correlations with mesial temporal glucose metabolism and Aβ binding [67]. The authors concluded that tau imaging does not just mirror [18F]FDG PET findings.
Another 2-arylquinoline, [18F]THK5351 has also been recently developed which also shows high tau binding affinity in AD brains. [18F]THK5351 PET has been trialled in ten healthy controls and ten AD patients, while two other patients received [18F]THK5117 and [18F]THK5351 for a direct comparison. [18F]THK5351 had similar grey matter but lower white matter and brainstem retention than [18F]THK5117, potentially allowing for better tau visualization, while faster uptake and washout kinetics may facilitate kinetic modelling [68].
The same group has also developed [11C]THK951, which has low lipophilicity (and therefore a higher signal to noise ratio), rapid brain uptake, and fast clearance. Uptake ratio in mouse brain was found to be superior to that of [18F]THK523, [18F]THK5105, and [18F]THK 5117, with slightly lower affinity to tau. In-vivo human testing has not yet been reported [69]. Figure 3 shows [18F]THK5351 in different stages of cognitive impairment.

Novel tau tracer [18F]THK-5351 in different stages of cognitive impairment. PET images of [18F]THK-5351 in a healthy control, an MCI subject (MMSE 25), and an AD subject (MMSE 16). There is increasing tracer retention as disease progresses. In the AD patient, a [11C]PIB PET scan shows amyloid deposition in discrete separate areas of cortex. Courtesy of Nobayaki Okamura, unpublished work
[18F] AV-1451 (T807) and [18F]T808
[18F]AV-1451 (T807) and [18F]T808 are tau-selective compounds recently synthesized by Hartmuth Kolb and his colleagues [70] and now tested in vivo [71]. [18F]T808 showed high binding affinity and good selectivity for tau over Aβ, with rapid uptake and washout in transgenic mice. MicroPET showed that it crossed the blood–brain barrier, with minimal white matter binding. However, studies in mice showed that 18F accumulated in bone, indicating defluorination was occurring, so the tracer has not been taken forward, even though it did not exhibit defluorination when tested in humans [72]. In-vivo tracer retention correlated closely with histological examination of tau deposition in another AD patient who died 2 weeks after PET images were obtained [73].
[18F]AV-1451 (T807) has demonstrated >25-fold selectivity for PHF-tau over Aβ (Kd = 14.6nM) on autoradiography and in vivo shows rapid brain extraction and washout, with no plasma metabolites entering the brain [74]. It is of low molecular weight (262.1 g/mol) and crosses the blood–brain barrier readily. The LogP of [18 F]AV-1451 is 1.67[74]. In mice, there was some accumulation in the bone noted, but the authors commented that radioactivity did not increase over time, and that brain homogenate in humans did not contain active metabolite [74]. Post-mortem validation of the tracer has shown high binding of the ligand to dystrophic neurites in AD [75]. In-vivo studies in humans show favourable [18F]AV-1451 uptake and washout kinetics, and tracer retention in AD mirrors the known distribution of tau in the brain. MCI cases showed lower uptake than AD patients, with tracer uptake patterns following Braak staging [55].
Pontecorvo et al. have reported preliminary findings in a PET study comparing Aβ and tau binding in subjects with MCI, AD, and cognitively normal controls. They noted highest tau deposition in patients with AD, followed by MCI, compared with low signal in normal controls. They also noted that cortical tau binding was significantly higher in Aβ-positive than in Aβ-negative individuals. In Aβ-negative individuals, hippocampal tau increased with age but no cortical deposition was detected [76].
Other studies have also shown increased binding of [18F]AV-1451 in MCI compared with controls which targeted all the association cortical areas. Worse cognitive performance (in terms of delayed recall) was associated with increased ligand retention in the entorhinal cortex [77]. Preliminary human studies using [18F]AV-1451 have been used to follow MCI and AD progression over relatively short periods of time (up to 19 months), and have shown significant rises in tau signals over time as symptoms progress, indicating the potential for tau tracers to detect disease progression [57].
[18F]AV-1451 (T807) also has the ability to distinguish variants of AD. In a patient with posterior cortical atrophy (PCA), binding was seen in primary visual and visual association cortices which correlated with decreased [18F]FDG uptake, whereas [11C]PIB uptake was globally elevated and showed no association with FDG metabolism [78]. The same group found that tau binding was increased in left parietal, temporal, and frontal regions in logopenic primary progressive aphasia [79].
[18F]AV-1451 PET has also detected increased signal in non-AD tauopathies such as PSP where the basal ganglia, thalamus, and frontal cortex were targeted. Its uptake has also been evaluated in patients with FTD, including the progressive aphasia and semantic dementia variants. The authors found that a symptomatic MAPT carrier showed increased ligand binding in the frontal, insular, and anterior temporal cortex, whereas in aphasic patients it was increased in left dorsolateral, prefrontal, and insular cortices. Patients with semantic dementia had highest uptake in the anterior temporal cortex with marked asymmetry [80].
Autoradiographic studies on brain specimens of patients with a range of disorders [AD, FTD-tau and PSP, CBD, Parkinsons disease (PD), dementia with Lewy bodies (DLB), and cerebral amyloid angiopathy (CAA)] have shown that while AD brains containing NFTs show high tracer binding, this is not evident in DLB, CAA, and FTD-TDP43. Fluorescent staining with disease specific tau antibodies revealed labelling of neurites and tangles in AD, PSP, CBD, and Pick’s disease, but not Lewy bodies or TDP43 [75]. Some preliminary work has shown binding of tracer in a distribution known to be compatible with PHF-tau distribution in PSP distinct from PD brains [81].
[18F]AV-1451 binding has been described in a retired American National Football League (NFL) player with cognitive decline and features suggestive of either CTE or PSP, and confirmed CTE based on binding patterns, demonstrating that tau imaging can help to differentiate between different types of dementia [82].
[18F]AV-1451 PET has also revealed insights concerning tau deposition in healthy cognitively normal individuals. Schultz et al. used Aβ and tau imaging to characterise in vivo the complex relationship between Aβ and tau pathologies. Seventy-five healthy elderly subjects were examined using longitudinal [11C]PIB and [18F]AV-1451 PET. A significant relationship was found between baseline Aβ burden and tau binding in the inferior temporal lobe, as well as an association between tau binding and the rate of Aβ accumulation, consistent with histology findings that Aβ pathology can influences tau ‘spreading’ throughout the cortex [83].
Sperling et al. compared Aβ with tau binding in cognitively healthy individuals, and showed that the presence of tau tangles on PET did not correlate with memory loss in healthy individuals unless Aβ plaques were also present. In the presence of plaques, however, there was a correlation between tau deposition and memory loss, again reinforcing the principle that the presence of Aβ accelerates tau pathology [84]. In cognitively normal elderly subjects, [18F]AV-1451 binding has been shown to correlate with levels of CSF tau [85].
Lockhart et al. used [18F]AV-1451 and[11C]PIB PET to study the effects of age on Aβ and tau aggregation in cognitively normal elders. They found significantly increased accumulation of tau in the basal ganglia, midbrain, hippocampus, and fornix of elderly normals, which extended to the neocortex as their age and the level of Aβ binding increased. The authors concluded that age and levels of Aβ binding could independently predict tau binding in healthy older people. Age predicted the level of tau accumulation in the medial temporal lobe, while levels of Aβ binding predicted tau deposition outside the medial temporal lobe [86]. Figure 4 shows [18F]AV-1451 (T807) in different stages of dementia and in longitudinal progression of disease.

Novel tau tracer [18F]AV-1451 (previously [18F]-T807. a [18F]T807 in a healthy control (top left), through increasing severity of cognitive impairment to severe AD (bottom right). Increased tracer retention is seen as disease progresses, with widespread neocortical deposition in severe disease. Reproduced from Chien 2013 [55]. b PET images from two cognitively normal individuals, and one with AD dementia, with amyloid PET images on the top row ([11C]PIB) and tau PET images on the bottom row ([18F] T807). From left to right, increasing amyloid deposition is seen in the neocortex, as well as increasing tau in the inferior temporal cortices. Reproduced from Sperling 2014) [56]. c [18F] T807 in a subject with MCI at baseline, and after 10 months, showing a significant increase in tracer deposition in the temporal and parietal lobes. This indicates the clinical utility of tau imaging in detecting disease progression over relatively short time periods. Reproduced from Mark Mintun, 2015 [57]
[11C]PBB3
Maruyama et al. have developed another family of ligands, the phenyl/pyridinyl-butadienyl-benzothiazoles/benzothiazoliums or PBBs, which bind strongly to NFTs in AD brains. In addition, ex-vivo examination of the brains and spinal cords of transgenic mice that had been injected with these compounds showed intense uptake in areas of tau accumulation [58]. [11C}PBB3 has a 40–50 times higher affinity for NFTs than for senile plaques, with affinity in the nanomolar range [58], readily crosses the blood–brain barrier (LogD = 3.3) [87], and is quickly washed out. There is minimal non-specific and white-matter binding [58].The tracer decays quickly to radioactive metabolites in preclinical models, but the radioactive metabolites have not been shown to enter the brain [87]. Nevertheless, simplified analysis methods such as Reference Tissue Models seem to agree with dual input compartment modeling [88].
[11C]PBB3 PET imaging of AD patients has shown increased tracer retention in the hippocampi in contrast to [11C]PIB. There was spreading of [11C]PBB3 binding throughout the cortex as the disease progressed [58, 89]. When [11C]PBB3 PET was performed in a patient with corticobasal degeneration, tracer retention was noted in the neocortical and subcortical structures, while [11C]PIB uptake was normal, highlighting the potential of tau tracers in non-AD tauopathies [58]. [11C] PBB3 has lower brain uptake than [18F]T807 or [11C]PIB due to this rapid metabolism and clearance, but the authors conclude that this may assist its selective binding to high-affinity, low-capacity sites on NFTs, compared to low-affinity, high-capacity sites on the more prevalent β-amyloid [87]. Figure 5 shows tau tracer [11C]-PBB in differing stages of cognitive impairment.

Tau tracer [11C]-PBB in differing stages of cognitive impairment. [11C]PBB3 and [11C]PIB in normal controls and AD patients with increasing severity of disease. The arrowheads indicate the hippocampi. While there is minimal tracer retention in the hippocampi of normal controls, there is increasing retention in the AD patients, especially as MMSE declines, with spread from the hippocampus to the neocortex, consistent with Braak staging. Reproduced from Maruyama 2013 [58]
Other compounds
Honer et al. have developed further compounds, RO6931643, RO6924963 and RO6958948, which are high-affinity binders at the [3H]T808 binding site on tau aggregates. All compounds have been noted to bind with high affinity and specificity to tau aggregates, and lack affinity to Aβ plaques. They also showed low non-specific binding in healthy brain tissues. In addition, there was macro- and micro-colocalisation of radioligand binding. Their pharmacokinetics showed rapid brain entry, washout, and safe metabolic patterns. These tracers are currently being tested in humans [90].
To date, several studies have used [18F]AV1451, which has demonstrated robust pharmacokinetics, high affinity for tau NFTs over amyloid, and minimal non-specific binding, as well as the ability to bind non AD tauopathies. Of the THK compounds, [18F] THK5117 has a very good pharmacokinetic profile, but with further data for [18F]THK5351 and [18F]THK951 awaited. [11C]PBB3 also offers good pharmacokinetics, with the ability to bind non-AD NFTs, but the short half-life of the 11C tracer may limit its use, and the rapid metabolism of the tracer may result in difficulty with data analysis.
Conclusions
Recent years have seen exciting progress in the molecular imaging of dementia, and the tauopathies in particular. In-vivo tau imaging provides further information about the start and progression of the neuropathology of neurodegenerative disorders and, combined with amyloid imaging and FDG, it will be a promising biomarker, both clinically, in supporting differential diagnosis, and also in research, where it will help select appropriate patients and provide proof of mechanism and efficacy in clinical trials. While Aβ imaging plays a key role in the evaluation of dementia, the closer correlation of tau with cognitive impairment and neuronal dysfunction makes it more suitable as a biomarker of disease progression.
Several novel tau tracers are under development, and a number of phase 3 clinical studies are ongoing, with results keenly anticipated. In addition, further work involving tau, Aβ, and further pathologies, performed at different stages of the disease process will yield yet further insights into disease pathogenesis. These novel imaging targets give a real opportunity to diagnose dementia accurately, and to evaluate multi-targeted therapy much more efficiently.
References
Spillantini MG, Goedert M. Tau pathology and neurodegeneration. Lancet Neurol. 2013;12:609–22. doi:10.1016/s1474-4422(13)70090-5.
Gomez-Isla. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol. 1997;41:17–24.
Ingelsson M, Fukumoto H, Newell K, Growdon W, Hedley-White E, Frosch M, et al. Early amyloid accumulation and progressive synaptic loss, gliosis and tangle formation in the Alzheimer’s disease brain. Neurology. 2004;62:925–31.
Lee HG, Perry G, Moreira PI, Garrett MR, Liu Q, Zhu X, et al. Tau phosphorylation in Alzheimer’s disease: pathogen or protector? Trends Mol Med. 2005;11:164–9. doi:10.1016/j.molmed.2005.02.008.
Giacobini E, Gold G. Alzheimer disease therapy—moving from amyloid-beta to tau. Nat Rev Neurol. 2013;9:677–86. doi:10.1038/nrneurol.2013.223.
Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8:663–72. doi:10.1038/nrn2194.
Herrup K, Carrillo MC, Schenk D, Cacace A, Desanti S, Fremeau R, et al. Beyond amyloid: getting real about nonamyloid targets in Alzheimer’s disease. Alzheimer’s Dement: J Alzheimer’s Assoc. 2013;9:452–8. doi:10.1016/j.jalz.2013.01.017. e1.
Stoothoff W, Johnson G. Tau phosphorylation: physiological and pathological consequences. Biochim Biophys Acta. 2005;280–97.
Grundke-Iqbal I, Iqbal K, Tung Y-C, Quinlan M, Wisniewski H, Binder L. Abnormal phosporylation of the microtubule-associated protein tau in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–7.
Kosik K, Joachim C, Selkoe D. Microtubule-associated protein Tau is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci. 1986;83:4044–8.
Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, et al. Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron. 2010;68:1067–81. doi:10.1016/j.neuron.2010.11.030.
Braak H, Braak E. Neuropathological staging of Alzheimer related changes in Alzheimer’s disease. Acta Neuropathol. 1991;82:239–59.
Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18:351–7.
Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer’s Disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10:698–712.
Rowe CC, Ellis KA, Rimajova M, Bourgeat P, Pike KE, Jones G, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging. 2010;31:1275–83. doi:10.1016/j.neurobiolaging.2010.04.007.
Becker JA, Hedden T, Carmasin J, Maye J, Rentz DM, Putcha D, et al. Amyloid-beta associated cortical thinning in clinically normal elderly. Ann Neurol. 2011;69:1032–42. doi:10.1002/ana.22333.
Villemagne V, Pike K, Chetelat G, Ellis K, Mulligan R, Bourgeat P, et al. Longitudinal assessment of Aβ and cognition in aging and Alzheimer disease. Ann Neurol. 2011;69:181–92. doi:10.1002/ana.22248.
Villemagne V, Burnham S, Bourgeat P, Brown B, Ellis K, Salvado O, et al. Amyloid B deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s Disease: a prospective cohort study. Lancet. 2013;12:357–67. doi:10.1016/S1474-4422(13)70044-9.
Engler H, Forsby A, Almqvist O, Blomquist G, Larsson E, Savitcheva I, et al. Two year follow up of amyloid deposition in patients with Alzheimer’s disease. Brain: J Neurol. 2006;129:2856–66.
Ittner LM, Gotz J. Amyloid-beta and tau—a toxic pas de deux in Alzheimer’s disease. Nat Rev Neurosci. 2011;12:65–72. doi:10.1038/nrn2967.
Price J, Morris J. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol. 1999;45:358–68.
Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Aβ 42 fibrils. Science. 2001;293:1491–5. doi:10.1126/science.1062097.
Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–91. doi:10.1126/science.1058189.
Bolmont T, Clavaguera F, Meyer-Luehmann M, Herzig MC, Radde R, Staufenbiel M, et al. Induction of tau pathology by intracerebral infusion of amyloid-beta-containing brain extract and by amyloid-beta deposition in APP x Tau transgenic mice. Am J Pathol. 2007;171:2012–20. doi:10.2353/ajpath.2007.070403.
Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284:12845–52. doi:10.1074/jbc.M808759200.
Novak P, Prcina M, Kontsekova E. Tauons and prions: infamous cousins? J Alzheimer’s Dis: JAD. 2011;26:413–30. doi:10.3233/JAD-2011-110194.
Boeve BF. Progressive supranuclear palsy. Parkinsonism Relat Disord. 2012;18:S192–4. doi:10.1016/s1353-8020(11)70060-8.
Takashima A. Tauopathies and tau oligomers. J Alzheimer’s Dis: JAD. 2013;37:565–8. doi:10.3233/JAD-130653.
Saito Y, Ruberu N, Sawabe M, Motoji M, Arai T, Tanaka N, et al. Staging of argyrophilic grains — an age-associated tauopathy. J Neuropathol Exp Neurol. 2004;63:911–8.
Yamada M, Itoh Y, Sodeyama N, Suematsu N, Otamo E, Matohushita M, et al. Senile dementia of the neurofibrillary tangle type : a comparison with Alzheimer’s disease. Dement Geriatr Cogn Disord. 2001;12:117–26.
Murray M, Kouri N, Lin W, Jack C, Dickson D, Vemuri P. Clinicopathologic assessment and imaging of tauopathies in neurodegenerative dementias. Alzheimer’s Res Ther. 2014;6(1):1.
Gandy S, Ikonomovic M, Mitsis E, Elder G, Ahlers S, Barth J, et al. Chronic traumatic encephalopathy: clinical–biomarker correlations and current concepts in pathogenesis. Mol Degener. 2014;9:37.
McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain: J Neurol. 2013;136:43–64. doi:10.1093/brain/aws307.
Liscic RM, Srulijes K, Groger A, Maetzler W, Berg D. Differentiation of progressive supranuclear palsy: clinical, imaging and laboratory tools. Acta Neurol Scand. 2013;127:362–70. doi:10.1111/ane.12067.
Kovacs G. Invited review: Neuropathology of tauopathies: principles and practice. Neuropathol Appl Neurobiol. 2015;41:3–23.
Rabano A, Rodal I, Cuadros R, Calero M, Hernandez F, Avila J. Argyrophilic grain pathology as a natural model of tau propagation. J Alzheimers Dis. 2014;40:S123–33.
Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging–Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement: J Alzheimer’s Assoc. 2011;7:280–92. doi:10.1016/j.jalz.2011.03.003.
Forlenza OV, Diniz BS, Teixeira AL, Stella F, Gattaz W. Mild cognitive impairment. Part 2: biological markers for diagnosis and prediction of dementia in Alzheimer’s disease. Rev Bras Psiquiatr. 2013;35:284–94. doi:10.1590/1516-4446-2012-3505.
Bucchave P, Minthon L, Zetterberg H, Wallin A, Blemar K, Hanson O. Cerebrospinal fluid levels of B-amyloid 1–42 but not of tau are fully changed 5–10 years before the onset of Alzheimer’s disease. Arch Gen Psychiatry. 2012;69:98–106.
Wallin AK, Blennow K, Andreasen N, Minthon L. CSF biomarkers for Alzheimer’s Disease: levels of beta-amyloid, tau, phosphorylated tau relate to clinical symptoms and survival. Dement Geriatr Cogn Disord. 2006;21:131–8. doi:10.1159/000090631.
Shah M, Catafau AM. Molecular imaging insights into neurodegeneration: focus on Tau PET radiotracers. J Nucl Med: Off Publi Soc Nucl Med. 2014;55:871–4. doi:10.2967/jnumed.113.136069.
Villemagne V, Fodero-Tavoletti M, Masters C, Rowe C. Tau imaging: early progress and future directions. Lancet Neurol. 2015;14:114–24.
Villemagne V, Furomoto S, Fodero-Tavoletto M, Harada R, Mulligan R, Kudo Y, et al. The challenges of Tau imaging. Future Neurol. 2012;7:409–21.
Okamura N, Harada R, Furumoto S, Arai H, Yanai K, Kudo Y. Tau PET imaging in Alzheimer’s disease. Curr Neurol Neurosci Rep. 2014;14:500. doi:10.1007/s11910-014-0500-6.
Villemagne VL, Okamura N. In vivo tau imaging: obstacles and progress. Alzheimer’s Dement: J Alzheimer’s Assoc. 2014;10:S254–64. doi:10.1016/j.jalz.2014.04.013.
Barrio J, Kepe V, Satyamurthy N, Huang S, Small G. Amyloid and tau imaging, neuronal losses and function in mild cognitive impairment. J Nutr Health Ageing. 2008;12:61(S)–5(S).
Small G, Siddarth P, Kepe V, Ercoli L, Burggren A, Bookheimer S, et al. Prediction of cognitive decline by positron emissions tomography of brain amyloid and Tau. Arch Neurol. 2012;69:215–20.
Nelson L, Siddarth P, Kepe V, Scheibel K, Huang S, Barrio J, et al. Positron emission tomography of brain B-amyloid and Tau levels in adults with Down Syndrome. Arch Neurol. 2011;68:768–74.
Smid L, Kepe V, Vinters H, Bresjanac M, Toyokuni T, Satyamurthy N, et al. Postmortem 3-D brain hemisphere cortical tau and amyloid-beta pathology mapping and quantification as a validation method of neuropathology imaging. J Alzheimer’s Dis: JAD. 2013;36:261–74. doi:10.3233/JAD-122434.
Agdeppa ED, Kepe V, Petri A, Satyamurthy N, Liu J, Huang SC, et al. In vitro detection of (S)-naproxen and ibuprofen binding to plaques in the Alzheimer’s brain using the positron emission tomography molecular imaging probe 2-(1-[6-[(2-[(18)F]fluoroethyl)(methyl)amino]-2-naphthyl]ethylidene)malononitrile. Neuroscience. 2003;117:723–30.
Yaqub M, Boellaard R, van Berckel BN, Tolboom N, Luurtsema G, Dijkstra AA, et al. Evaluation of tracer kinetic models for analysis of [18F]FDDNP studies. Mol Imaging Biol. 2009;11:322–33. doi:10.1007/s11307-009-0208-1.
Small G, Kepe V, Ercoli L, Siddarth P, Bookheimer S, Miller K, et al. Positron emission tomography of brain amyloid and Tau in mild cognitive impairment. N Engl J Med. 2006;355:2652–63.
Villemagne VL, Furumoto S, Fodero-Tavoletti MT, Mulligan RS, Hodges J, Harada R, et al. In vivo evaluation of a novel tau imaging tracer for Alzheimer’s Disease. Eur J Nucl Med Mol Imaging. 2014;41:816–26.
Okamura N, Furumoto S, Fodero-Tavoletti MT, Mulligan RS, Harada R, Yates P, et al. Non-invasive assessment of Alzheimer’s disease neurofibrillary pathology using 18F-THK5105 PET. Brain: JNeurol. 2014;137:1762–71. doi:10.1093/brain/awu064.
Chien DT, Bahri S, Szardenings AK, Walsh JC, Mu F, Su MY, et al. Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807. J Alzheimer’s Dis: JAD. 2013;34:457–68. doi:10.3233/JAD-122059.
Sperling RMM, Johnson K. The Evolutions of Preclinical Alzheimer’s Disease: Implications for Prevention Trials. Neuron. 2014;84:608–22.
Mintun M, Devous M, Pontecorvo M, Joshi A, Siderowf A, Johnson K, et al. Potential for PET Imaging Tau Tracer 18F-AV-1451 (also known as 18F-T807) to Detect Neurodegenerative Progression in Alzheimer’s Disease. Human Amyloid Imaging Conference Book of Abstracts 2015 Submission ID 95. 2015:87.
Maruyama M, Shimada H, Suhara T, Shinotoh H, Ji B, Maeda J, et al. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron. 2013;79:1094–108. doi:10.1016/j.neuron.2013.07.037.
Okamura NST, Furumoto S, Suzuki M, Shimadzu H, Akatsu H, Yamamoto T, et al. Quinoline and benzimidazole derivatives: candiate probes for in vivo imaging of tau pathology in Alzheimer’s Disease. J Neurosci. 2005;25:10857–62.
Villemagne V, Furumoto S, Fodero-Tavoletti M, Mulligan R, Hodges J, Harada R, et al. In vivo evaluation of a novel tau imaging tracer for Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2014;41:816–26. doi:10.1007/s00259-013-2681-7.
Harada R, Okamura N, Furumoto S, Tago T, Maruyama M, Higuchi M, et al. Comparison of the binding characteristics of [18F]THK523 and other amyloid imaging tracers to Alzheimer’s Disease pathology. Eur J Nucl Med Mol Imaging. 2013;40:125–32.
Fodero-Tavoletti M, Okamura N, Furumoto S, Mulligan R, Connor A, McLean C, et al. 18F-THK523: a novel in vivo tau imaging ligand for Alzheimer’s disease. Brain: J Neurol. 2011;134:1089–100. doi:10.1093/brain/awr038.
Fodero-Tavoletti M, Furomoto S, Taylor L, McLean C, Mulligan R, Buchal I, et al. Assessing THK523 selectivity for Tau deposits in Alzheimer’s disease and non-Alzheimer’s disease tauopathies. Alzheimer’s Res Ther. 2014;6.
Okamura N, Furumoto S, Harada R, Tago T, Yoshikawa T, Fodero-Tavoletti M, et al. Novel 18F-labeled arylquinoline derivatives for noninvasive imaging of tau pathology in Alzheimer disease. J Nucl Med: Off Publ Soc Nucl Med. 2013;54:1420–7. doi:10.2967/jnumed.112.117341.
Lemoine L, Saint-Aubert L, Marutle A, Antoni G, Eriksson JP, Ghetti B, et al. Visualization of regional tau deposits using (3)H-THK5117 in Alzheimer brain tissue. Acta Neuropathol Commun. 2015;3:40. doi:10.1186/s40478-015-0220-4.
Nordberg A, Chiotis K, Aubert L, Wall A, Jonasson M, Lubberink M, et al. Tau Imaging with [18F]-(S)-THK5117 in MCI and AD Patients Using a PET Multii-Tracer Concept. Human Amyloid Imaging Conference Book of Abstracts 2015 ID submission 106. 2015:124.
Saint-Aubert L, Lemoine L, Marutle A, Antoni G, Eriksson J, Okamura N, et al. Relationship between Post-Mortem THK5117 Binding and In-Vivo PET Biomarkers Uptake in Alzheimer’s Disease. Human Amyloid Imaging Conference Book of Abstracts 2015 ID submission 94. 2015:94.
Harada R, Okamura N, Furumoto S, Tago T, Furukawa K, Ishiki A, et al. First-in-human PET Study of a Novel Tau Tracer [18F]THK5351. Human Amyloid Imaging Conference Book of Abstracts 2015 Submission ID 24. 2015.
Tago TFS, Okamura N, Harada R, Ishikawa Y, Arai H, Yanai K, et al. Synthesis and preliminary evaluation of 2-arylhydroxyquinoline derivatives for tau imaging. J Labelled Comp Radiopharm. 2014;57:18–24.
Gao M, Wang M, Zheng QH. Concise and high-yield synthesis of T808 and T808P for radiosynthesis of [(18)F]-T808, a PET tau tracer for Alzheimer’s disease. Bioorg Med Chem Lett. 2014;24:254–7. doi:10.1016/j.bmcl.2013.11.025.
Zhang W, Arteaga J, Cashion DK, Chen G, Gangadharmath U, Gomez LF, et al. A highly selective and specific PET tracer for imaging of tau pathologies. J Alzheimer’s Dis: JAD. 2012;31:601–12. doi:10.3233/JAD-2012-120712.
Chien DT, Szardenings AK, Bahri S, Walsh JC, Mu F, Xia C, et al. Early clinical PET imaging results with the novel PHF-tau radioligand [F18]-T808. J Alzheimer’s Dis: JAD. 2014;38:171–84. doi:10.3233/JAD-130098.
Kolb H, Attardo G, Mintun M, Chien D, Elizarov A, Conti P, et al. First case report: image to autopsy correlation for tau imaging with [18F]-T808 (AV-680). Alzheimers Dement. 2013;9:P844–5. doi:10.1016/j.jalz.2013.08.008.
Xia CF, Arteaga J, Chen G, Gangadharmath U, Gomez LF, Kasi D, et al. [(18)F]T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimer’s Dement: J Alzheimer’s Assoc. 2013;9:666–76. doi:10.1016/j.jalz.2012.11.008.
Marquie M, Normandin M, Vanderburg C, Costantino I, Dickerson B, Frosch M, et al. Towards the Validation of Novel PET tracer T807 on Postmortem Human Brain Tissue Samples. Abstract Online: Human Amyloid Imaging Book of Abstracts 2015 Submission ID 89. 2015:28.
Pontecorvo M, Devous M, Joshi A, Lu M, Siderowf A, Arora A, et al. Relationships between Florbetapir PET Amyloid and 18F AV-1451 (aka 18F-T807) PET Tau Binding in Cognitively Normal Subjects and Patients with Cognitive Impairment Suspected of Alzheimer’s Disease. Human Amyloid Imaging Conference Book of Abstracts 2015 ID submission 98. 2015:131.
Johnson K, Becker J, Sepulcre J, Rentz D, Schultz A, Pepin L, et al. Tau PET: Initial Experience with F18 T807. Human Amyloid Imaging Conference Book of Abstracts 2014 ID submission 103. 2014:22.
Ossenkoppele R, Schonhaut D, Baker S, O’Neil J, Janabi M, Ghosh P, et al. Tau, amyloid, and hypometabolism in a patient with posterior cortical atrophy. Ann Neurol. 2014. doi:10.1002/ana.
Ossenkoppele R, Schonhaut D, Baker S, Lazaris A, Ayakte N, Cantwell A, et al. Distinct [18F]AV1451 Retention Patterns in Clinical Variants of Alzheimer’s Disease. Human Amyloid Imaging Conference Book of Abstracts 2015 P55 Submission 113 2015:62.
Dickerson B, McGinnis S, Gomperts S, Caso C, Makaretz S, Kelly K, et al. [18F]T807 of Frontotemporal Lobar Degeneration. Human Amyloid Imaging Conference Book of Abstracts 2015 P15 Submission ID 61. 2015:37.
Gomperts S, Makaretz S, Schultz A, Caso C, McGinnis S, Growdon J, et al. [18F] T807 PET Imaging of Hyperphosphorylated Tau to Differentiate PSP from PD. Human Amyloid Imaging Conference Book of Abstracts 2015 ID submission118. 2015:111.
Mitsis EM, Riggio S, Kostakoglu L, Dickstein DL, Machac J, Delman B, et al. Tauopathy PET and amyloid PET in the diagnosis of chronic traumatic encephalopathies: studies of a retired NFL player and of a man with FTD and a severe head injury. Transl Psychiatry. 2014;4, e441. doi:10.1038/tp.2014.91.
Schultz A, Chhatwal J, Mormino E, LaPoint M, Dagley A, Sperling R, et al. Relationship between longitudinal amyloid accumulation and T807-Tau. Human Amyloid Imaging Conference Book of Abstracts 2015 P65 Submission ID 101. 2015:68.
Sperling R, Mormino E, Rentz D, Schultz A, Becker A, Hedden J, et al. T807 PET Measures of Tau Pathology Associated with Episodic Memory Performance among Clinically Normal Older Individuals. Human Amyloid Imaging Conference Book of Abstracts 2015 ID submission 123. 2015:93.
Chhatwal J, Schultz A, Marshall G, Boot B, Gomez-Isla T, Dumurgier J, et al. Entorhinal, Parahippocampal, and Inferior Temporal F18-T807 SUVR Correlates with CSF Total Tau and Tau T181P in Cognitively Normal Elderly. Human Amyloid Imaging Conference Book of Abstracts 2015 Submission ID 115. 2015:88.
Lockhart S, Vogel J, Schonhaut D, Baker S, Schwimmer H, Scholl M, et al. Effects of Age and B-Amyloid on Tau Deposition Measured Using [18F]AV-1451 in Normal Older People. Human Amyloid Imaging Conference Book of Abstracts 2015 P41 Submission ID 55. 2015:54.
Hashimoto H, Kawamura K, Igarashi N, Takei M, Fujishiro T, Aihara Y, et al. Radiosynthesis, photoisomerization, biodistribution, and metabolite analysis of 11C-PBB3 as a clinically useful PET probe for imaging of tau pathology. J Nucl Med: Off Publ Soc Nucl Med. 2014;55:1532–8. doi:10.2967/jnumed.114.139550.
Kimura Y, Ichise M, Ito H, Shimada H, Ikoma Y, Seki C, et al. PET quantification of Tau pathology in human brain with 11C-PBB3. J Nucl Med: Off Publ Soc Nucl Med. 2015. doi:10.2967/jnumed.115.160127.
Shimada H, Higuchi M, Ikoma Y, Shinotoh H, Furukawa S, Moriguchi S, et al. In vivo visualization of tau pathology. Alzheimers Dement. 2013;9:P101–2. doi:10.1016/j.jalz.2013.05.172.
Honer M, Knust H, Gobbi L, Kuwubara H, Muri D, Valentine H, et al. In Vitro and In vivo Evaluation of RO6931643, RO6924963 and RO6958948 Candidate PET Tracers for Aggregated Tau. Human Amyloid Imaging Conference Book of Abstracts 2015 ID submission 34. 2015:114.
Acknowledgments
The authors thank Nobuyuki Okamura for providing the PET image of [18F]THK5351
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Dr. Edison was funded by the Medical Research Council and now by Higher Education Funding Council for England (HEFCE). He has also received grants from Alzheimer’s Research, UK, Alzheimer’s Drug Discovery Foundation, Alzheimer’s Society, UK, Novo Nordisk and GE Healthcare. Prof. Brooks has received research grants and non-financial support from the Medical Research Council, grants from Alzheimer’s Research Trust, during the conduct of the study; other than from GE Healthcare, personal fees from AstraZeneca, personal fees from Cytox, personal fees from Shire, personal fees from Novartis, personal fees from GSK, Holland, personal fees from Navidea, personal fees from UCB, personal fees from Acadia, grants from Michael J Fox Foundation, grants from European Commission, outside the submitted work.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institution and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Dani, M., Brooks, D.J. & Edison, P. Tau imaging in neurodegenerative diseases. Eur J Nucl Med Mol Imaging 43, 1139–1150 (2016). https://doi.org/10.1007/s00259-015-3231-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00259-015-3231-2