
Abstract.
Vascular endothelial growth factors (VEGFs) regulate blood and lymphatic vessel development and homeostasis but also have profound effects on neural cells. VEGFs are predominantly produced by endothelial, hematopoietic and stromal cells in response to hypoxia and upon stimulation with growth factors such as transforming growth factors, interleukins or platelet-derived growth factor. VEGFs bind to three variants of type III receptor tyrosine kinases, VEGF receptor 1, 2 and 3. Each VEGF isoform binds to a particular subset of these receptors giving rise to the formation of receptor homo- and heterodimers that activate discrete signaling pathways. Signal specificity of VEGF receptors is further modulated upon recruitment of coreceptors, such as neuropilins, heparan sulfate, integrins or cadherins. Here we summarize the knowledge accumulated since the discovery of these proteins more than 20 years ago with the emphasis on the signaling pathways activated by VEGF receptors in endothelial cells during cell migration, growth and differentiation.
Similar content being viewed by others


Historical background
In higher organisms, blood and lymphatic vasculature is formed by two distinct processes: vasculogenesis and angiogenesis. Vasculogenesis is the de novo formation of vessels from hematopoietic precursor cells and predominantly takes place in the developing embryo. Angiogenesis, on the other hand, is the formation of vessels from preexisting vasculature by processes such as sprouting, pruning and intussusception [1] and is an important biological process throughout the life of an organism, both under normal conditions and in disease. Impaired vessel function is the cause of many illnesses such as atherosclerosis, diabetic retinopathy, psoriasis, arthritis, malignant cell growth, neurodegenerative disease and a placental insufficiency, preeclampsia [2]. Hematopoietic precursor cells are programmed by soluble factors, extracellular matrix (ECM) components as well as cell-cell contacts to mature to their final functional states during vessel formation. Research in this field has been profoundly stimulated by the discovery of a plethora of growth factors that instruct primordial cells to migrate, divide and differentiate and to give rise to endothelial cells that ultimately form blood and lymphatic vessels. Vascular endothelial growth factors, (VEGFs) are among the most important players that regulate vessel formation during embryonic development, in wound healing and in maintaining vessel homeostasis in adult organisms. In addition, impaired vessel function resulting from defects in VEGF ligands or receptors is the cause of many diseases. VEGF was originally described as vascular permeability factor (VPF), an activity released by tumor cells that promotes vascular leakage [3–12]. It is now clear that VPF represented a biological activity attributable to a family of polypeptide growth factors that are encoded by several genes. VEGFs specifically interact with hematopoietic cells, endothelial precursor cells, such as the angioblasts, and with differentiating and mature endothelial cells. The best studied variant is VEGF-A, which activates a plethora of signaling pathways through VEGF receptor-2 (VEGFR-2) and regulates vessel morphogenesis through VEGFR-1. The importance of VEGF-A and its receptors in vascular development has been best illustrated in knockout mice. Both VEGF-A [13] and VEGFR-2 knockouts [14, 15] are lethal due to a deficiency in blood vessel formation, while VEGFR-1 knockouts show overgrowth of immature vessels that blocks the development of a fully developed functional vasculature and leads to embryonic death [16]. The function of VEGFs in vessel formation is complemented by additional factors, such as basic fibroblast growth factor (bFGF) [17], transforming growth factor β (TGFβ) [18], platelet-derived growth factors (PDGFs) [19] and angiopoietins [20].
VEGF homologs also exist in arthropods, where they regulate hemocyte development upon binding to PDGF/VEGF receptors (PVRs). Apparently, in such simpler organisms, a single growth factor performs the tasks performed by PDGF and VEGF in higher organisms [21–23]. Apart from their role in vessel development and homeostasis, VEGF family proteins play diverse roles in other organs such as the neural system, bones, the hematopoietic system and the reproductive organs, which have been excellently reviewed recently [24].
Biological function of VEGF receptors and their ligands
VEGF family proteins
VEGF polypeptides belong to the PDGF family of growth factors. They are dimeric cysteine-linked secreted glycoproteins with an Mr of approximately 40 kDa. In mammals, VEGFs are encoded by a family of genes that includes VEGF-A, -B, -C, -D [25] and PlGF [26]. Highly related proteins called VEGF-E are encoded by pox viruses of the Orf family [27–29] and additional variants, collectively called VEGF-F, have been isolated from snake venoms [30–35]. Alternative splicing and proteolytic processing of VEGFs give rise to a number of functionally distinct isoforms with different signaling properties [36, 37]. For example, Bates and colleagues described an interesting variant, VEGF-A165b, closely related to VEGF-A165, that carries sequences encoded by exon 9, instead of exon 8, at the carboxy terminus [38, 39]. When added together with VEGF-A165 to endothelial cells, this variant inhibited VEGF signaling. The concept emerging from such studies is that multiple isoforms of VEGFs, binding their receptors with similar affinity, yet eliciting distinct signaling properties, are responsible for the bewildering complexity of VEGF-induced signal output.
VEGFs are expressed in response to hypoxia and when cells encounter specific growth and differentiation factors and are produced by many cell types, in particular by hematopoietic, stromal and endothelial cells [40–42]. One of the most prominent examples is the stimulation of VEGF expression by cancer cells that cannot form tumors bigger than a few millimeters unless they produce angiogenic growth factors [2, 43].
VEGF receptors
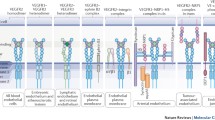
The biological functions of VEGF polypeptides are mediated upon binding to type III receptor tyrosine kinases (RTKs), VEGFR-1 (Flt-1), VEGFR-2 (KDR/Flk-1) and VEGFR-3 (Flt-4) [2, 44–47]. VEGF receptors are closely related to Fms, Kit and PDGF receptors. They consist of seven extracellular immunoglobulin (Ig)-like domains, a transmembrane (TM) domain, a regulatory juxtamembrane domain, an intracellular tyrosine kinase domain interrupted by a short peptide, the kinase insert domain, followed by a sequence carrying several tyrosine residues involved in recruiting downstream signaling molecules. These receptors are expressed on the cell surface of many bone-marrow-derived cells such as hematopoietic cells [48], macrophages and endothelial cells [49], on some malignant cells [50] and on vascular smooth muscle cells (VSMCs) [51]. Mutation analysis of the extracellular domains of VEGFR-1 and -2 showed that the second and third Ig-like domains constitute the high-affinity ligand-binding domain for VEGF with the first and fourth Ig domains apparently regulating ligand binding and receptor dimerization, respectively [52–54]. VEGFs show distinct patterns of receptor specificity as indicated in figure 1. VEGF-A binds to VEGFR-1 and -2 and to receptor heterodimers, while VEGF-C and -D bind VEGFR-2 and -3. Receptor-specific interactions have been described for some VEGF variants: PlGF [55, 56] and VEGF-B [57] exclusively bind VEGFR-1 and VEGF-E interacts only with VEGFR-2 [27, 58]. VEGF-F variants interact with either VEGFR-1 or -2, e.g. VR-1 and Vammin bind only to VEGFR-2 [32, 33]. VEGF-A, -B and PlGF are predominantly required for blood vessel formation, while VEGF-C and -D are essential for the formation of lymphatic vessels [59, 60].

Schematic representation of VEGF family ligands and their receptors.
Regulation of receptor activity
RTKs are activated upon ligand-mediated receptor dimerization [61–63]. Earlier published work suggested that ligand binding is not directly responsible for receptor dimerization but may induce conformational changes in the extracellular Ig domain 4 which then promote receptor dimerization [64–67]. Ligand-induced dimerization leads to structural changes transduced to the intracellular kinase domain upon rearrangement of the transmembrane and the juxtamembrane domain. The molecular mechanisms responsible for activation of the intracellular kinase domain of these receptors are poorly understood at present. Kit and PDGF receptors mutated in the trans- or juxtamembrane domains are constitutively active and often oncogenic [68, 69]. Similarly, a role of the TM domain in receptor activation has been elegantly documented for PDGF receptors carrying artificially engineered dimerization motifs [70]. These data clearly establish that the TM and the juxtamembrane domains are involved in regulating receptor kinase activity.
All three VEGF receptors contain tyrosine phosphorylation sites that have an either regulatory or signaling function. While phosphorylation of sites in the juxtamembrane domain and the lower lobe of the kinase domain presumably modulates receptor structure and kinase activity, others act as docking sites for SH2-domain-containing signaling molecules. Four experimental approaches have been used to study the biological function of these phosphorylation sites: (i) mutation analysis in which specific tyrosine residues were mutated and receptor-associated signaling molecules and biological output were determined, (ii) phosphopeptide mapping of receptors isolated from resting and ligand-stimulated cells labeled with inorganic phosphate in vivo, (iii) phosphopeptide mapping of in-vitro-phosphorylated receptors, (iv) determination of the phosphorylation state of specific residues with phosphotyrosine-specific antibodies. Each of these methods has its shortcomings and published data on receptor activity and binding of downstream signaling molecules are therefore often difficult to compare. Mutational analysis, for example, might change receptor structure and therefore biological activity, while results from in vivo and in vitro phosphorylation analysis are biased by the different turnover of tyrosine phosphates labeled under these conditions.
VEGFR-1 regulates blood vessel morphogenesis
VEGFR-1 is an 180-kDa glycoprotein expressed in many hematopoietic cells. The receptor is required for normal blood vessel development during embryogenesis, since homozygous deletion of VEGFR-1 is lethal in mice at embryonic day E8.5 due to severe malformation of the vasculature [16]. A VEGFR-1 splice variant lacking the intracellular tyrosine kinase and the transmembrane domain, sVEGFR-1 or sFlt-1, has been shown to be deficient in signaling, yet is expressed in many tissues during normal embryonic development. This molecule apparently acts as a decoy for VEGF ligands [71–73] and is clinically associated with a placental insufficiency, called preeclampsia, observed in some patients late in pregnancy [74]. The view that VEGFR-1 kinase activity is dispensable for vessel development at particular developmental stages is further supported by the finding that a kinase-inactive VEGFR-1 mutant rescues VEGFR-1 null mutant mice [75]. More recent data indicate that the kinase activity of VEGFR-1 plays an essential role during pathological angiogenesis and in wound healing, by potentiating VEGFR-2 signaling [76–78], however, the molecular details for this receptor cross-talk have not yet been elucidated. Undisputed is the role of kinase-active VEGFR-1 in recruiting hematopoietic cells from bone marrow precursors [79, 80].
VEGFR-1 has poor kinase activity compared with VEGFR-2 due to the presence of a repressor motif in the juxtamembrane domain, making studies on receptor phosphorylation difficult [81]. A wide variety of signaling molecules has been shown to be activated by VEGFR-1 upon recruitment to specific phosphorylation sites [82–86]. Tyr1213 and 1333 serve as binding sites for adaptor molecules such as Nck, Crk, Grb-2 [84, 87], Sck [88], the regulatory p85 subunit of phosphatidylinositol (PI) 3-kinase [85] and the phosphatase SHP-2 [87]. Phospholipase Cγ-1 (PLCγ-1) has been shown to associate with VEGFR-1 via Tyr794 and 1169 [86, 89]. Tyr1242 and 1327 are minor phosphorylation sites with no interacting intermediates described to date. The downstream signaling pathways activated by VEGFR-1 are not well characterized and only weak proliferative or migratory effects are mediated by this receptor in endothelial cells. Conflicting reports indicate that VEGFR-1 has mitogenic potential in a PLCγ-1- and phosphokinase C (PKC)-dependent or independent manner [89–91]. Taking into account more recent results from P. Carmeliet’s group, these data may arise from receptor cross-talk with VEGFR-2 [76]. A single study shows activation of Fyn and Yes, two members of Src family kinases [92], but no biological function has been attributed to these interactions. The function of VEGFR-1 is best established in monocyte migration and differentiation [79, 93], in the recruitment of endothelial cell progenitors from bone marrow [80, 94, 95], the migration and invasion of carcinoma cells [96] , the production of growth factors by liver sinusoidal endothelial cells [97] and the adhesion of natural killer cells to endothelial cells [98].
VEGFR-2 is the predominant receptor in angiogenic signaling
VEGFR-2 is a 200-kDa glycoprotein expressed in hematopoietic [48, 99, 100], neural [101–103] and retinal cells [104]. VEGFR-2 regulates endothelial cell migration, proliferation, differentiation and survival as well as vessel permeability and dilation. Among the 19 tyrosine residues present in the intracellular domain of VEGFR-2, seven putative phosphorylation sites have been described in some detail to date: Tyr801, 951, 996, 1054, 1059, 1175 and 1214. A comprehensive study of receptor activity based on in vitro phosphorylation of immunoprecipitated VEGFR-2, on receptor mutagenesis and on in vivo mapping with phosphorylation site-specific antibodies identified Tyr951, 1054, 1059, 1175 and 1214 as the most prominent phosphorylation sites and Tyr1305, 1309 and 1319 as minor sites, while Tyr801 and 996 phosphorylation was not detected in this study [105]. Tyr1175 is clearly the most important site implicated in activation of many pathways via PLCγ-1 [106]. A role for Tyr1175 in endothelial and hematopoietic cell signaling was also shown in knockin mice expressing a Tyr1175 mutant. In this study, animals expressing the mutant receptor died in utero from vascular defects similar to those observed in VEGFR-2 null mice [107]. A mutagenesis study led to the identification of Tyr1008 as an additional site involved in activation of PLCγ-1, promoting endothelial cell tubulogenesis and differentiation but not cell proliferation [108].
The mechanism of receptor activation is not understood in molecular detail. Tyr1054 and 1059 in the kinase domain, which are homologous to regulatory residues present in all protein kinases, were tentatively identified as autophosphorylation sites [109, 110]. Similar to other RTKs, the putative phosphorylation sites Tyr801 and 822 located in the juxtamembrane domain may be phosphorylated following dimerization of VEGFR-2 and maintain the receptor in an active conformation [111]. Mutation analysis also showed that Tyr996 and 1214 in the kinase and the carboxy-terminal domain are essential for receptor activation; however, one should bear in mind that such results may derive from structural changes in these mutant receptors [109, 112].
VEGFR-2 is downregulated and dephosphorylated upon internalization into endocytic vesicles [109, 113]. Both inactivation by directly associated phosphatases as well as dephosphorylation upon association with other membrane receptors carrying associated phosphatases such as SHP-1 and -2 have been described [114, 115]. A particularly interesting case is the downregulation of VEGFR-2 by tumor necrosis factor α (TNFα), which recruits SHP-1 to the plasma membrane [116, 117]. Finally, a variety of less well characterized signaling molecules have been shown to associate with the activated receptor and to transmit downstream signals. These include the Ras GT-Pase-activating protein GAP [118], the adaptor proteins Nck [118], Grb-2, Grb-10 and Sck [88, 114, 119, 120] and the human cellular protein tyrosine phosphatase A (HCTPA) [121].
VEGFR-2 promotes mitogenesis
VEGF-mediated endothelial cell proliferation depends on the activation of multiple pathways downstream from VEGFR-2. Similar to other members of the RTK family, VEGFR-2 activates the classical Ras-dependent signaling cascade impinging on MAP kinases such as ERK1 and 2 [122]. In this pathway, VEGFR-2 recruits Grb-2, either by a direct interaction involving the putative docking site at Tyr1214 [123], or via association with the adaptor protein Shc [114]. Phosphorylation of Grb-2 leads to activation of the nucleotide exchange factor Sos followed by activation of Ras and stimulation of the Raf1/MEK/ERK signaling cascade. Cross-talk between ERK1 and 2 and another MAP kinase family member, c-Jun N-terminal protein kinase (JNK), has also been described, rendering difficult the assignment of specific biological roles to the individual MAP kinase family members [124]. Activated MAP kinases translocate to the nucleus where they phosphorylate transcription factors and regulate gene expression.
Most studies on VEGF signaling agree that receptor recruitment of PLCγ-1 upon phosphorylation of Tyr1175 is essential for stimulation of mitogenesis. Activation of PLCγ-1 promotes phosphatidylinositol 4,5-bisphosphate (PIP2) hydrolysis giving rise to 1,2-diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). Production of DAG activates PKC resulting in Ras-independent Raf activation which leads to stimulation of ERK activity [106, 125–128]. Treating cells with PKC inhibitors blocked mitogenic signaling by VEGF supporting a role for PKC in this pathway [129]. Receptor association of PLCγ-1 via Tyr801 has been reported using receptor mutants [86] and mutation analysis of VEGFR-2 showed that Tyr801 and 1175 are putative docking sites for the p85 subunit of PI 3-kinase [130]. PI 3-kinase has also been implicated in mitogenic signaling by VEGFR-2, based on treatment of cells with specific inhibitors [131]. This lipid kinase regulates the S6 kinase/Akt pathway which has been shown in many cell types to stimulate cell growth. Conflicting results suggest, however, that PI 3-kinase is not required for VEGFR-2-mediated mitogenesis [122, 126]. Taken together, these data, mostly based on mutagenesis studies, should be interpreted with some care since changes in receptor structure that alter association with signaling molecules may affect receptor readout.
Finally, c-Src and nitric oxide (NO) [132] have been identified as intracellular mediators of VEGF signaling, and heparan sulfate and components of the ECM act as extracellular modulators in mitogenic signaling [133, 134], while the classical mechanism of ‘contact inhibition of growth’, described for epithelial cells or fibroblasts and mediated by VE-cadherin in endothelial cells, is reponsible for regulation of cell proliferation in a density-dependent manner [135].
VEGFR-2 regulates cytoskeleton organization and cell migration
The motogenic signals transduced by VEGFR-2 impinge on focal adhesion kinase (FAK), which regulates focal adhesion assembly and disassembly and actin organization. VEGF regulates FAK phosphorylation and activity and leads, together with paxillin and actin-anchoring proteins such as talin or vinculin, to recruitment of this kinase to focal adhesions [136, 137]. FAK activation has been shown to require signaling by PKC [136]. In addition, Nck/PAK (p21-activated kinase) was shown to promote FAK phosphorylation and cell migration [138–141]. Src-dependent FAK phosphorylation was reported to regulate cell migration and survival [142] and, consistent with these data, the Src kinase inhibitor M475271 blocked VEGF-induced endothelial cell migration [143]. Besides its role in cellular proliferation, Tyr1175 phosphorylation has been shown to regulate stress fiber formation, focal adhesion assembly and cell migration upon recruitment of the adaptor protein Shb and subsequent activation of PI 3-kinase and FAK [144]. More recently, Tyr951 located in the kinase insert domain and presumably phosphorylated by an associated kinase when VEGFR-2 is activated, was shown to recruit the adaptor molecule VRAP/TSAd (VEGF-receptor-associated protein/T-cell-specific adaptor molecule) in a subfraction of endothelial cells [105]. This adapter molecule associates with Src, PI 3-kinase and PLCγ-1 and regulates actin organization and cell migration, but it also plays a crucial role in tumor angiogenesis [105, 145].
VEGF-induced endothelial cell migration is also mediated by stress activated protein kinase 2, SAPK/p38 [139, 146]. SAPK/p38 activity is regulated by Src and related focal adhesion protein kinase/proline-rich tyrosine kinase 2 (RAFTK/Pyk2) [147]. Ca2+ mobilization and activation of Cdc42 have been shown to lead to SAPK/p38 phosphorylation [147, 148] and to induce phosphorylation of MAPKAP kinases 2 and 3 and the small heat shock protein HSP27 [146, 149]. This leads to the release of phosphorylated HSP27 from capped actin filaments, actin reorganization and the formation of stress fibers and lamellipodia which promote cell migration [150]. Furthermore, a role for the small GTPases Rho and Rac, which modulate actin dynamics and cell contraction in endothelial cell migration and which are regulated by G proteins such as Gq/11 and Gbg and by PLC, is well documented [151].
Signaling by VEGFR-2 is essential for cell survival
VEGF also protects endothelial cells against apoptosis in vitro and in vivo. In vivo, the role of VEGF in preventing apoptosis is restricted to immature vessels that lack pericytes, as indicated by experiments showing that VEGF is essential for endothelial cell survival and blood vessel development in early postnatal life, but not in adult mice [152–154]. Activation of the PI 3-kinase/Akt pathway by VEGF protects cultured cells against apoptosis induced by serum starvation [155, 156]. Signal output from VEGFR-2 maintains adequate levels of active PI 3-kinase and thereby regulates the production of the second messenger phosphoinositide(3,4,5)trisphosphate (PIP3) which is required for activation of the serine/threonine kinase Akt [157]. VEGF was also shown to induce the expression of anti-apoptotic molecules including the caspase inhibitors Bcl-2 and A1 [158] and IAP (inhibitors of apoptosis) family proteins [159]. Finally, survival signaling by VEGFR-2, PI 3-kinase and Akt depends on the integrity of adherens junctions that contain a transient tetrameric complex composed of VEGFR-2, PI 3-kinase, VE-cadherin and β-catenin [160, 161]. Disruption of this multimeric complex by loss or truncation of VE-cadherin induced endothelial cell apoptosis and blocked the transmission of PI 3-kinase-dependent survival signals.
VEGFR-2 regulates vessel permeability
VEGF also regulates vascular permeability, and indeed was initially described as a vascular permeability factor [3]. Increased vascular permeability is observed shortly after VEGF administration concomitant with the formation of so-called vesicular-vacuolar organelles, VVOs, [162] and fenestrae [163–165]. These specialized regions in the plasma membrane of endothelial cells are highly permeable for macromolecules. How these membrane structures are formed is still unclear, but based on data from mutant mice lacking both c-Src and c-Yes, which showed reduced vascular permeability after VEGF administration, a role for Src family kinases has been suggested [166]. Addition of VEGF to endothelial cells induces expression of the plasmalemmal vesicule-associated protein (PLVAP), a component of diaphragmed endothelial fenestrations. PLVAP expression is regulated by PI 3-kinase and SAPK/p38 [167]. In mature vessels, VEGF also regulates vascular permeability by loosening the junctions between endothelial cells, giving rise to the formation of transcellular gaps. Phosphorylation of major components of tight, adherens and gap junctions, such as VE-cadherin [168–170], β-catenin [168, 171], occludin and zonula occluden 1 [172, 173] and of connexin 43 [174] have been reported in response to VEGF. Vessel dilation and permeability are also regulated by NO which is upregulated by Akt upon induction of endothelial NO synthase (eNOS) expression [175, 176]. Blocking eNOS and cyclooxygenase with specific inhibitors prevents the release of NO and prostacyclin (PGI2) and blocks VEGF-induced vessel hyperpermeability [177].
VEGFR-3 regulates lymphangiogenesis
VEGFR-3 is synthesized as a 195-kDa precursor protein consisting of seven extracellular Ig-like domains, a TM and an intracellular kinase domain. The protein is proteolytically processed in the fifth Ig domain giving rise to a 125- and a 75-kDa chain held together by a disulfide bond [178, 179]. Expression of this receptor starts at E8.5 of mouse development in all embryonic endothelial cells. After E8.5, VEGFR-3 expression is only seen on developing veins and lymphatics but not on arteries [59, 180]. Later in development, the expression gradually becomes restricted to lymphatic vessels. In VEGFR-3 null mice, vascular remodeling and maturation are abnormal in larger vessels with a defective lumens causing fluid accumulation in the pericardial cavity and embryonic death at E9.5 [181]. Recent reports also showed expression of the receptor on blood vessels in the vicinity of tumors as well as on several benign and malignant tumor cells [182, 183]. A role for VEGFR-3 in cell-to-cell signaling in adult blood vessel angiogenesis, where the expression of this receptor and its ligands seems to be induced by VEGF-A, has also been suggested [184]. VEGFR-3 is activated by VEGF-C and -D, and proteolytic processing of these ligands gives rise to variants also interacting with VEGFR-2, although with lower affinity than with VEGFR-3. VEGFR-3 apparently also heterodimerizes with VEGFR-2 in lymphatic endothelial cells, expanding the repertoire of signaling pathways activated by this receptor [185]. Paracrine expression of VEGF-C at sites of lymphatic sprouting further supports a role for VEGFR-3 in the development of lymphatic vessels [186]. VEGFR-3 promotes cell migration and survival in lymphatic endothelia by inducing PKC-dependent MAP kinase activation and via a wortmannin-sensitive pathway requiring PI 3-kinase and Akt [187, 188]. In addition, a splice variant of this receptor has been shown to bind SHC [189]. That signaling via VEGFR-3 is a prerequisite for lymphangiogenesis is further supported by data showing that a soluble, kinase-deficient receptor variant blocks lymphatic vessel formation [190, 191].
VEGF receptors form multiprotein complexes with various coreceptors
VEGF signaling is complicated by the fact that the ligands and their receptors interact with additional cellular proteins such as neuropilins, heparan sulfate, integrins and cadherins. These interactions allow coordination of signal strength, timing and specificity with extracellular cues arising from soluble ligands, cell-cell and cell-substratum interactions.
Association with heparan sulfate and neuropilins
The short form of VEGF-A, VEGF-A121, encoded by exons 2–5 and 8, consists of a receptor-binding domain specific for VEGFR-1 and -2 and is a poor mitogen. The longer VEGF-A165 isoform, that also contains sequences encoded by exon 7, binds the receptors with similar affinity as VEGF-A121 but displays increased signaling potential [192–194]; the underlying mechanism remains unclear at present. All isoforms carrying exon 7, or 6 plus 7, such as VEGF-A165, VEGF-A183, VEGF-A189 and VEGF-A203, interact with proteins of the neuropilin family and with heparan sulfate [193–202]. Neuropilin-1 and -2 interact with VEGFR-2 and -1, respectively, stimulating signaling by these receptors. Interaction of VEGF-A165 with neuropilin-1 is particularly important for endothelial tip cell guidance where regulation of cell migration is the predominant signal output [203, 204]. VEGF-A165 has also been shown to interact with neuropilin-1 and VEGFR-2 expressed separately on adjacent cells. This may be particularly important during endothelial cell guidance when vessels are formed along tracks predetermined by neural cells [205–207].
Association with integrins
Integrins play an important role in cell signaling linking intracellular signaling pathways activated by soluble factors to output elicited by cellular interactions with the ECM and with neighboring cells. Specific integrins bind to the extracellular domain of VEGFR-2 and augment receptor signaling [208]. Integrins of the β 3 subfamily specifically bind to the extracellular domain of VEGFR-2 resulting in increased receptor activation upon VEGF stimulation [209–211]. Direct interaction between β 3 integrin and VEGFR-2 is restricted to α v β 3 and was shown to be either ligand independent [210] or dependent [211]. VEGFR-2 signaling in the context of α 1 β 1 and α 2 β 1 integrins has been shown to regulate lymphangiogenesis during tissue repair, further demonstrating how output from VEGF receptors is modulated by cellular interactions with the ECM [212]. VEGFR-2-mediated angiogenesis is also directly regulated by integrins as proposed by work performed in knockout mice lacking β 3 or β 5 integrin [213, 214]. Animals that did not express these integrins showed increased VEGFR-2 activity and tumor vascularization. Taken together these studies suggest that integrins act as ‘gatekeepers’, preventing aberrant stimulation of resting endothelial cells under non-pathological conditions, or as ‘caretakers’, which facilitate angiogenesis during vessel repair in disease [215].
VEGF bound to the ECM promotes integrin-dependent cell spreading, migration and survival that do not require signaling by VEGF receptors [216]. Similarly, sVEGFR-1 interacts with α 5 β 1 integrin thus becoming part of the ECM and promoting cell migration and spreading [217]. In addition, interaction of VEGF-A with fibronectin, an ECM component interacting predominantly with α 5 β 1 and, to a minor extent, with the vitronectin receptor α v β 3, has been reported recently [218]. Finally, in cells grown on fibronectin instead of vitronectin or collagen, increased biological activity of VEGF and augmented endothelial cell migration and MAP kinase activity were reported [219–221].
Association with cadherins
Cadherins are involved in the formation of adherens junctions in endothelial and epithelial cells and play an essential role in VEGF signaling [160, 222–225]. Interaction of VEGFR-2 with VE-cadherin is regulated by β-catenin. At high cell density, the phosphatase PTP1/Dep1/CD148 associates with VE-cadherin and attenuates tyrosine phosphorylation of VEGFR-2 thereby suppressing signaling via PI 3-kinase, MAP kinases and PLCγ-1 [135]. At low cell density, such as at the tip of developing blood vessels, VEGFR-2 associates with integrin α v β 3 instead of cadherin and signal output is directed toward stimulation of cell migration and mitogenesis [215]. Reduced turnover of VEGFR-2 at high cell density has also been demonstrated to depend on cadherin and to enhance VEGF-mediated activation of MAP kinases [226]. To a large extent, vessel homeostasis is determined by cell-to-cell junctions that play an essential role during formation of a mechanosensory complex that regulates the response of endothelial cells to fluid shear stress [227].
Structure of VEGF receptors and their ligands
The structures of VEGF-A [228, 229], PlGF [230] and the snake venom components Vammin and VR-1 [31] have been solved. These molecules form homodimers and fold into a cysteine-knot structure also described for other growth factors [231]. The structure of Ig domain 2, which is part of the ligand-binding domain of VEGFR-1, has also been determined [232] and there are structural data available for complexes formed between VEGF-A and PlGF with domain 2 of VEGFR-1 [233–235]. These structural studies were complemented with biochemical investigation of receptor-blocking peptides [236, 237]. Biochemical analysis also led to the identification of specific residues determining receptor selectivity [56, 229, 238, 239].
The extracellular Ig-like domain 2 is sufficient for high-affinity binding of VEGF-A to VEGFR-1, while domains 2 and 3 are required for binding to VEGFR-2 [54]. VEGF mutants with altered loops L1 and L3 were used to determine the molecular basis for receptor selectivity of VEGF polypeptides [239, 240]. We created similar chimeric molecules based on our recently solved structure of the VEGFR-2-specific VEGF-E NZ2 variant. Loops L1 and L3 are tightly associated at their base and apparently function as a structural entity determining receptor selectivity, while L2 is required for interaction with both receptors [unpublished data].
The structure of the heparan-sulfate- and neuropilin-1-binding domain of VEGF-A, encoded by exons 7 and 8, has been solved by nuclear magnetic resonance spectroscopy [241]. Analysis of the electrostatic surface potential shows an extended patch of positively charged residues on one side of the molecule that is a candidate binding site for negatively charged heparan sulfate side chains. All VEGFs carrying sequences encoded by exon 7 bind to heparan sulfate both in vitro and in vivo [242]. However, high-affinity interaction with heparan sulfate, and particularly with neuropilin-1, also requires the short carboxy-terminal peptide encoded by exon 8 as shown by our laboratory [unpublished data].
Clearly, additional in-depth structural information is required for a comprehensive understanding of VEGF interaction with VEGFR-1 and -2, neuropilin-1 and heparan sulfate. Similarly challenging is the task to unravel the structural changes in the intracellular kinase domain following ligand binding to the extracellular domain. A first step in this direction is the resolution of a partial structure of the kinase domain of VEGFR-2 [243]. Such information will be useful to dissect the activation mechanism of VEGF receptor kinases and to engineer more specific reagents interfering with receptor activation, with the goal to block or stimulate VEGF signaling in disease.
Conclusions
Endothelial cells integrate signals elicited by cell-cell contacts, cell-extracellular matrix interactions and angiogenic growth factors. The final signal output results from the formation of context-specific signaling modules in distinct membrane compartments where receptor activity is tuned to the specific needs of a particular cell and aberrant signaling is suppressed [244]. Signal specificity of VEGF receptors arises from combinatorial activation of multiple cellular pathways. Each receptor subtype assembles a distinct set of signaling molecules in a spatially and temporally controlled manner giving rise to the formation of specific signal transduction modules or ‘signalosomes’ at the plasma membrane. In vivo, cells release VEGFs into their neighborhood in a temporally and spatially well-defined manner that allows gradual formation of receptor-ligand complexes. This significantly differs from tissue culture models where growth factors are usually administered as a single bolus, and great caution is required when extrapolating such in vitro data to the in vivo situation. Signal output is also determined by competition among the various VEGF receptors for VEGFs that interact with more than one receptor isoform and is influenced by the kinetics with which receptors are activated by different ligands. Finally, the exact three-dimensional structure of each ligand-receptor-coreceptor complex determines the efficacy with which intracellular tyrosine residues are phosphorylated and subsequently exposed to downstream signaling molecules. This has an impact on the strength and the kinetics with which individual signaling pathways are activated and execute their tasks.
References
Kurz H., Burri P. H. and Djonov V. G. (2003) Angiogenesis and vascular remodeling by intussusception: from form to function. News Physiol Sci. 18: 65–70
Ferrara N. (2004) Vascular endothelial growth factor: basic science and clinical progress. Endocr. Rev. 25: 581–611
Senger D. R., Galli S. J., Dvorak A. M., Perruzzi C. A., Harvey V. S. and Dvorak H. F. (1983) Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 219: 983–985
Senger D. R., Perruzzi C. A., Feder J. and Dvorak H. F. (1986) A highly conserved vascular permeability factor secreted by a variety of human and rodent tumor cell lines. Cancer Res. 46: 5629–5632
Senger D. R., Connolly D. T., Van De W. L., Feder J. and Dvorak H. F. (1990) Purification and NH2-terminal amino acid sequence of guinea pig tumor-secreted vascular permeability factor. Cancer Res. 50: 1774–1778
Keck P. J., Hauser S. D., Krivi G., Sanzo K., Warren T., Feder J. et al. (1989) Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science 246: 1309–1312
Lobb R. R., Key M. E., Alderman E. M. and Fett J. W. (1985) Partial purification and characterization of a vascular permeability factor secreted by a human colon adenocarcinoma cell line. Int. J. Cancer 36: 473–478
Bruce J. N., Criscuolo G. R., Merrill M. J., Moquin R. R., Blacklock J. B. and Oldfield E. H. (1987) Vascular permeability induced by protein product of malignant brain tumors: inhibition by dexamethasone. J. Neurosurg. 67: 880–884
Criscuolo G. R., Merrill M. J. and Oldfield E. H. (1988) Further characterization of malignant glioma-derived vascular permeability factor. J. Neurosurg. 69: 254–262
Connolly D. T., Heuvelman D. M., Nelson R., Olander J. V., Eppley B. L., Delfino J. J. et al. (1989) Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J. Clin. Invest 84: 1470–1478
Rosenthal R. A., Megyesi J. F., Henzel W. J., Ferrara N. and Folkman J. (1990) Conditioned medium from mouse sarcoma 180 cells contains vascular endothelial growth factor. Growth Factors 4: 53–59
Clauss M., Gerlach M., Gerlach H., Brett J., Wang F., Familletti P. C. et al. (1990) Vascular permeability factor: a tumor-derived polypeptide that induces endothelial cell and monocyte procoagulant activity, and promotes monocyte migration. J. Exp. Med. 172: 1535–1545
Carmeliet P., Ferreira V., Breier G., Pollefeyt S., Kieckens L., Gertsenstein M. et al. (1996) Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 380: 435–439
Shalaby F., Rossant J., Yamaguchi T. P., Gertsenstein M., Wu X. F., Breitman M. L. et al. (1995) Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376: 62–66
Shalaby F., Ho J., Stanford W. L., Fischer K. D., Schuh A. C., Schwartz L. et al. (1997) A requirement for Flk1 in primitive and definitive hematopoiesis and vasculogenesis. Cell 89: 981–990
Fong G. H., Rossant J., Gertsenstein M. and Breitman M. L. (1995) Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 376: 66–70
Javerzat S., Auguste P. and Bikfalvi A. (2002) The role of fibroblast growth factors in vascular development. Trends Mol. Med. 8: 483
Pepper M. S. (1997) Transforming growth factor-beta: vasculogenesis, angiogenesis, and vessel wall integrity. Cytokine Growth Factor Rev. 8: 21–43
Benjamin L. E., Hemo I. and Keshet E. (1998) A plasticity window for blood vessel remodelling is defined by pericyte coverage of the preformed endothelial network and is regulated by PDGF-B and VEGF. Development 125: 1591–1598
Jones N., Iljin K., Dumont D. J. and Alitalo K. (2001) Tie receptors: new modulators of angiogenic and lymphangiogenic responses. Nat. Rev. Mol. Cell Biol. 2: 257–267
Bruckner K., Kockel L., Duchek P., Luque C. M., Rorth P. and Perrimon N. (2004) The PDGF/VEGF receptor controls blood cell survival in Drosophila. Dev. Cell 7: 73–84
Cho N. K., Keyes L., Johnson E., Heller J., Ryner L., Karim F. et al. (2002) Developmental control of blood cell migration by the Drosophila VEGF pathway. Cell 108: 865–876
Duchek P., Somogyi K., Jekely G., Beccari S. and Rorth P. (2001) Guidance of cell migration by the Drosophila PDGF/VEGF receptor. Cell 107: 17–26
Tjwa M., Luttun A., Autiero M. and Carmeliet P. (2003) VEGF and PlGF: two pleiotropic growth factors with distinct roles in development and homeostasis. Cell Tissue Res. 314: 5–14
Tammela T., Enholm B., Alitalo K. and Paavonen K. (2005) The biology of vascular endothelial growth factors. Cardiovasc. Res. 65: 550–563
Maglione D., Guerriero V., Viglietto G., Delli-Bovi P. and Persico M. G. (1991) Isolation of a human placenta cDNA coding for a protein related to the vascular permeability factor. Proc. Natl. Acad. Sci. USA 88: 9267–9271
Meyer M., Clauss M., Lepple W. A., Waltenberger J., Augustin H. G., Ziche M. et al. (1999) A novel vascular endothelial growth factor encoded by Orf virus, VEGF-E, mediates angiogenesis via signaling through VEGFR-2 (KDR) but not VEGFR-1 (Flt-1) receptor tyrosine kinases. EMBO J. 18: 363–374
Ogawa S., Oku A., Sawano A., Yamaguchi S., Yazaki Y. and Shibuya M. (1998) A novel type of vascular endothelial growth factor, VEGF-E (NZ-7 VEGF), preferentially utilizes KDR/Flk-1 receptor and carries a potent mitotic activity without heparin-binding domain. J. Biol. Chem. 273: 31273–31282
Wise L. M., Veikkola T., Mercer A. A., Savory L. J., Fleming S. B., Caesar C. et al. (1999) Vascular endothelial growth factor (VEGF)-like protein from orf virus NZ2 binds to VEGFR2 and neuropilin-1. Proc. Natl. Acad. Sci. USA 96: 3071–3076
Junqueira-De-Azevedo Id Ide, Silva M. B., Chudzinski-Tavassi A. M. and Ho P. L. (2004) Identification and cloning of snake venom vascular endothelial growth factor (svVEGF) from Bothrops erythromelas pitviper. Toxicon 44: 571–575
Suto K., Yamazaki Y., Morita T. and Mizuno H. (2004) Crystal structures of novel VEGFs from snake venoms. J. Biol. Chem. 280: 2126–2131
Takahashi H., Hattori S., Iwamatsu A., Takizawa H. and Shibuya M. (2004) A novel snake venom vascular endothelial growth factor (VEGF) predominantly induces vascular permeability through preferential signaling via VEGF receptor-1. J. Biol. Chem. 279: 46304–46314
Yamazaki Y., Takani K., Atoda H. and Morita T. (2003) Snake venom VEGFs exhibit potent activity through their specific recognition of KDR (VEGF receptor 2). J. Biol. Chem. 278: 51985–51988
Tokunaga Y., Yamazaki Y. and Morita T. (2005) Specific distribution of VEGF-F in Viperinae snake venoms: isolation and characterization of a VEGF-F from the venom of Daboia russelli siamensis. Arch. Biochem. Biophys. 439: 241–247
Yamazaki Y., Matsunaga Y., Nakano Y. and Morita T. (2005) Identification of VEGF receptor-binding protein in the venom of eastern cottonmouth: a new role of snake venom myotoxic Lys49-phospholipase A2. J. Biol. Chem. 280: 29989–29992
Robinson C. and Stringer S. (2001) The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J. Cell Sci. 114: 853–865
Lee S., Jilani S. M., Nikolova G. V., Carpizo D. and Iruela-Arispe M. L. (2005) Processing of VEGF-A by matrix metalloproteinases regulates bioavailability and vascular patterning in tumors. J. Cell Biol. 169: 681–691
Bates D. O., Cui T. G., Doughty J. M., Winkler M., Sugiono M., Shields J. D. et al. (2002) VEGF(165)b, an inhibitory splice variant of vascular endothelial growth factor, is down-regulated in renal cell carcinoma. Cancer Res. 62: 4123–4131
Woolard J., Wang W. Y., Bevan H. S., Qiu Y., Morbidelli L., Pritchard-Jones R. O. et al. (2004) VEGF165b, an inhibitory vascular endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res. 64: 7822–7835
Forsythe J. A., Jiang B. H., Iyer N. V., Agani F., Leung S. W., Koos R. D. et al. (1996) Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 16: 4604–4613
Brogi E., Schatteman G., Wu T., Kim E. A., Varticovski L., Keyt B. et al. (1996) Hypoxia-induced paracrine regulation of vascular endothelial growth factor receptor expression. J. Clin. Invest. 97: 469–476
Namiki A., Brogi E., Kearney M., Kim E. A., Wu T., Couffinhal T. et al. (1995) Hypoxia induces vascular endothelial growth factor in cultured human endothelial cells. J. Biol. Chem. 270: 31189–31195
Ferrara N. (2002) Timeline: VEGF and the quest for tumour angiogenesis factors. Nat. Rev. Cancer 2: 795–803
Shibuya M., Yamaguchi S., Yamane A., Ikeda T., Tojo A., Matsushime H. et al. (1990) Nucleotide sequence and expression of a novel human receptor-type tyrosine kinase gene (flt) closely related to the fms family. Oncogene 5: 519–524
Terman B. I., Carrion M. E., Kovacs E., Rasmussen B. A., Eddy R. L. and Shows T. B. (1991) Identification of a new endothelial cell growth factor receptor tyrosine kinase. Oncogene 6: 1677–1683
Pajusola K., Aprelikova O., Korhonen J., Kaipainen A., Pertovaara L., Alitalo R. et al. (1992) FLT4 receptor tyrosine kinase contains seven immunoglobulin-like loops and is expressed in multiple human tissues and cell lines. Cancer Res. 52: 5738–5743
Matthews W., Jordan C. T., Gavin M., Jenkins N. A., Copeland N. G. and Lemischka I. R. (1991) A receptor tyrosine kinase cDNA isolated from a population of enriched primitive hematopoietic cells and exhibiting close genetic linkage to ckit. Proc. Natl. Acad. Sci. USA 88: 9026–9030
Kabrun N., Buhring H. J., Choi K., Ullrich A., Risau W. and Keller G. (1997) Flk-1 expression defines a population of early embryonic hematopoietic precursors. Development 124: 2039–2048
Ferrara N. and Davis-Smyth T. (1997) The biology of vascular endothelial growth factor. Endocr. Rev. 18: 4–25
Bellamy W. T. (2002) Vascular endothelial growth factor as a target opportunity in hematological malignancies. Curr. Opin. Oncol. 14: 649–656
Ishida A., Murray J., Saito Y., Kanthou C., Benzakour O., Shibuya M. et al. (2001) Expression of vascular endothelial growth factor receptors in smooth muscle cells. J. Cell Physiol. 188: 359–368
Davis-Smyth T., Presta L. G. and Ferrara N. (1998) Mapping the charged residues in the second immunoglobulin-like domain of the vascular endothelial growth factor/placenta growth factor receptor Flt-1 required for binding and structural stability. J. Biol. Chem. 273: 3216–3222
Fuh G., Li B., Crowley C., Cunningham B. and Wells J. A. (1998) Requirements for binding and signaling of the kinase domain receptor for vascular endothelial growth factor. J. Biol. Chem. 273: 11197–11204
Shinkai A., Ito M., Anazawa H., Yamaguchi S., Shitara K. and Shibuya M. (1998) Mapping of the sites involved in ligand association and dissociation at the extracellular domain of the kinase insert domain-containing receptor for vascular endothelial growth factor. J. Biol. Chem. 273: 31283–31288
De Falco S., Gigante B. and Persico M. (2002) Structure and function of placental growth factor. Trends Cardiovasc. Med. 12: 241–246
Errico M., Riccioni T., Iyer S., Pisano C., Acharya K. R., Persico G. M. et al. (2004) Identification of placental growth factor determinants for binding and activation of Flt-1 receptor. J. Biol. Chem. 279: 43929–43939
Olofsson B., Korpelainen E., Pepper M. S., Mandriota S. J., Aase K., Kumarr et al. (1998) Vascular endothelial growth factor B (VEGF-B) binds to VEGF receptor-1 and regulates plasminogen activator activity in endothelial cells. Proc. Natl. Acad. Sci. USA 95: 11709–11714
Wise L. M., Ueda N., Dryden N. H., Fleming S. B., Caesar C., Roufail S. et al. (2003) Viral vascular endothelial growth factors vary extensively in amino acid sequence, receptor-binding specificities, and the ability to induce vascular permeability yet are uniformly active mitogens. J. Biol. Chem. 278: 38004–38014
Jussila L. and Alitalo K. (2002) Vascular growth factors and lymphangiogenesis. Physiol. Rev. 82: 673–700
Takahashi H. and Shibuya M. (2005) The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin. Sci. (Lond) 109: 227–241
Hubbard S. R. (1999) Structural analysis of receptor tyrosine kinases. Prog. Biophys. Mol. Biol. 71: 343–358
Jiang G. and Hunter T. (1999) Receptor signaling: when dimerization is not enough. Curr. Biol. 9: R568–R571
Lemmon M. A. and Schlessinger J. (1998) Transmembrane signaling by receptor oligomerization. Methods Mol. Biol. 84: 49–71
Blechman J. M., Lev S., Barg J., Eisenstein M., Vaks B., Vogel Z. et al. (1995) The fourth immunoglobulin domain of the stem cell factor receptor couples ligand binding to signal transduction. Cell 80: 103–113
Blechman J. M. and Yarden Y. (1995) Structural aspects of receptor dimerization: c-kit as an example. Ann. N. Y. Acad. Sci. 766: 344–362
Herren B., Rooney B., Weyer K. A., Iberg N., Schmid G. and Pech M. (1993) Dimerization of extracellular domains of platelet-derived growth factor receptors: a revised model of receptor-ligand interaction. J. Biol. Chem. 268: 15088–15095
Tao Q., Backer M. V., Backer J. M. and Terman B. I. (2001) Kinase insert domain receptor (kdr) extracellular immunoglobulin-like domains 4–7 contain structural features that block receptor dimerization and vascular endothelial growth factor-induced signaling. J. Biol. Chem. 276: 21916–21923
Corless C. L., McGreevey L., Haley A., Town A. and Heinrich M. C. (2002) KIT mutations are common in incidental gastrointestinal stromal tumors one centimeter or less in size. Am. J. Pathol. 160: 1567–1572
Heinrich M. C., Corless C. L., Duensing A., McGreevey L., Chen C. J., Joseph N. et al. (2003) PDGFRA activating mutations in gastrointestinal stromal tumors. Science 299: 708–710
Bell C. A., Tynan J. A., Hart K. C., Meyer A. N., Robertson S. C. and Donoghue D. J. (2000) Rotational coupling of the transmembrane and kinase domains of the neu receptor tyrosine kinase. Mol. Biol. Cell 11: 3589–3599
Kendall R. L. and Thomas K. A. (1993) Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc. Natl. Acad. Sci. USA 90: 10705–10709
Goldman C. K., Kendall R. L., Cabrera G., Soroceanu L., Heike Y., Gillespie G. Y. et al. (1998) Paracrine expression of a native soluble vascular endothelial growth factor receptor inhibits tumor growth, metastasis, and mortality rate. Proc. Natl. Acad. Sci. USA 95: 8795–8800
Hornig C. and Weich H. A. (1999) Soluble VEGF receptors. Angiogenesis 3: 33–39
Luttun A. and Carmeliet P. (2003) Soluble VEGF receptor Flt1: the elusive preeclampsia factor discovered? J. Clin. Invest. 111: 600–602
Hiratsuka S., Minowa O., Kuno J., Noda T. and Shibuya M. (1998) Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc. Natl. Acad. Sci. USA 95: 9349–9354
Autiero M., Waltenberger J., Communi D., Kranz A., Moons L., Lambrechts D. et al. (2003) Role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat. Med. 9: 936–943
Hiratsuka S., Maru Y., Okada A., Seiki M., Noda T. and Shibuya M. (2001) Involvement of Flt-1 tyrosine kinase (vascular endothelial growth factor receptor-1) in pathological angiogenesis. Cancer Res. 61: 1207–1213
Park J. E., Chen H. H., Winer J., Houck K. A. and Ferrara N. (1994) Placenta growth factor: potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to Flt-1 but not to Flk-1/KDR. J. Biol. Chem. 269: 25646–25654
Barleon B., Sozzani S., Zhou D., Weich H. A., Mantovani A. and Marmé D. (1996) Migration of human monocytes in response to vascular endothelial growth factor (VEGF) is mediated via the VEGF receptor flt-1. Blood 87: 3336–3343
Hattori K., Heissig B., Wu Y., Dias S., Tejada R., Ferris B. et al. (2002) Placental growth factor reconstitutes hematopoiesis by recruiting VEGFR1(+) stem cells from bone-marrow microenvironment. Nat. Med. 8: 841–849
Gille H., Kowalski J., Yu L., Chen H., Pisabarro M. T., Davis S. T. et al. (2000) A repressor sequence in the juxtamembrane domain of Flt-1 (VEGFR-1) constitutively inhibits vascular endothelial growth factor-dependent phosphatidylinositol 3′-kinase activation and endothelial cell migration. EMBO J. 19: 4064–4073
Huang K., Andersson C., Roomans G. M., Ito N. and Claesson-Welsh L. (2001) Signaling properties of VEGF receptor-1 and −2 homo- and heterodimers. Int. J. Biochem. Cell Biol. 33: 315–324
Ito N., Huang K. and Claesson-Welsh L. (2001) Signal transduction by VEGF receptor-1 wild type and mutant proteins. Cell. Signal. 13: 849–854
Ito N., Wernstedt C., Engstrom U. and Claesson-Welsh L. (1998) Identification of vascular endothelial growth factor receptor-1 tyrosine phosphorylation sites and binding of SH2 domain-containing molecules. J. Biol. Chem. 273: 23410–23418
Cunningham S. A., Waxham M. N., Arrate P. M. and Brock T. A. (1995) Interaction of the Flt-1 tyrosine kinase receptor with the p85 subunit of phosphatidylinositol 3-kinase. Mapping of a novel site involved in binding. J. Biol. Chem. 270: 20254–20257
Cunningham S. A., Arrate M. P., Brock T. A. and Waxham M. N. (1997) Interactions of FLT-1 and KDR with phospholipase C gamma: identification of the phosphotyrosine binding sites. Biochem. Biophys. Res. Commun. 240: 635–639
Igarashi K., Isohara T., Kato T., Shigeta K., Yamano T. and Uno I. (1998) Tyrosine 1213 of Flt-1 is a major binding site of Nck and SHP-2. Biochem. Biophys. Res. Commun. 246: 95–99
Igarashi K., Shigeta K., Isohara T., Yamano T. and Uno I. (1998) Sck interacts with KDR and Flt-1 via its SH2 domain. Biochem. Biophys. Res. Commun. 251: 77–82
Sawano A., Takahashi T., Yamaguchi S. and Shibuya M. (1997) The phosphorylated 1169-tyrosine containing region of flt-1 kinase (VEGFR-1) is a major binding site for PLCgamma. Biochem. Biophys. Res. Commun. 238: 487–491
Landgren E., Schiller P., Cao Y. and Claesson-Welsh L. (1998) Placenta growth factor stimulates MAP kinase and mitogenicity but not phospholipase C-gamma and migration of endothelial cells expressing Flt 1. Oncogene 16: 359–367
Seetharam L., Gotoh N., Maru Y., Neufeld G., Yamaguchi S. and Shibuya M. (1995) A unique signal transduction from FLT tyrosine kinase, a receptor for vascular endothelial growth factor VEGF. Oncogene 10: 135–147
Waltenberger J., Claesson-Welsh L., Siegbahn A., Shibuya M. and Heldin C. H. (1994) Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J. Biol. Chem. 269: 26988–26995
Clauss M., Weich H., Breier G., Knies U., Rockl W., Waltenberger J. et al. (1996) The vascular endothelial growth factor receptor Flt-1 mediates biological activities: implications for a functional role of placenta growth factor in monocyte activation and chemotaxis. J. Biol. Chem. 271: 17629–17634
Carmeliet P. and Luttun A. (2001) The emerging role of the bone marrow-derived stem cells in (therapeutic) angiogenesis. Thromb. Haemost. 86: 289–297
Lyden D., Hattori K., Dias S., Costa C., Blaikie P., Butros L. et al. (2001) Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat. Med. 7: 1194–1201
Wey J. S., Fan F., Gray M. J., Bauer T. W., McCarty M. F., Somcio R. et al. (2005) Vascular endothelial growth factor receptor-1 promotes migration and invasion in pancreatic carcinoma cell lines. Cancer 104: 427–438
LeCouter J., Moritz D. R., Li B., Phillips G. L., Liang X. H., Gerber H. P. et al. (2003) Angiogenesis-independent endothelial protection of liver: role of VEGFR-1. Science 299: 890–893
Chen W. S., Kitson R. P. and Goldfarb R. H. (2002) Modulation of human NK cell lines by vascular endothelial growth factor and receptor VEGFR-1 (FLT-1). In Vivo 16: 439–445
Choi K., Kennedy M., Kazarov A., Papadimitriou J. C. and Keller G. (1998) A common precursor for hematopoietic and endothelial cells. Development 125: 725–732
Gille H., Kowalski J., Li B., LeCouter J., Moffat B., Zioncheck T. F. et al. (2001) Analysis of biological effects and signaling properties of Flt-1 (VEGFR-1) and KDR (VEGFR-2): a reassessment using novel receptor-specific vascular endothelial growth factor mutants. J. Biol. Chem. 276: 3222–3230
Jin K. L., Mao X. O. and Greenberg D. A. (2000) Vascular endothelial growth factor: direct neuroprotective effect in in vitro ischemia. Proc. Natl. Acad. Sci. USA 97: 10242–10247
Omolara O., Ogunshola O. O., Antic A., Donoghue M. J., Fan S.-Y., Kim H. et al. (2002) Paracrine and autocrine functions of neuronal VEGF in the CNS. J. Biol. Chem. 277: 11410–11415
Shiote M., Nagano I., Ilieva H., Murakami T., Narai H., Ohta Y. et al. (2005) Reduction of a vascular endothelial growth factor receptor, fetal liver kinase-1, by antisense oligonucleotides induces motor neuron death in rat spinal cord exposed to hypoxia. Neuroscience 132: 175–182
Yang K. and Cepko C. L. (1996) Flk-1, a receptor for vascular endothelial growth factor (VEGF), is expressed by retinal progenitor cells. J. Neurosci. 16: 6089–6099
Matsumoto T., Bohman S., Dixelius J., Berge T., Dimberg A., Magnusson P. et al. (2005) VEGF receptor-2 Y951 signaling and a role for the adapter molecule TSAd in tumor angiogenesis. EMBO J. 24: 2342–2353
Takahashi T., Yamaguchi S., Chida K. and Shibuya M. (2001) A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J. 20: 2768–2778
Sakurai Y., Ohgimoto K., Kataoka Y., Yoshida N. and Shibuya M. (2005) Essential role of Flk-1 (VEGF receptor 2) tyrosine residue 1173 in vasculogenesis in mice. Proc. Natl. Acad. Sci. USA 102: 1076–1081
Meyer R. D., Latz C. and Rahimi N. (2003) Recruitment and activation of phospholipase Cgamma1 by vascular endothelial growth factor receptor-2 are required for tubulogenesis and differentiation of endothelial cells. J. Biol. Chem. 278: 16347–16355
Dougher M. and Terman B., I. (1999) Autophosphorylation of KDR in the kinase domain is required for maximal VEGF-stimulated kinase activity and receptor internalization. Oncogene 18: 1619–1627
Kendall R. L., Rutledge R. Z., Mao X., Tebben A. J., Hungate R. W. and Thomas K. (1999) Vascular endothelial growth factor receptor KDR tyrosine kinase activity is increased by autophosphorylation of two activation loop tyrosine residues. J. Biol. Chem. 274: 6453–6460
Hubbard S. R. (2004) Juxtamembrane autoinhibition in receptor tyrosine kinases. Nat. Rev. Mol. Cell Biol. 5: 464–471
Meyer R. D., Dayanir V., Majnoun F. and Rahimi N. (2002) The presence of a single tyrosine residue at the carboxyl domain of vascular endothelial growth factor receptor-2/FLK-1 regulates its autophosphorylation and activation of signaling molecules. J. Biol. Chem. 277: 27081–27087
Duval M., Bedard-Goulet S., Delisle C. and Gratton J. P. (2003) Vascular endothelial growth factor-dependent down-regulation of Flk-1/KDR involves Cbl-mediated ubiquitination: consequences on nitric oxide production from endothelial cells. J. Biol. Chem. 278: 20091–20097
Kroll J. and Waltenberger J. (1997) The vascular endothelial growth factor receptor KDR activates multiple signal transduction pathways in porcine aortic endothelial cells. J. Biol. Chem. 272: 32521–32527
Gallicchio M., Mitola S., Valdembri D., Fantozzi R., Varnum B., Avanzi G. C. et al. (2004) Inhibition of vascular endothelial growth factor receptor 2-mediated endothelial cell activation by Axl tyrosine kinase receptor. Blood 105: 1970–1976
Guo D. Q., Wu L. W., Dunbar J. D., Ozes O. N., Mayo L. D., Kessler K. M. et al. (2000) Tumor necrosis factor employs a protein-tyrosine phosphatase to inhibit activation of KDR and vascular endothelial cell growth factor-induced endothelial cell proliferation. J. Biol. Chem. 275: 11216–11221
Nakagami H., Cui T. X., Iwai M., Shiuchi T., Takeda-Matsubara Y., Wu L. et al. (2002) Tumor necrosis factor-alpha inhibits growth factor-mediated cell proliferation through SHP-1 activation in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 22: 238–242
Guo D., Jia Q., Song H. Y., Warren R. S. and Donner D. B. (1995) Vascular endothelial cell growth factor promotes tyrosine phosphorylation of mediators of signal transduction that contain SH2 domains: association with endothelial cell proliferation. J. Biol. Chem. 270: 6729–6733
Warner A. J., Lopez-Dee J., Knight E. L., Feramisco J. R. and Prigent S. A. (2000) The Shc-related adaptor protein, Sck, forms a complex with the vascular-endothelial-growth-factor receptor KDR in transfected cells. Biochem. J. 347: 501–509
Giorgetti-Peraldi S., Murdaca J., Mas J. C. and Van Obberghen E. (2001) The adapter protein, Grb10, is a positive regulator of vascular endothelial growth factor signaling. Oncogene 20: 3959–3968
Huang L., Sankar S., Lin C., Kontos C. D., Schroff A. D., Cha E. H. et al. (1999) HCPTPA, a protein tyrosine phosphatase that regulates vascular endothelial growth factor receptor-mediated signal transduction and biological activity. J. Biol. Chem. 274: 38183–38188
Meadows K. N., Bryant P. and Pumiglia K. M. (2001) VEGF-induction of the angiogenic phenotype requires Ras activation. J. Biol. Chem. 276: 49289–49298
Songyang Z., Sheolson S. E., Chaudhuri M., Gish G., Pawson T., Haser W. G. et al. (1993) SH2 domains recognize specific phosphopeptide sequences. Cell 72: 767–778
Pedram A., Razandi M. and Levin E. R. (1998) Extracellular signal-regulated protein kinase/Jun kinase cross-talk underlies vascular endothelial cell growth factor-induced endothelial cell proliferation. J. Biol. Chem. 273: 26722–26728
Doanes A. M., Hegland D. D., Sethi R., Kovesdi I., Bruder J. T. and Finkel T. (1999) VEGF stimulates MAPK through a pathway that is unique for receptor tyrosine kinases. Biochem. Biophys. Res. Commun. 255: 545–548
Takahashi T., Ueno H. and Shibuya M. (1999) VEGF activates protein kinase C-dependent, but Ras-independent Raf- MEKMAP kinase pathway for DNA synthesis in primary endothelial cells. Oncogene 18: 2221–2230
Wu L. W., Mayo L. D., Dunbar J. D., Kessler K. M., Baerwald M. R., Jaffe E. A. et al. (2000) Utilization of distinct signaling pathways by receptors for vascular endothelial cell growth factor and other mitogens in the induction of endothelial cell proliferation. J. Biol. Chem. 275: 5096–5103
Xia P., Aiello L. P., Ishii H., Jiang Z. Y., Park D. J., Robinson G. S. et al. (1996) Characterization of vascular endothelial growth factor's effect on the activation of protein kinase C, its isoforms, and endothelial cell growth. J. Clin. Invest. 98: 2018–2026
Gliki G., Abu-Ghazaleh R., Jezequel S., Wheeler-Jones C. and Zachary I. (2001) Vascular endothelial growth factor-induced prostacyclin production is mediated by a protein kinase C (PKC)-dependent activation of extracellular signal-regulated protein kinases 1 and 2 involving PKC-delta and by mobilization of intracellular Ca2+. Biochem. J. 353: 503–512
Dayanir V., Meyer R. D., Lashkari K. and Rahimi N. (2001) Identification of tyrosine residues in vascular endothelial growth factor receptor-2/FLK-1 involved in activation of phosphatidylinositol-3 kinase and cell proliferation. J. Biol. Chem. 276: 17686–17692
Yu Y. and Sato J. D. (1999) MAP kinases, phosphatidylinositol 3-kinase, and p70 S6 kinase mediate the mitogenic response of human endothelial cells to vascular endothelial growth factor. J. Cell Physiol. 178: 235–246
He H., Venema V. J., Gu X., Venema R. C., Marrero M. B. and Caldwell R. B. (1999) Vascular endothelial growth factor signals endothelial cell production of nitric oxide and prostacyclin through flk-1/KDR activation of c-Src. J. Biol. Chem. 274: 25130–25135
Miralem T., Steinberg R., Price D. and Avraham H. (2001) VEGF(165) requires extracellular matrix components to induce mitogenic effects and migratory response in breast cancer cells. Oncogene 20: 5511–5524
Wang A., Miralem T. and Templeton D. M. (1999) Heparan sulfate chains with antimitogenic properties arise from mesangial cell-surface proteoglycans. Metabolism 48: 1220–1229
Grazia Lampugnani M., Zanetti A., Corada M., Takahashi T., Balconi G., Breviario F. et al. (2003) Contact inhibition of VEGF-induced proliferation requires vascular endothelial cadherin, beta-catenin, and the phosphatase DEP-1/CD148. J. Cell Biol. 161: 793–804
Abedi H. and Zachary I. (1997) Vascular endothelial growth factor stimulates tyrosine phosphorylation and recruitment to new focal adhesions of focal adhesion kinase and paxillin in endothelial cells. J. Biol. Chem. 272: 15442–15451
Kanno S., Oda N., Abe M., Terai Y., Ito M., Shitara K. et al. (2000) Roles of two VEGF receptors, Flt-1 and KDR, in the signal transduction of VEGF effects in human vascular endothelial cells. Oncogene 19: 2138–2146
Kiosses W. B., Daniels R. H., Otey C., Bokoch G. M. and Schwartz M. A. (1999) A role for p21-activated kinase in endothelial cell migration. J. Cell Biol. 147: 831–844
Rousseau S., Houle F., Kotanides H., Witte L., Waltenberger J., Landry J. et al. (2000) Vascular endothelial growth factor (VEGF)-driven actin-based motility is mediated by VEGFR2 and requires concerted activation of stress-activated protein kinase 2 (SAPK2/p38) and geldanamycin-sensitive phosphorylation of focal adhesion kinase. J. Biol. Chem. 275: 10661–10672
Stoletov K. V., Ratcliffe K. E., Spring S. C. and Terman B. I. (2001) NCK and PAK participate in the signaling pathway by which VEGF stimulates the assembly of focal adhesions. J. Biol. Chem. 276: 22748–22755
Stoletov K. V., Gong C. and Terman B. I. (2004) Nck and Crk mediate distinct VEGF-induced signaling pathways that serve overlapping functions in focal adhesion turnover and integrin activation. Exp. Cell Res. 295: 258–268
Abu-Ghazaleh R., Kabir J., Jia H., Lobo M. and Zachary I. (2001) Src mediates stimulation by vascular endothelial growth factor of the phosphorylation of focal adhesion kinase at tyrosine 861, and migration and anti-apoptosis in endothelial cells. Biochem. J. 360: 255–264
Ali N., Yoshizumi M., Fujita Y., Izawa Y., Kanematsu Y., Ishizawa K. et al. (2005) A novel Src kinase inhibitor, M475271, inhibits VEGF-induced human umbilical vein endothelial cell proliferation and migration. J. Pharmacol. Sci. 98: 130–141
Holmqvist K., Cross M. J., Rolny C., Hagerkvist R., Rahimi N., Matsumoto T. et al. (2004) The adaptor protein Shb binds to tyrosine 1175 in the VEGFR-2 and regulates VEGF-dependent cellular migration. J. Biol. Chem. 279: 22267–22275
Wu L. W., Mayo L. D., Dunbar J. D., Kessler K. M., Ozes O. N., Warren R. S. et al. (2000) VRAP is an adaptor protein that binds KDR, a receptor for vascular endothelial cell growth factor. J. Biol. Chem. 275: 6059–6062
Rousseau S., Houle F., Landry J. and Huot J. (1997) p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene 15: 2169–2177
McMullen M., Keller R., Sussman M. and Pumiglia K. (2003) Vascular endothelial growth factor-mediated activation of p38 is dependent upon Src and RAFTK/Pyk2. Oncogene 23: 1275–1282
Lamalice L., Houle F., Jourdan G. and Huot J. (2004) Phosphorylation of tyrosine 1214 on VEGFR2 is required for VEGF-induced activation of Cdc42 upstream of SAPK2/p38. Oncogene 23: 434–445
Huot J., Houle F., Marceau F. and Landry J. (1997) Oxidative stress-induced actin reorganization mediated by the p38 mitogen-activated protein kinase/heat shock protein 27 pathway in vascular endothelial cells. Circ. Res. 80: 383–392
Rousseau S., Houle F. and Huot J. (2000) Integrating the VEGF signals leading to actin-based motility in vascular endothelial cells. Trends Cardiovasc. Med. 10: 321–327
Zeng H., Zhao D. and Mukhopadhyay D. (2002) KDR stimulates endothelial cell migration through heterotrimeric G proteins Gq/11-mediated activation of a small GTPase Rho A. J. Biol. Chem. 277: 46791–46798
Alon T., Hemo I., Itin A., Peer J., Stone J. and Keshet E. (1995) Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat. Med. 1: 1024–1028
Gerber H. P., Hillan K. J., Ryan A. M., Kowalski J., Keller G. A., Rangell L. et al. (1999) VEGF is required for growth and survival in neonatal mice. Development 126: 1149–1159
Benjamin L. E., Golijanin D., Itin A., Pode D. and Keshet E. (1999) Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J. Clin. Invest 103: 159–165
Gerber H. P., McMurtrey A., Kowalski J., Yan M., Keyt B. A., Dixit V. et al. (1998) Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway: requirement for Flk-1/KDR activation. J. Biol. Chem. 273: 30336–30343
Fujio Y. and Walsh K. (1999) Akt mediates cytoprotection of endothelial cells by vascular endothelial growth factor in an anchorage-dependent manner. J. Biol. Chem. 274: 16349–16354
Brazil D. P. and Hemmings B. A. (2001) Ten years of protein kinase B signaling: a hard Akt to follow. Trends Biochem. Sci. 26: 657–664
Gerber H. P., Dixit V. M. and Ferrara N. (1998) Vascular endothelial growth factor induces expression of the antiapoptotic proteins Bcl-2 and A1 in vascular endothelial cells. J. Biol. Chem. 273: 13313–13316
Tran J., Rak J., Sheehan C., Saibil S. D., LaCasse E., Korneluk R. G. et al. (1999) Marked induction of the IAP family antiapoptotic proteins survivin and XIAP by VEGF in vascular endothelial cells. Biochem. Biophys. Res. Commun. 264: 781–788
Carmeliet P., Lampugnani M. G., Moons L., Breviario F., Compernolle V., Bono F. et al. (1999) Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell 98: 147–157
Spagnuolo R., Corada M., Orsenigo F., Zanetta L., Deuschle U., Sandy P. et al. (2004) Gas1 is induced by VE-cadherin and vascular endothelial growth factor and inhibits endothelial cell apoptosis. Blood 103: 3005–3012
Dvorak A. M. and Feng D. (2001) The vesiculo-vacuolar organelle (VVO): a new endothelial cell permeability organelle. J. Histochem. Cytochem. 49: 419–432
Esser S., Wolburg K., Wolburg H., Breier G., Kurzchalia T. and Risau W. (1998) Vascular endothelial growth factor induces endothelial fenestrations in vitro. J. Cell Biol. 140: 947–959
Roberts W. G. and Palade G. E. (1995) Increased microvascular permeability and endothelial fenestration induced by vascular endothelial growth factor. J. Cell Sci. 108: 2369–2379
Roberts W. G. and Palade G. E. (1997) Neovasculature induced by vascular endothelial growth factor is fenestrated. Cancer Res. 57: 765–772
Eliceiri B. P., Paul R., Schwartzberg P. L., Hood J. D., Leng J. and Cheresh D. A. (1999) Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol. Cell 4: 915–924
Strickland L. A., Jubb A. M., Hongo J. A., Zhong F., Burwick J., Fu L. et al. (2005) Plasmalemmal vesicle-associated protein (PLVAP) is expressed by tumour endothelium and is upregulated by vascular endothelial growth factor-A (VEGF). J. Pathol. 206: 466–475
Cruz A., DeFouw L. M. and DeFouw D. O. (2000) Restrictive endothelial barrier function during normal angiogenesis in vivo: partial dependence on tyrosine dephosphorylation of beta-catenin. Microvasc. Res. 59: 195–203
DeJana E., Bazzoni G. and Lampugnani M. G. (1999) Vascular endothelial (VE)-cadherin: only an intercellular glue? Exp. Cell Res. 252: 13–19
Esser S., Lampugnani M. G., Corada M., DeJana E. and Risau W. (1998) Vascular endothelial growth factor induces VE-cadherin tyrosine phosphorylation in endothelial cells. J. Cell Sci. 111: 1853–1865
Cohen A. W., Carbajal J. M. and Schaeffer R. C. Jr (1999) VEGF stimulates tyrosine phosphorylation of beta-catenin and small-pore endothelial barrier dysfunction. Am. J. Physiol 277: H2038–H2049
Kevil C. G., Payne D. K., Mire E. and Alexander J. S. (1998) Vascular permeability factor/vascular endothelial cell growth factor- mediated permeability occurs through disorganization of endothelial junctional proteins. J. Biol. Chem. 273: 15099–15103
Antonetti D. A., Barber A. J., Hollinger L. A., Wolpert E. B. and Gardner T. W. (1999) Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occluden 1: a potential mechanism for vascular permeability in diabetic retinopathy and tumors. J. Biol. Chem. 274: 23463–23467
Suarez S. and Ballmer-Hofer K. (2001) VEGF transiently disrupts gap junctional communication in endothelial cells. J. Cell Sci. 114: 1229–1235
Fulton D., Gratton J. P., McCabe T. J., Fontana J., Fujio Y., Walsh K. et al. (1999) Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 399: 597–601
Michell B. J., Griffiths J. E., Mitchelhill K., I, Rodriguez C., I, Tiganis T., Bozinovski S. et al. (1999) The Akt kinase signals directly to endothelial nitric oxide synthase. Curr. Biol. 9: 845–848
Murohara T., Horowitz J. R., Silver M., Tsurumi Y., Chen D., Sullivan A. et al. (1998) Vascular endothelial growth factor/vascular permeability factor enhances vascular permeability via nitric oxide and prostacyclin. Circulation 97: 99–107
Pajusola K., Aprelikova O., Armstrong E., Morris S. and Alitalo K. (1993) Two human FLT4 receptor tyrosine kinase isoforms with distinct carboxy terminal tails are produced by alternative processing of primary transcripts. Oncogene 8: 2931–2937
Petrova T. V., Makinen T. and Alitalo K. (1999) Signaling via vascular endothelial growth factor receptors. Exp. Cell Res. 253: 117–130
Kaipainen A., Korhonen J., Mustonen T., van Hinsbergh V., Fang G. H., Dumont D. et al. (1995) Expression of the fmslike tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. Proc. Natl. Acad. Sci. USA 92: 3566–3570
Dumont D. J., Jussila L., Taipale J., Lymboussaki A., Mustonen T., Pajusola K. et al. (1998) Cardiovascular failure in mouse embryos deficient in VEGF receptor-3. Science 282: 946–949
Valtola R., Salven P., Heikkila P., Taipale J., Joensuu H., Rehn M. et al. (1999) VEGFR-3 and its ligand VEGF-C are associated with angiogenesis in breast cancer. Am. J. Pathol. 154: 1381–1390
Partanen T. A., Alitalo K. and Miettinen M. (1999) Lack of lymphatic vascular specificity of vascular endothelial growth factor receptor 3 in 185 vascular tumors. Cancer 86: 2406–2412
Witmer A. N., Blijswijk B. C. van, Dai J., Hofman P., Partanen T. A., Vrensen G. F. et al. (2001) VEGFR-3 in adult angiogenesis. J. Pathol. 195: 490–497
Dixelius J., Makinen T., Wirzenius M., Karkkainen M., Wernstedt C., Alitalo K. et al. (2003) Ligand-induced vascular endothelial growth factor receptor-3 (VEGFR-3) heterodimerization with VEGFR-2 in primary lymphatic endothelial cells regulates tyrosine phosphorylation sites. J. Biol. Chem. 278: 40973–40979
Kukk E., Lymboussaki A., Taira S., Kaipainen A., Jeltsch M., Joukov V. et al. (1996) VEGF-C receptor binding and pattern of expression with VEGFR-3 suggests a role in lymphatic vascular development. Development 122: 3829–3837
Wang J. F., Zhang X. and Groopman J. E. (2004) Activation of vascular endothelial growth factor receptor-3 and its downstream signaling promote cell survival under oxidative stress. J. Biol. Chem. 279: 27088–27097
Makinen T., Veikkola T., Mustjoki S., Karpanen T., Catimel B., Nice E. C. et al. (2001) Isolated lymphatic endothelial cells transduce growth, survival and migratory signals via the VEGF-C/D receptor VEGFR-3. EMBO J. 20: 4762–4773
Fournier E., Rosnet O., Marchetto S., Turck C. W., Rottapel R., Pelicci P. G. et al. (1996) Interaction with the phosphotyrosine binding domain/phosphotyrosine interacting domain of SHC is required for the transforming activity of the FLT4/VEGFR3 receptor tyrosine kinase. J. Biol. Chem. 271: 12956–12963
Lin J., Lalani A. S., Harding T. C., Gonzalez M., Wu W. W., Luan B. et al. (2005) Inhibition of lymphogenous metastasis using adeno-associated virus-mediated gene transfer of a soluble VEGFR-3 decoy receptor. Cancer Res. 65: 6901–6909
Makinen T., Jussila L., Veikkola T., Karpanen T., Kettunen M. I., Pulkkanen K. J. et al. (2001) Inhibition of lymphangiogenesis with resulting lymphedema in transgenic mice expressing soluble VEGF receptor-3. Nat. Med. 7: 199–205
Keyt B. A., Berleau L. T., Nguyen H. V., Chen H., Heinsohn H., Vandlen R. et al. (1996) The carboxyl-terminal domain (111–165) of vascular endothelial growth factor is critical for its mitogenic potency. J. Biol. Chem. 271: 7788–7795
Tessler S., Rockwell P., Hicklin D., Cohen T., Levi B. Z., Witte L. et al. (1994) Heparin modulates the interaction of VEGF165 with soluble and cell associated flk-1 receptors. J. Biol. Chem. 269: 12456–12461
Whitaker G. B., Limberg B. J. and Rosenbaum J. S. (2001) Vascular endothelial growth factor receptor-2 and neuropilin-1 form a receptor complex that is responsible for the differential signaling potency of VEGF(165) and VEGF(121). J. Biol. Chem. 276: 25520–25531
Chiang M. K. and Flanagan J. G. (1995) Interactions between the Flk-1 receptor, vascular endothelial growth factor, and cell surface proteoglycan identified with a soluble receptor reagent. Growth Factors 12: 1–10
Gitay-Goren H., Cohen T., Tessler S., Soker S., Gengrinovitch S., Rockwell P. et al. (1996) Selective binding of VEGF121 to one of the three vascular endothelial growth factor receptors of vascular endothelial cells. J. Biol. Chem. 271: 5519–5523
Soker S., Fidder H., Neufeld G. and Klagsbrun M. (1996) Characterization of novel vascular endothelial growth factor (VEGF) receptors on tumor cells that bind VEGF165 via its exon 7-encoded domain. J. Biol. Chem. 271: 5761–5767
Fuh G., Garcia K. C. and Vos A. M. de (2000) The interaction of neuropilin-1 with vascular endothelial growth factor and its receptor flt-1. J. Biol. Chem. 275: 26690–26695
Gluzman-Poltorak Z., Cohen T., Herzog Y. and Neufeld G. (2000) Neuropilin-2 is a receptor for the vascular endothelial growth factor (VEGF) forms VEGF-145 and VEGF-165. J. Biol. Chem. 275: 18040–18045
Soker S., Takashima S., Miao H. Q., Neufeld G. and Klagsbrun M. (1998) Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 92: 735–745
Soker S., Miao H. Q., Nomi M., Takashima S. and Klagsbrun M. (2002) VEGF(165) mediates formation of complexes containing VEGFR-2 and neuropilin-1 that enhance VEGF(165)-receptor binding. J. Cell Biochem. 85: 357–368
Klagsbrun M., Takashima S. and Mamluk R. (2002) The role of neuropilin in vascular and tumor biology. Adv. Exp. Med. Biol. 515: 33–48
Gerhardt H., Golding M., Fruttiger M., Ruhrberg C., Lundkvist A., Abramsson A. et al. (2003) VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 161: 1163–1177
Gerhardt H., Ruhrberg C., Abramsson A., Fujisawa H., Shima D. and Betsholtz C. (2004) Neuropilin-1 is required for endothelial tip cell guidance in the developing central nervous system. Dev. Dyn. 231: 503–509
Kawasaki T., Kitsukawa T., Bekku Y., Matsuda Y., Sanbo M., Yagi T. et al. (1999) A requirement for neuropilin-1 in embryonic vessel formation. Development 126: 4895–4902
Neufeld G., Cohen T., Shraga N., Lange T., Kessler O. and Herzog Y. (2002) The neuropilins: multifunctional semaphorin and VEGF receptors that modulate axon guidance and angiogenesis. Trends Cardiovasc. Med. 12: 13–19
Schwarz Q., Gu C., Fujisawa H., Sabelko K., Gertsenstein M., Nagy A. et al. (2004) Vascular endothelial growth factor controls neuronal migration and cooperates with Sema3A to pattern distinct compartments of the facial nerve. Genes Dev. 18: 2822–2834
Eliceiri B. P. and Cheresh D. A. (2001) Adhesion events in angiogenesis. Curr. Opin. Cell Biol. 13: 563–568
Hall H. and Hubbell J. A. (2004) Matrix-bound sixth Ig-like domain of cell adhesion molecule L1 acts as an angiogenic factor by ligating alphavbeta3-integrin and activating VEGFR2. Microvasc. Res. 68: 169–178
Borges E., Jan Y. and Ruoslahti E. (2000) Platelet-derived growth factor receptor beta and vascular endothelial growth factor receptor 2 bind to the beta 3 integrin through its extracellular domain. J. Biol. Chem. 275: 39867–39873
Soldi R., Mitola S., Strasly M., Defilippi P., Tarone G. and Bussolino F. (1999) Role of alphavbeta3 integrin in the activation of vascular endothelial growth factor receptor-2. EMBO J. 18: 882–892
Hong Y. K., Lange-Asschenfeldt B., Velasco P., Hirakawa S., Kunstfeld R., Brown L. F. et al. (2004) VEGF-A promotes tissue repair-associated lymphatic vessel formation via VEGFR-2 and the alpha1beta1 and alpha2beta1 integrins. FASEB J. 18: 1111–1113
Reynolds A. R., Reynolds L. E., Nagel T. E., Lively J. C., Robinson S. D., Hicklin D. J. et al. (2004) Elevated Flk1 (vascular endothelial growth factor receptor 2) signaling mediates enhanced angiogenesis in beta3-integrin-deficient mice. Cancer Res. 64: 8643–8650
Reynolds L. E., Wyder L., Lively J. C., Taverna D., Robinson S. D., Huang X. et al. (2002) Enhanced pathological angiogenesis in mice lacking beta3 integrin or beta3 and beta5 integrins. Nat. Med. 8: 27–34
Carmeliet P. (2002) Integrin indecision. Nat. Med. 8: 14–16
Hutchings H., Ortega N. and Plouet J. (2003) Extracellular matrix-bound vascular endothelial growth factor promotes endothelial cell adhesion, migration, and survival through integrin ligation. FASEB J. 17: 1520–1522
Orecchia A., Lacal P. M., Schietroma C., Morea V., Zambruno G. and Failla C. M. (2003) Vascular endothelial growth factor receptor-1 is deposited in the extracellular matrix by endothelial cells and is a ligand for the alpha 5 beta 1 integrin. J. Cell Sci. 116: 3479–3489
Wijelath E. S., Murray J., Rahman S., Patel Y., Ishida A., Strand K. et al. (2002) Novel vascular endothelial growth factor binding domains of fibronectin enhance vascular endothelial growth factor biological activity. Circ. Res. 91: 25–31
Eliceiri B. P., Klemke R., Stromblad S. and Cheresh D. A. (1998) Integrin alphavbeta3 requirement for sustained mitogen-activated protein kinase activity during angiogenesis. J. Cell Biol. 140: 1255–1263
Eliceiri B. P., Puente X. S., Hood J. D., Stupack D. G., Schlaepfer D. D., Huang X. Z. et al. (2002) Src-mediated coupling of focal adhesion kinase to integrin alpha(v)beta5 in vascular endothelial growth factor signaling. J. Cell Biol. 157: 149–160
Friedlander M., Brooks P. C., Shaffer R. W., Kincaid C. M., Varner J. A., Cheresh D. A. et al. (1995) Definition of two angiogenic pathways by distinct alpha v integrins. Science 270: 1500–1502
Rahimi N. and Kazlauskas A. (1999) A role for cadherin-5 in regulation of vascular endothelial growth factor receptor 2 activity in endothelial cells. Mol. Biol. Cell 10: 3401–3407
Shay-Salit A., Shushy M., Wolfovitz E., Yahav H., Breviario F., DeJana E. et al. (2002) VEGF receptor 2 and the adherens junction as a mechanical transducer in vascular endothelial cells. Proc. Natl. Acad. Sci. USA 99: 9462–9467
Corada M., Zanetta L., Orsenigo F., Breviario F., Lampugnani M. G., Bernasconi S. et al. (2002) A monoclonal antibody to vascular endothelial-cadherin inhibits tumor angiogenesis without side effects on endothelial permeability. Blood 100: 905–911
Zanetti A., Lampugnani M. G., Balconi G., Breviario F., Corada M., Lanfrancone L. et al. (2002) Vascular endothelial growth factor induces shc association with vascular endothelial cadherin: a potential feedback mechanism to control vascular endothelial growth factor receptor-2 signaling. Arterioscler. Thromb. Vasc. Biol. 22: 617–622
Calera M. R., Venkatakrishnan A. and Kazlauskas A. (2004) VE-cadherin increases the half-life of VEGF receptor 2. Exp. Cell Res. 300: 248–256
Tzima E., Irani-Tehrani M., Kiosses W. B., DeJana E., Schultz D. A., Engelhardt B. et al. (2005) A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437: 426–431
Muller Y. A., Christinger H. W., Keyt B. A. and de-Vos A. M. (1997) The crystal structure of vascular endothelial growth factor (VEGF) refined to 1.93 A resolution: multiple copy flexibility and receptor binding. Curr. Biol. 5: 1325–1338
Muller Y. A., Li B., Christinger H. W., Wells J. A., Cunningham B. C. and Vos A. M. de (1997) Vascular endothelial growth factor: crystal structure and functional mapping of the kinase domain receptor binding site. Proc. Natl. Acad. Sci. USA 94: 7192–7197
Iyer S., Leonidas D.D., Swaminathan G. J., Maglione D., Battisti M., Tucci M. et al. (2001) The crystal structure of human placenta growth factor-1 (PlGF-1), an angiogenic protein, at 2.0 Å resolution. J. Biol. Chem. 276: 12153–12161
Schlunegger M. P. and Grutter M. G. (1992) An unusual feature revealed by the crystal structure at 2. 2 A resolution of human transforming growth factor-beta 2. Nature 358: 430–434
Christinger H. W., Muller Y. A., Berleau L. T., Keyt B. A., Cunningham B. C., Ferrara N. et al. (1996) Crystallization of the receptor binding domain of vascular endothelial growth factor. Proteins 26: 353–357
Wiesmann C., Fuh G., Christinger H. W., Eigenbrot C., Wells J. A. and Vos A. de (1997) Crystal structure at 1. 7 A resolution of VEGF in complex with domain 2 of the Flt-1 receptor. Cell 91: 695–704
Christinger H. W., Fuh G., Vos A. M. de and Wiesmann C. (2003) The crystal structure of PlGF in complex with domain 2 of VEGFR1. J. Biol. Chem. 279: 10382–10388
Starovasnik M. A., Christinger H. W., Wiesmann C., Champe M. A., Vos A. M. de and Skelton N. J. (1999) Solution structure of the VEGF-binding domain of Flt-1: comparison of its free and bound states. J. Mol. Biol. 293: 531–544
Wiesmann C., Christinger H. W., Cochran A. G., Cunningham B. C., Fairbrother W. J., Keenan C. J. et al. (1998) Crystal structure of the complex between VEGF and a receptor-blocking peptide. Biochemistry 37: 17765–17772
Pan B., Li B., Russell S. J., Tom J. Y., Cochran A. G. and Fairbrother W. J. (2002) Solution structure of a phage-derived peptide antagonist in complex with vascular endothelial growth factor. J. Mol. Biol. 316: 769–787
Ma L., Wang X., Zhang Z., Zhou X., Chen A. and Yao L. (2001) Identification of the ligand-binding domain of human vascular-endothelial-growth-factor receptor Flt-1. Biotechnol. Appl. Biochem. 34: 199–204
Keyt B. A., Nguyen H. V., Berleau L. T., Duarte C. M., Park J., Chen H. et al. (1996) Identification of vascular endothelial growth factor determinants for binding KDR and FLT-1 receptors: generation of receptor-selective VEGF variants by site-directed mutagenesis. J. Biol. Chem. 271: 5638–5646
Kiba A., Yabana N. and Shibuya M. (2003) A set of loop-1 and -3 structures in the novel VEGF family member, VEGF-ENZ-7, is essential for the activation of VEGFR-2 signaling. J. Biol. Chem. 278: 13453–13461
Fairbrother W. J., Champe M. A., Christinger H. W., Keyt B. A. and Starovasnik M. A. (1998) Solution structure of the heparin-binding domain of vascular endothelial growth factor. Structure 6: 637–648
El Sheikh A., Liu C., Huang H. and Edgington T. S. (2002) A novel vascular endothelial growth factor heparin-binding domain substructure binds to glycosaminoglycans in vivo and localizes to tumor microvascular endothelium. Cancer Res. 62: 7118–7123
McTigue M. A., Wickersham J. A., Pinko C., Showalter R. E., Parast C., V, Tempczyk R. A. et al. (1999) Crystal structure of the kinase domain of human vascular endothelial growth factor receptor 2: a key enzyme in angiogenesis. Structure 7: 319–330
Bussolino F., Serini G., Mitola S., Bazzoni G. and DeJana E. (2001) Dynamic modules and heterogeneity of function: a lesson from tyrosine kinase receptors in endothelial cells. EMBO Rep. 2: 763–767
Acknowledgement
This work was supported by grants from the Swiss National Foundation (3100A0-100204, 3100B0-10345/1 and 3100-054441), from Schweizerische Krebsliga (KLS-01220-02-2002), from the Hauptabteilung für die Sicherheit der Kernanlagen des Bundesamtes für Energiewirtschaft and by grants from the Paul Scherrer Institut. We are grateful to M. Pieren for critical reading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Received 15 September 2005; received after revision 11 November; accepted 24 November 2005
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Cébe-Suarez, S., Zehnder-Fjällman, A. & Ballmer-Hofer, K. The role of VEGF receptors in angiogenesis; complex partnerships. Cell. Mol. Life Sci. 63, 601 (2006). https://doi.org/10.1007/s00018-005-5426-3
Published:
DOI: https://doi.org/10.1007/s00018-005-5426-3