

Abstract
The use of radiolabeled antibodies, immunoglobulin fragments, and other proteins are an increasingly important sector of research for diagnostic imaging and targeted radiotherapy in nuclear medicine. As with all radiopharmaceuticals, efficient radiochemistry is a prerequisite to clinical translation. For proteins, variations in the primary amino acid sequence, the secondary structures, and tertiary folds, as well as differences in the size, charge, polarity, lipophilicity, and the presence of posttranslational modifications, add complexity to the system. The choice of radionuclide or chelate, and its impact on the thermodynamic, kinetic, and metabolic stability of a radiotracer, has attracted much attention but the chemistry by which the radionuclide is conjugated to the protein scaffold is of equal importance. Recently, a wealth of creative advances in protein ligation methods based on chemical, photochemical, and enzyme-mediated processes has emerged. As radiochemists explore alternative bioconjugation strategies, this article considers their potential impact on radiotracer design.
- bioconjugation
- immuno-PET
- antibodies
- protein ligation
- enzymes
- photoradiochemistry
It has long been established that the precise nature of the chemical linker used to couple a small molecule or peptide to a radiometal complex can have a profound impact on the properties of a radiotracer (1). Our recent work on coupling the 68Ga-DFO-complex via two different linker strategies to the urea-based Glu-NH-C(O)-NH-Lys group for imaging prostate-specific membrane antigen (PSMA) illustrates this point (2,3). When compared with the clinically translated agent 68Ga-PSMA-11, dramatic differences were observed in the cellular binding, tumor targeting, and whole-body distribution and excretion profiles of 68Ga-DFO-Nsucc-PSMA and 68Ga-DFO-pNCS-Bn-PSMA—radiotracers constructed using N-succinyl and thiourea-based chemistries, respectively. Specifically, the small and less lipophilic N-succinyl linker reduced PSMA binding affinity with a dissociation constant from cellular assays (Kd value) of 26.4 ± 7.8 nM compared with a value of 13.6 ± 2.6 nM for the compound with the pNCS-Bn linker and 2.89 ± 0.55 nM for 68Ga-PSMA-11. These differences in PSMA binding affinity also led to pronounced differences the uptake, retention, and excretion of these three radiotracers in mice bearing LNCaP tumors.
For radiotracers based on antibodies, and other proteins, less is known about the impact that linker methodology may have on the stability, pharmacokinetics, and target specificity of the ensuing radiotracer. Until recently, our collective experience with radiolabeled antibodies has relied on a small handful of conjugation routes. Classic chemistries typically involve reactions of native cysteine or lysine residues (4–11). For example, thiolate groups of cysteine side chains readily undergo Michael addition reactions with maleimide groups, and the ε-NH2 group of lysine side chains participates in nucleophilic reactions using activated esters or isothiocyanates. An overview of the essential structure of an antibody and various options for performing conjugation chemistry is shown in Figure 1 (4,12). Note that highlighted functionalization sites (positions 1 to 6) are not intended to be a comprehensive list of all chemically accessible features but rather give a flavor of the types of conjugation reactions that are currently being explored. The selection does not include the wide array of click methods for which the reader is referred to the detailed review by Meyer et al. (13). The following sections discuss some of the features of recent chemical, photochemical, and enzyme-mediated conjugation reactions, with a particular emphasis on applications with antibodies (14,15). Some of the methods described are yet to be applied in radiosynthesis but illustrate the scope of reactions that harbor potential for future use.

Structural features of a generic IgG1 human antibody showing various domains of heavy and light chains, antigen binding (CDR = complementary determining region), Fab fragment, crystallizable fragment (Fc), and location of glycosylated residues. Reaction sites include (but are not limited to): (1) cysteine or reduced disulfides; (2) lysine functionalization; (3) C- or N-terminal chemistry, including use of exogenous engineered sequences such as His6, Myc-tag, HaloTag, or SNAP-tags etc.; (4) transglutaminase coupling; (5) enzyme-mediated glycan modification; and (6) bispecific antibodies with one arm selective for a metal ion complex and the other for the target of interest.
CHEMICAL FUNCTIONALIZATION METHODS
Despite the diverse range of new bioconjugation methods, it remains true that most synthetic transformations rely on the native chemistry of cysteine (sulfhydryl) and lysine (primary amine) side chains. Some of the reactions of these two amino acids are presented in Figure 2.

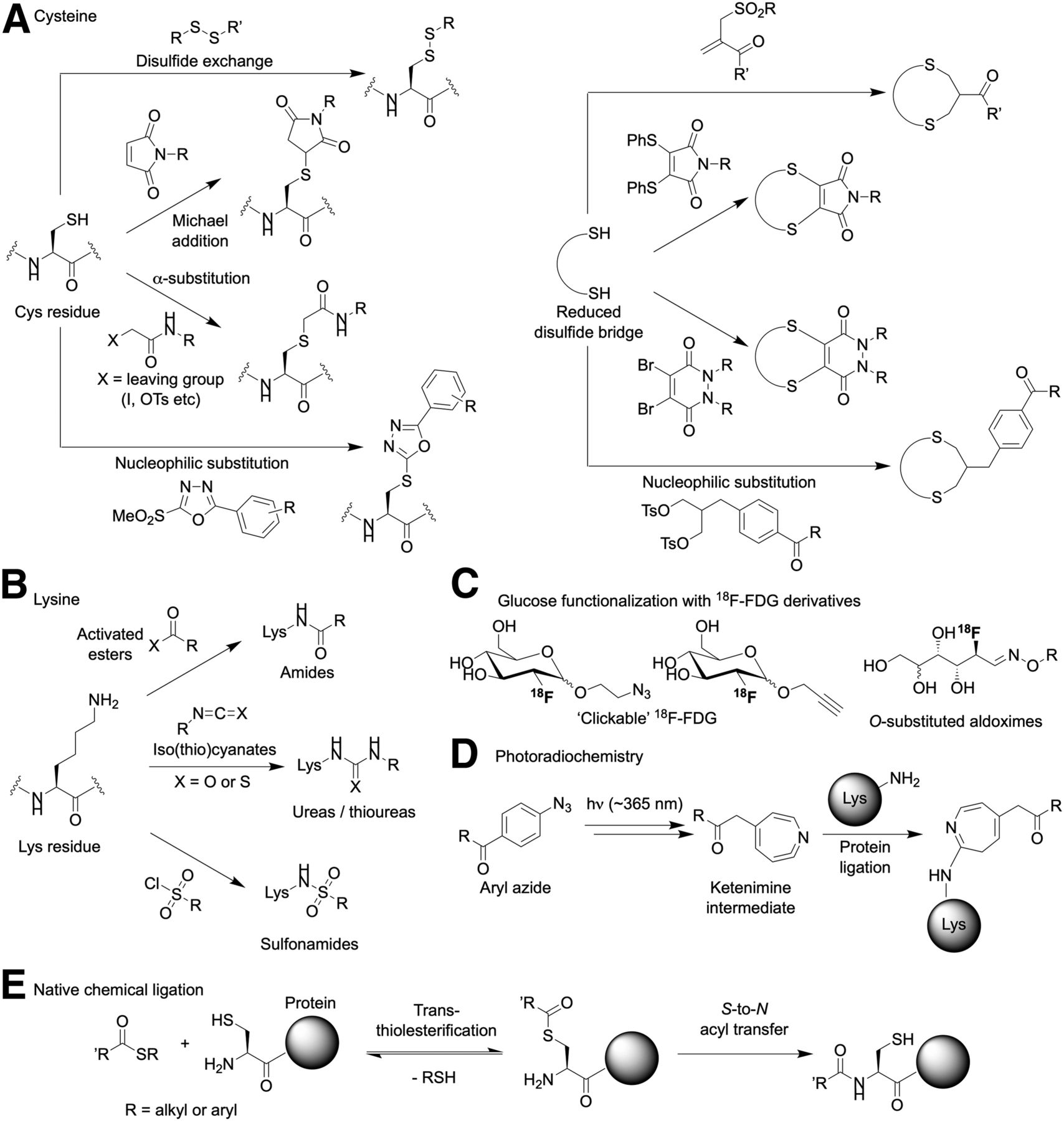
(A–E) Illustration of some of the classic and modern chemical reactions for protein ligation. Note that the figure is not intended to be comprehensive but rather gives a flavor of the different types of reagents and transformations that are potentially useful in protein-based radiotracers synthesis.
In general, reactions at cysteines involve either derivatization of a single residue or sequential reduction of disulfides followed by substrate-induced rebridging (Fig. 2A). In all cases, the chemistry features nucleophilic substitution or addition–elimination mechanisms using reagents bearing labile leaving groups. Conjugation with maleimide groups has been a mainstay of conjugation chemistry for many decades. Indeed, two U.S. Food and Drug Administration–approved antibody–drug conjugates—brentuximab vedotin (Adcetris; Seattle Genetics) and trastuzumab emtansine (Kadcyla; Genentech/Roche)—use this conjugation strategy. However, in cysteine-rich environments, the reaction is potentially reversible, and experience has found that such conjugates are susceptible to chemical and metabolic instability in vivo, leading to questions over long-term stability (16). Stability is of particular concern for radiotracers based on antibodies that require extended circulation times for peak accumulation in tumor lesions and clearance from background organs. Notably, the stability of maleimide conjugates in vivo can be improved by ensuring that the 5-membered ring hydrolyses to the ring-opened succinic acid thioether—the rate of which can be increased by the use of electron-withdrawing substituents on the nitrogen atom (16). Related agents based on 18F have also been introduced (17). Because two bonds are usually better than one (from a thermodynamic and kinetic perspective), disulfide rebridging is also an attractive strategy to potentially overcome stability-related issues associated with cysteine functionalization (18). Rebridging can be achieved with the reagents shown (Fig. 2A [right]), but variants of the photochemically induced thiol-ene and thiol-yne radical–based coupling reactions are also potential options (15).
In many aspects, lysine-based conjugation reactions are more readily accessible than cysteine functionalization. A typical IgG1 human antibody has around 90 lysines, many of which are solvent-exposed and chemically accessible (19). In contrast, IgG1 antibodies such as trastuzumab have 16 cysteine residues, all of which are involved in inter- and intrachain disulfide bridges. Only the 4 interchain disulfides are easily modified but require reduction before the thiolate residues become accessible for functionalization. Hence, lysine-based reactions are the most prevalent method used for chemical modification of antibodies (Fig. 2B). Radiolabeling antibodies at lysine residues takes one of two mechanistic forms: either direct conjugation of the radiolabel to the ε-NH2 group, or a “tag-and-modify” approach in which the side chain is first tagged with another reactive group that facilitates bioorthogonal coupling to a tertiary reagent (compare with numerous click-based conjugation and pretargeting studies (13,20–22)). From a clinical perspective, the high reproducibility and efficiency of lysine modification using activated esters or isothiocyanate-based reagents make these the most prevalent methods used in the synthesis of clinical-grade radiolabeled antibodies (23).
An interesting variation on the tag-and-modify route involves the use of radiolabeled sugars to functionalize proteins (24–27). For instance, in 2015 Rashidan et al. reported that 18F-FDG could be substituted at the anomeric (C1) position using tetrazine reagents bearing an RO-NH2 group (Fig. 2C) (25). Reaction of the amine with the ring-opened aldehyde isomer of 18F-FDG (catalyzed by the addition of p-phenylenediamine) gave a stable oxime intermediate that was subsequently used in site-specific radiolabeling of an anti–class II major histone complex antibody. This work also featured the use of site-specific enzyme-mediated tagging of the antibody using sortase-A (vide infra) to incorporate the transcyclooctene partner for strain-promoted inverse electron demand Diels Alder click conjugation.
A related tag-and-modify approach was reported by Vugts et al. who used traceless Staudinger ligation to label antibodies that were premodified with an azide group (28). A series of phosphine-based reagents was synthesized to allow conjugation of the chelates DFO and DOTA or an 123I-radiolabeled cubyl-phosphine to the mAb via the traceless approach. Use of the traceless Staudinger ligation was also explored in the context of antibody pretargeting. Although derivatization of proteins using Staudinger ligation chemistry was successful, several drawbacks including slow biomolecular conjugation kinetics and metabolic instability of the phosphine groups in vivo will likely preclude further use of this chemistry for pretargeting (29).
As an alternative conjugation method, our group recently reported the development of a photochemically mediated conjugation and radiolabeling reaction based on chelates bearing aryl azide groups (Fig. 2D) (30–32). The process is distinct from almost all other conjugation chemistries in that the active reagent is formed in situ by irradiating the samples with ultraviolet light (∼365 nm). Under irradiation, aryl azides spontaneously release N2(g) and undergo rapid isomerization to give a ketenimine intermediate. The unique feature of this mechanism is that the ketenimine intermediate acts as a preferential electrophile for primary and secondary amines, thereby facilitating chemo-selective, lysine-based conjugation. Conceptually, photochemistry is an attractive foundation for bioconjugation reactions because photoactivation can occur at wavelengths at which most proteins do not absorb and also on a short time scale that minimizes potential damage to the underlying protein; productive protein ligation occurs with high quantum efficiency and under ambient conditions so no heating is required; the aryl azide reagents are inexpensive, easy to synthesize, and thermally stable up to about 60°C, permitting long-term storage; and photoradiochemistry permits the simultaneous, one-pot conjugation and radiolabeling of non-purified antibodies (i.e., antibodies in their standard good manufacturing practice formulation) in less than 20 min. A combination of stability measurements, cellular assays, temporal PET imaging, and biodistribution studies confirmed that the 89Zr-DFO-azepin-trastuzumab obtained by photoradiochemistry was biochemically equivalent to 89Zr-DFO-Nsucc-trastuzumab produced by a conventional thermochemical conjugation route (Fig. 3) (32,33). Recent work by Poot et al. introduced fully automated radiolabeling and purification of 89Zr-radiolabeled antibodies, but as with manual synthesis, the approach still relies on the use of preconjugated protein (34). An additional advantage of our photoradiochemical method is that the conjugation step is amenable to full automation, which opens the possibility of using nonmodified proteins as starting materials.
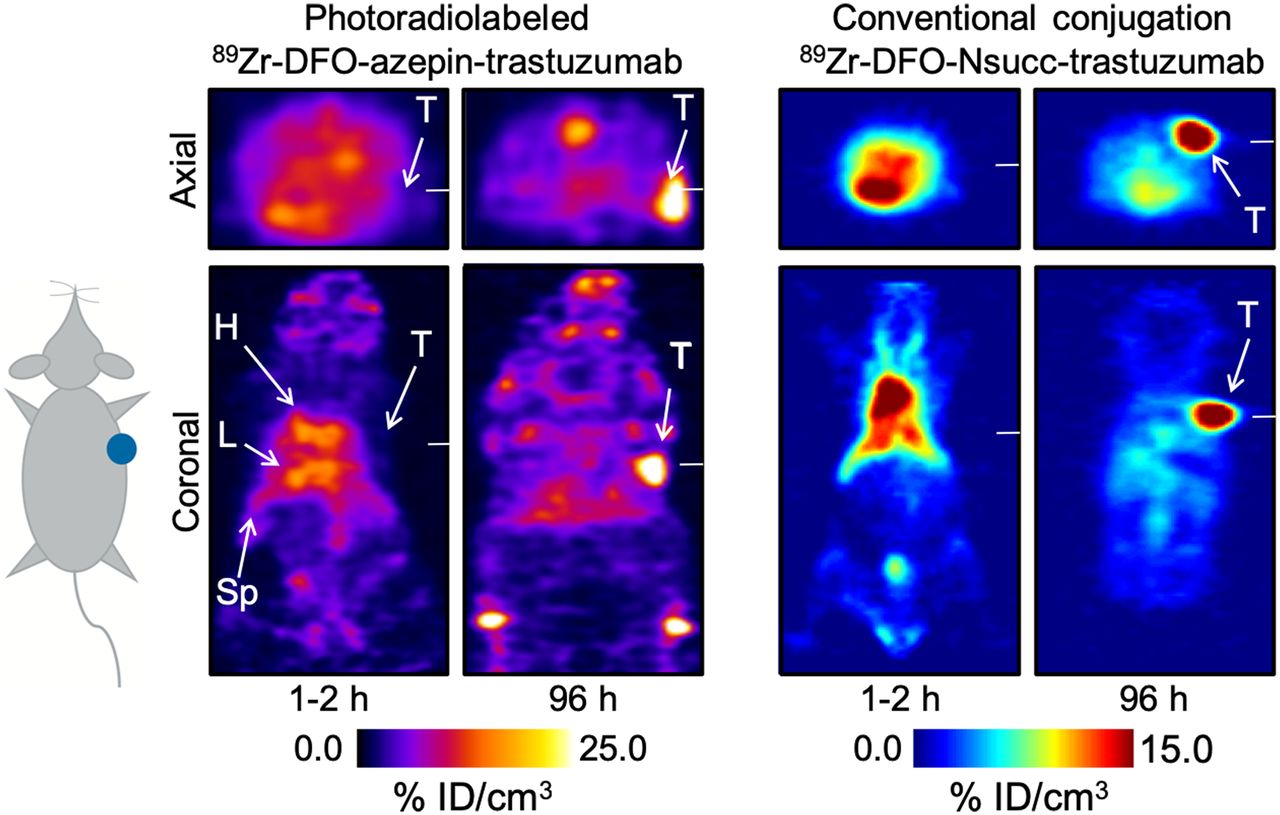
Comparison of PET images recorded in athymic nude mice that were administered either 89Zr-DFO-azepin-trastuzumab synthesized by a photochemical conjugation and radiolabeling process (unpublished data) (left) or 89Zr-DFO-Nsucc-trastuzumab synthesized via a conventional thermochemical route using N-succinyl (Nsucc) activated ester chemistry (right) (33). No discernable differences were observed when comparing the PET images or the biodistribution data for the 2 radiotracers produced via different photochemical or thermochemical routes.
Native chemical ligation was originally introduced in the 1990s by Kent et al. (Fig. 2E) (14,35,36). The native chemical ligation process is straightforward and involves a reversible transesterification reaction initiated by nucleophilic attack of an N-terminal Cys-thiolate on an engineered C-terminal thioester. The resulting activated thioester then undergoes a spontaneous and irreversible, intramolecular S-to-N acyl shift under physiologic conditions to yield a peptide bond linkage. This approach is an elegant, chemo-specific and regio-specific method for facile derivatization of proteins, but to the best of our knowledge has yet to be applied in radiotracer design. The caveat is that C-terminal thioesters are required, but recent efforts have improved access to these reagents (14,37).
ENZYME-MEDIATED FUNCTIONALIZATION METHODS
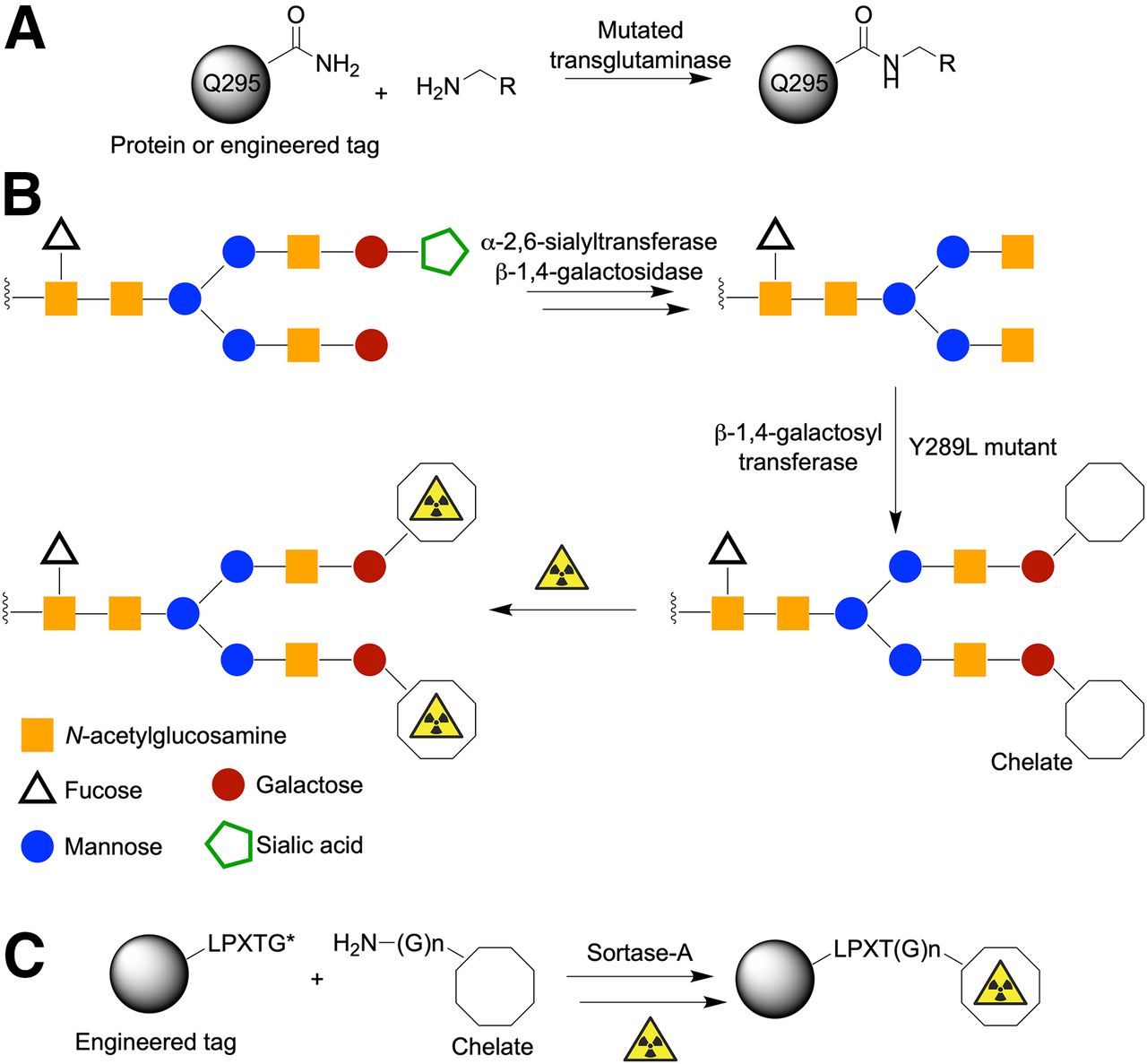
Beyond chemically mediated transformations, several groups have begun to adapt the exquisite specificity and selectivity of enzymes for antibody (protein) functionalization and radiotracer synthesis. A wide array of enzyme-mediated reactions are available (14), but three examples that have already been used in radiochemistry include applications of transglutaminase (4,38); glycan modification (8,39–42) with multi-step transformations using α-2,6-sialyltransferase, β-1,4-galactosidase, and mutated β-1,4-galactosyltransferase; and sortase-A–mediated conjugation using proteins engineered to express the LPXTG tags (Fig. 4) (25,43,44).
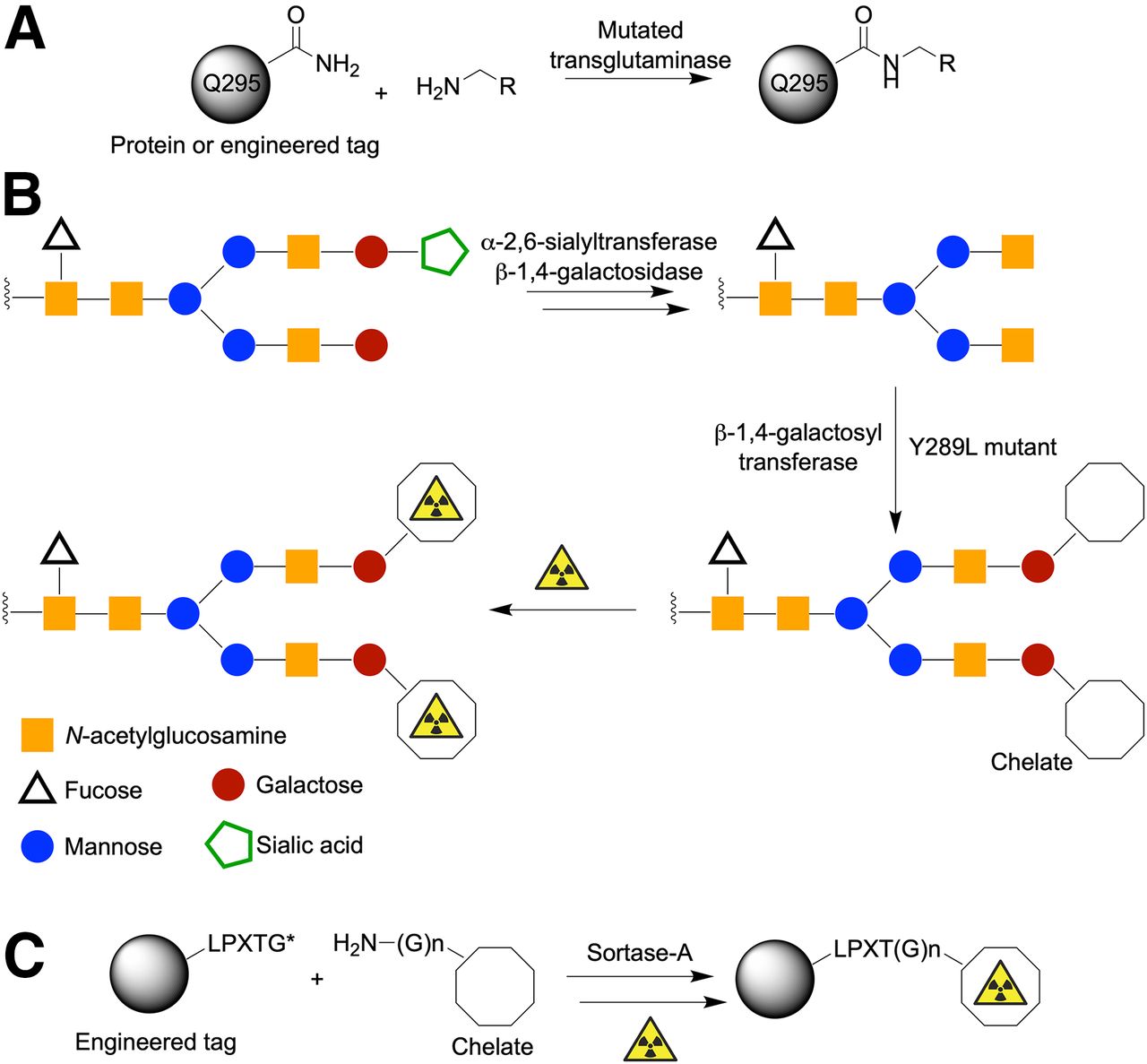
Representative examples of enzyme-mediated protein ligation reactions. (A) Transglutaminase reaction using primary amine substrates and either natural or engineered glutamine residues. (B) Multistep glycan modification. (C) Site-specific sortase-A–mediated conjugation using LPXTG tag (where X = any amino acid and coupled substrate contains N-terminal poly-glycine handle).
Conjugation reactions involving transglutaminase can occur at native Q295 glutamine residues in the CH2 domain of IgG1 antibodies (Fig. 4A). Alternatively, site-specific derivatization can be achieved at engineered protein tags (so-called Q-tags), which harbor a recognition sequence and an accessible glutamine residue that acts as a substrate for mutated transglutaminase enzymes (45).
Most antibodies are inherently glycoproteins (Fig. 1). Therefore, glycoengineering offers a fascinating approach for site-selective modification of antibodies. Different strategies for modifying glycans exist but the general features include an enzymatic cleaning step to homogenize the presentation of glycans on the antibody sample, followed by enzyme-mediated conjugation using a nonnative substrate bearing either a bioorthogonal reactive group or a chelate for subsequent radiolabeling (Fig. 4B). Using this approach, Zeglis et al. reported several interesting applications ranging from pretargeted immuno-PET through to the synthesis of functionalized antibodies for multimodal (PET and optical) imaging (41).
There is no doubt that the aforementioned examples of enzyme-mediated chemistry are elegant approaches to functionalize antibodies. However, for future clinical applications, it is crucial to ensure complete removal of the enzyme from the desired protein conjugate. Arguably one of the most exciting strategies involves the use of sortase-A transpeptidase (Fig. 4C). This approach is conceptually more challenging, because from the outset, it involves the use of engineered proteins that display a specific LPXTG sequence. However, this tag guarantees site-selective functionalization. For example, Paterson et al. reported the use of sortase-A to couple a sarcophogine chelate (derivatized with a short polyethylene glycol linker and an N-terminal triglycine (Gly)3 group) to an scFv fragment that targets the ligand-induced binding sites on the glycoprotein receptor GPIIb/IIIa (43). An additional feature of sortase-A conjugation is that the reactive components, including the sortase-A enzyme, can be coengineered to display a (His)6-tag sequence, which facilitates purification using standard affinity chromatography (44). If successful protein conjugation occurs, the (His)6-tag can be designed to be automatically cleaved from the product (e.g., by using augmented recognition C-terminal sequences such as LPXTG-(His)6). At the same time, unreacted reagents retain their original (His)6-tag, and byproducts receive a (His)6-tag via an exchange mechanism. Hence, purification of the desired protein from reagents and unwanted byproducts is a built-in part of the sortase-A process.
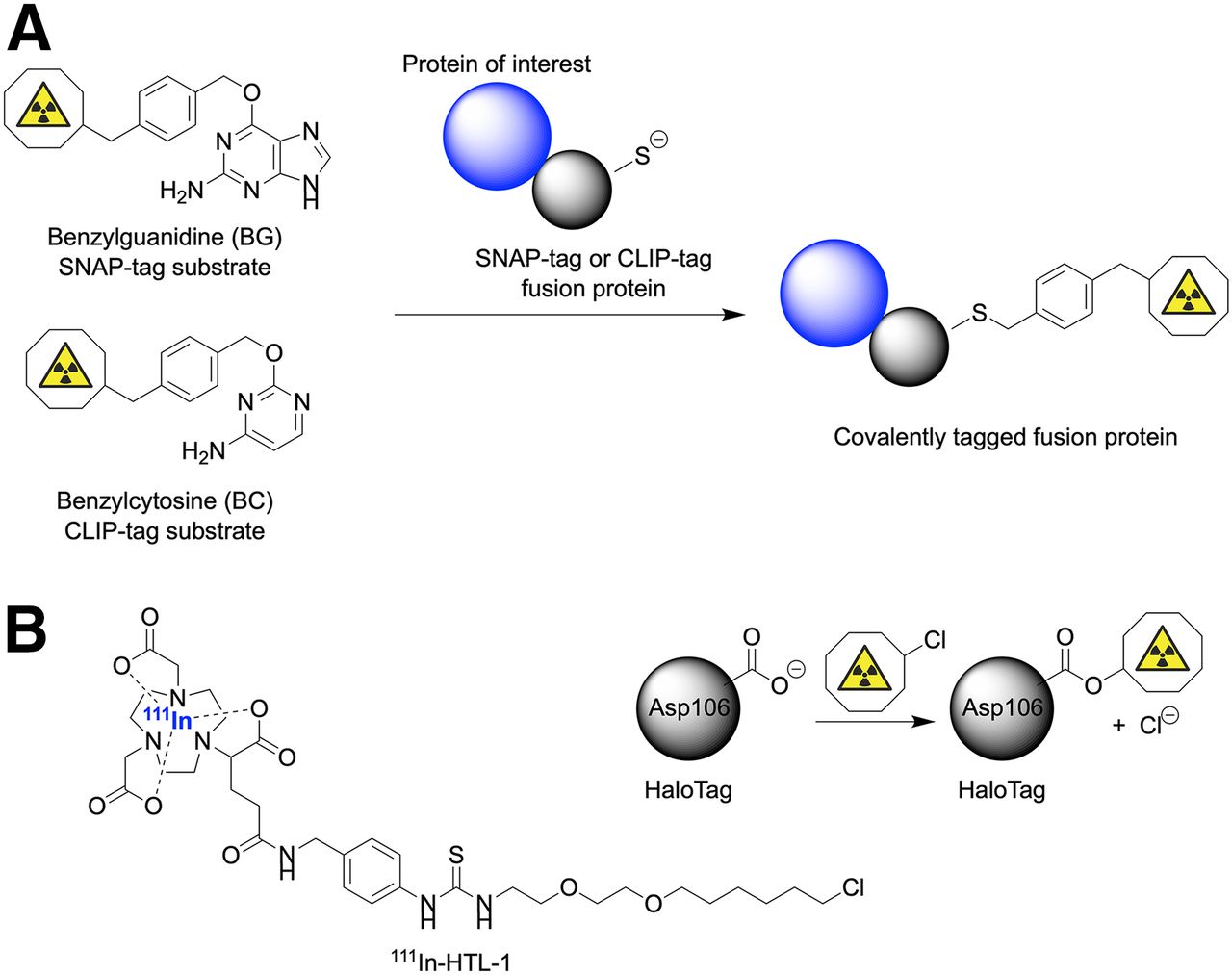
Finally, it is worth noting that alternative conjugation strategies involving engineered fusion proteins exist (46–48). Here, the most familiar example involves fusion of streptavidin (53 kDa) to a protein of interest followed by site-specific conjugation with a substrate bearing a biotin tag. However, other promising approaches include the use of SNAP-tag (∼19 kDa), CLIP-tag (∼19 kDa), and HaloTag (33 kDa) enzymes (Fig. 5). SNAP-tags and CLIP-tags work via the same mechanism (Fig. 5A). Knight et al. used the HaloTag approach combined with an 111In-radiolabeled complex to pretarget the noninternalizing TAG-72 biomarker expressed on LS174T colorectal cancer cells (48). Metal binding chelates featuring a fluorescence label and an alkylchloride group—which forms a covalent bond with the HaloTag enzyme—were developed. From a pretargeting perspective, HaloTags were found to be sufficiently reactive to allow specific accumulation in tumors. However, incorporation of long-chain aliphatic handles on the chelate component may potentially alter the lipophilicity and pharmacokinetic distribution of the reagents in vivo. Fusion of larger proteins to an antibody has the potential to alter the distribution profile. Nevertheless, excellent tumor localization of the radioactivity was achieved in tumor-bearing mice. With further optimization, HaloTags and related fusion proteins will undoubtedly allow expansion of this technology to other substrates and targeting vectors.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Illustration of substrates and covalent bioconjugation reactions on fusion proteins involving use of SNAP-tags or CLIP-tags with benzylguanosyl (BG) and benzylcytosyl (BC) substrates, respectively (A) and covalent bond formation between Asp106 residue in mutated active site of HaloTag and 111In-radiolabeled alkylchloride substrate (111In-HTL-1) (B) (48).
CONCLUSIONS AND FUTURE DIRECTIONS
A wide range of alternative conjugation methods are making their way into the radiochemical sciences. Strategies involving bioorthogonal chemistry, photochemistry, and enzyme-mediated processes all show promise for future radiotracer design. These new methods allow researchers to construct peptide- and protein-based radiotracers with unprecedented control over the chemo-selectivity and regio-selectivity of the conjugation step. Advances in site-specific conjugation allow new immuno-PET tracers to be synthesized while minimizing the risk of compromising the distribution profile and immunoreactivity of the antibody or biomolecule by avoiding modification at undesirable sites. As new technologies are developed, it is worth keeping in mind that existing methods (namely, non-specific cysteine and lysine functionalization) are simple, work efficiently for standard antibodies, and are already part of routine clinical translation. Therefore, any alternative conjugation method should deliver advantages that standard non-specific chemistry cannot provide. Ideally, new methods should also be applicable to a diverse range of cancer-targeting proteins. In this respect, selected chemistries described in this article have the potential to change the way that radiotracers are designed and produced, and may expand the range of immuno-PET for clinical imaging. It will be exciting to see how these technologies advance in the coming years.
DISCLOSURE
This project has received funding from the European Union’s Horizon 2020 research and innovation programme/from the European Research Council under the grant agreement no. 676904, ERC-StG-2015, NanoSCAN. Jason P. Holland thanks the Swiss National Science Foundation (SNSF Professorship PP00P2_163683), the Swiss Cancer League (Krebsliga Schweiz; KLS-4257-08-2017), and the University of Zurich (UZH) for financial support. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank all members of the Radiochemistry and Imaging Science group at UZH for helpful discussions.
Footnotes
Published online Mar. 22, 2019.
- © 2019 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.
- 12.
- 13.
- 14.
- 15.
- 16.
- 17.
- 18.
- 19.
- 20.
- 21.
- 22.
- 23.
- 24.
- 25.
- 26.
- 27.
- 28.
- 29.
- 30.
- 31.
- 32.
- 33.
- 34.
- 35.
- 36.
- 37.
- 38.
- 39.
- 40.
- 41.
- 42.
- 43.
- 44.
- 45.
- 46.
- 47.
- 48.
- Received for publication February 7, 2019.
- Accepted for publication March 18, 2019.