


Abstract
The field of radioligand therapy has advanced greatly in recent years, driven largely by β-emitting therapies targeting somatostatin receptor–expressing tumors and the prostate-specific membrane antigen. Now, more clinical trials are under way to evaluate α-emitting targeted therapies as potential next-generation theranostics with even higher efficacy due to their high linear energy and short range in human tissues. In this review, we summarize the important studies ranging from the first Food and Drug Administration–approved α-therapy, 223Ra-dichloride, for treatment of bone metastases in castration-resistant prostate cancer, including concepts in clinical translation such as targeted α-peptide receptor radiotherapy and 225Ac-PSMA-617 for treatment of prostate cancer, innovative therapeutic models evaluating new targets, and combination therapies. Targeted α-therapy is one of the most promising fields in novel targeted cancer therapy, with several early- and late-stage clinical trials for neuroendocrine tumors and metastatic prostate cancer already in progress, along with significant interest and investment in additional early-phase studies. Together, these studies will help us understand the short- and long-term toxicity of targeted α-therapy and potentially identify suitable therapeutic combination partners.
The use of α-emitters has evolved over the past few years. They offer two advantages over treatments using β-emitters: first, α-radiation has a short range in tissues, resulting in irradiation of only a few cell diameters (<0.1 mm), allowing for selective treatment of cancer cells, and second, the high linear energy transfer of several megaelectron volts of α-radiation results in effective cell killing via DNA double-strand breaks (Fig. 1) (1). Therefore, close binding to the target is crucial to ensure therapeutic efficacy and safety.

Comparison of radiobiologic effects of 225Ac-PSMA and 177Lu-PSMA. LET = linear energy transfer.
To date, several clinical experimental α-treatments and an approved treatment for prostate cancer exist. 223Ra-dichloride for treatment of bone metastases in castration-resistant prostate cancer was the first agent for which a survival benefit of 3 mo versus placebo was proven in a prospective phase 3 randomized clinical trial. It paved the way for the clinical acceptance of targeted α-therapies (TATs) in vivo and is so far the only Food and Drug Administration–approved α-therapy (2,3). Additionally, for several other diseases, targets have been identified for α-therapy. Among them are bladder carcinoma showing overexpression of epidermal growth factor receptor (4), metastatic prostate cancer showing overexpression of prostate-specific membrane antigen, metastases of neuroendocrine tumors (NETs) with upregulated somatostatin receptors (5), glioma with substance P as a molecular target, and specific targets such as HuM195 in patients with leukemia (6).
Most of these TATs use either 213Bi or 225Ac as α-emitters, but many other radionuclides are currently discussed (149Tb, 211At, 212Pb [for 212Bi], 226/227Th, and 230U) (7). Because of the short half-life of 46 min, 213Bi-labeled agents have to be synthetized on site. In contrast, 225Ac has 4 times more α-decays and a longer half-life (10 d), qualifying it as an attractive therapeutic nuclide–emitting energy of between 5.8 and 8.4 MeV (8). It has been proposed that because of the higher linear energy transfer of α-emitters, therapy resistance to β-emitters can be overcome. No relevant side effects were reported for local α-emitter application, such as in bladder cancer patients (4); however, systemic administration such as intravenous application with, for example, 223Ra-dichloride have side effects, notably on bone marrow (2).
We present an overview of 3 promising clinical applications of TAT administered systemically, as well as future directions.
THE FRONT RUNNER: 223RA-DICHLORIDE
Bone pain therapy using β-emitters for osteoblastic metastases has been established in the palliative setting for many years now. However, the only Food and Drug Administration–approved radiopharmaceutical with a positive impact on overall survival (OS) is the α-emitter 223Ra (9). 223Ra has a half-life of 11.4 d and decays into the stable daughter nuclide 207Pb via 4 α-emissions (5.0–7.5 MeV). The median penetration range in soft tissue is 0.04–0.05 mm. Radium has physiologic similarities to calcium and selectively accumulates in bones, especially in areas with high bone metabolism such as marginal areas of bone metastases (10).
223Ra-dichloride (Xofigo; Bayer) was Food and Drug Administration–approved in November 2013 for men with prostate cancer and bone metastases in whom the usual hormone blockade is no longer effective. The approval was based on the results of the randomized, phase 3 ALSYMPCA trial (2). In this study, 921 patients who had received, were not eligible to receive, or declined chemotherapy with docetaxel and with at least 2 or more symptomatic bone metastases with no known visceral metastases were randomized 2:1 to receive 6 cycles of 223Ra every 4 wk with the best standard of care or 6 infusions of placebo with the best standard of care. The OS was significantly longer in the 223Ra group (14.9 vs. 11.3 mo; P < 0.001), the frequency of skeleton-related events was reduced, and the median time to a skeleton-related event was longer (15.6 vs. 9.8 mo; P < 0.001). No clinically significant differences in the frequency of grade 3 or 4 adverse events were observed between the groups (2). Nonhematologic toxicities are mild to moderate in intensity. The most common side effects are diarrhea, fatigue, nausea, vomiting, and bone pain, some of which are dose-related. These side effects are easily manageable with symptomatic and supportive treatments. Grade 3 and 4 thrombocytopenia was reported in 6% of patients on 223Ra and in 2% of patients on placebo (2). 223Ra is also accompanied by quality-of-life benefits (11).
NOTEWORTHY
After approvals of 177Lu-DOTATATE and 177Lu-vipivotide tetraxetan, TATs represent the next generation of theranostics.
TATs offer advantages over other treatments due to their short range in tissues, allowing selective treatment of targeted cancer cells, and high linear energy transfer, leading to highly effective cell killing.
223Ra-dichloride is approved for the treatment of bone metastases in castration-resistant prostate cancer; α-peptide receptor radiotherapy for the treatment of NETs and 225Ac-PSMA therapies for prostate cancer are other promising clinical applications.
These therapy models can be further investigated with new targets, as therapy combinations, and in patients with different stages of disease.
Jiang et al. reported the 5-y real-world outcome of 223Ra therapies for 228 patients. The median OS was 11.1 mo. The OS in chemotherapy-naïve patients was significantly longer than in patients with a history of chemotherapy (12.3 vs. 8.1 mo; P = 0.02). No significant survival differences were observed between pre- and postabiraterone and prednisolone or enzalutamide patients. The fracture rate in the postabiraterone and prednisolone group was 24%, seemingly high (12).
Around the time of the ALSYMPCA trial, the hormone therapies abiraterone and enzalutamide were not part of the routine therapeutic approach, and therefore the feasibility of a therapy with 223Ra before or after or even at the same time as these agents could not be studied at the time of approval of 223Ra. The approval was changed in September 2018 because of the results of the ERA 223 study. In this prospective, phase 3 study, 806 patients were randomly assigned to receive 223Ra (n = 401) or placebo (n = 405) in addition to abiraterone acetate plus prednisone or prednisolone. The study was prematurely unmasked because of the increased risk of fractures and a trend toward increased mortality in the group of patients who received 223Ra in combination with abiraterone and prednisone. There was an increased incidence of fractures (29% vs. 11.4%) and a possible reduction in median OS (30.7 vs. 33.3 mo; P = 0.13) (13). Since then, according to the European Medicines Agency (14), 223Ra therapy should be used only as monotherapy or in combination with a luteinizing hormone-releasing hormone analog for the treatment of patients with symptomatic bone metastases and no known visceral metastases, who have progressed after at least 2 prior lines of systemic therapy for metastatic castration-resistant prostate cancer (mCRPC) or are ineligible for any available systemic mCRPC treatment. It is also not recommended in patients with a low level of osteoblastic bone metastases or in patients with only asymptomatic bone metastases (15). The recommended regimen per the European Medicines Agency is 6 treatments of 55 kBq/kg every 4 wk.
An assessment of skeletal tumor burden on bone scintigraphy or PET before 223Ra therapy is a valuable approach for the prognostication of OS and hematologic toxicity (16). In addition, tumor-specific prostate-specific membrane antigen imaging could be useful for selecting the most eligible patients (17).
There are limited data regarding the time interval between therapy with abiraterone and 223Ra. Based on the half-lives of 223Ra and abiraterone, it is recommended that subsequent treatment with 223Ra be started no earlier than 5 d after the last administration of abiraterone. Subsequent systemic cancer therapy should be initiated no earlier than 30 d after the last dose of 223Ra. Concurrent use of bisphosphonates or denosumab has been found to reduce the incidence of fractures in patients treated with 223Ra (15).
In the case of disease progression after the initial 6 cycles of 223Ra therapies, a rechallenge with a second course of 6 223Ra injections seems to be feasible, with minimal hematologic toxicity and sustained benefit in terms of OS (18).
α-PEPTIDE RECEPTOR RADIOTHERAPY: READY FOR PRIME TIME?
Somatostatin receptors are overexpressed in NETs and because of highly efficient in vivo agonist-induced internalization, they are a perfect target for peptide receptor radiotherapy. Consecutively, 213Bi-DOTATOC became the first somatostatin-receptor TAT demonstrating proof of mechanism in a clinical application: 8 patients without a sufficient response to previous β-peptide receptor radiotherapy demonstrated remarkable antitumor activity with α-peptide receptor radiotherapy (19). However, 2 y after therapy, patient glomerular filtration rate decreased significantly from an average of 115 mL/min to 83 mL/min (−30%). Also, the limited number of appropriate patients, challenging logistics related to the invasive arterial catheter placement and labeling procedure, and the costly demand of a gigabecquerel-sized 225Ac/213Bi generator on-site prevented broader clinical application.
225Ac-DOTATOC was suggested to overcome these challenges (20). Preclinical results appeared promising, showing an added tumor size decrease sequencing cold DOTATOC, 177Lu-DOTATOC, and 225Ac-DOTATOC. Additionally, a maximum tolerable dose (applicable to mice) was determined, with tubular necrosis presenting the dose-limiting toxicity (20). The promising therapeutic range between antitumor activity and modest renal toxicity was recently confirmed by an independent study on mice (21). Preliminary dosimetry attempts—not published for 213Bi- vs. 225Ac-DOTATOC but for 213Bi- vs. 225Ac-PSMA-617 (a shuttle molecule with similar tracer pharmacokinetics)—done by the Heidelberg group demonstrated that in humans the longer effective half-life of 225Ac-labeled small molecules in tumor versus kidney probably improves the therapeutic range of TAT (22). Nevertheless, neglecting microdosimetry, translocation effects of daughter nuclides and using average literature values for relative biological effectiveness, it was not possible to predict appropriate treatment activities for clinical application without additional empiric data. In 2015, the group presented its quasi-escalation experience with 225Ac-DOTATOC (5). As demonstrated in Figure 2, antitumor activity could be observed at all dose levels; a maximum tolerable single-cycle dose of 40 MBq or 4-mo intervals with 25 MBq or 2-mo intervals with less than 18.5 MBq were suggested to be tolerable regarding acute hematologic toxicity. However, follow-up was too short to draw a final conclusion regarding chronic nephrotoxicity. In 2020, the first prospective clinical trial for 225Ac-DOTATATE examined 32 patients with previous exposure to 177Lu-DOTATATE who were treated with a 100 kBq/kg dose of 225Ac-DOTATATE (23). Fifteen patients achieved partial remission, and 9 patients had stable disease. However, median follow-up was only 8 mo (range, 2–13 mo); thus, no conclusion could be drawn about long-term tolerability. However, 5-y follow-up data for the Heidelberg cohort revealed a dose-dependent acute hematologic toxicity at single doses above 40 MBq or repeated doses greater than approximately 20 MBq 225Ac-DOTATOC at 4-mo intervals. Treatment-related kidney failure occurred in 2 patients after a delay of greater than 4 y but was independent of administered radioactivity, and other clinical risk factors were important contributors (24).
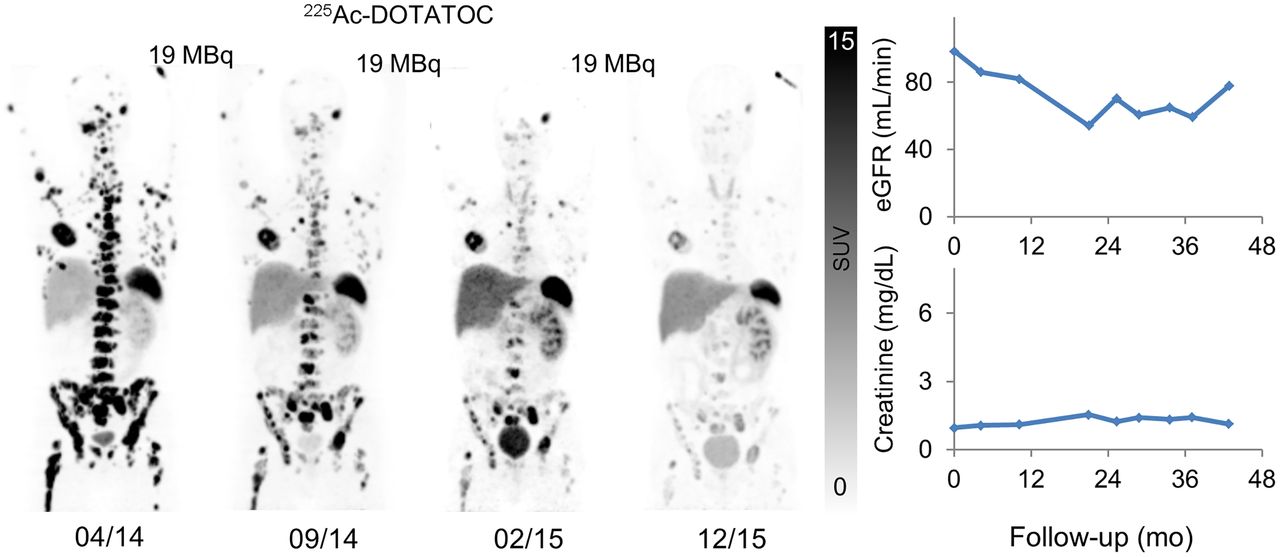
Patient with G2 (Ki-67, 5%) NET of right (resected) kidney received 3 cycles of 19 MBq of 225Ac-DOTATOC. Maximum-intensity projections of 68Ga-DOTATOC PET/CT done in advance, between cycles, and after last cycle demonstrate antitumor activity (left). Despite remaining solitary left kidney, cumulative 47 MBq of 225Ac-DOTATOC did not lead to increase in serum creatinine (right, bottom) or estimated glomerular filtration rate (right, top) during 3 y of follow-up. eGFR = estimated glomerular filtration rate.
Another α-emitter with appropriate decay characteristics for use in α-peptide receptor radiotherapy is 212Pb (half-life, 10.6 h). In 2019, the preclinical characterization of 212Pb-DOTAMTATE became available (25) and was well in line with the previous preclinical data obtained with 213Bi- or 225Ac-DOTATOC. A favorable outcome was observed, when the nontoxic cumulative dose of 1.7 MBq was fractionated into 3 × 0.6 MBq or 3 × 0.4 MBq in combination with chemotherapy. Another group has published preliminary results from the first-in-humans phase 1 dose escalation trial evaluating 212Pb-DOTAMTATE in 20 patients with somatostatin receptor–positive NETs with no prior history of 177Lu/90Y/111In peptide receptor radiotherapy (NCT03466216) (26). The study was a single-ascending-dose/multiple-ascending-dose trial using a 3 + 3 dose-escalation scheme with an 8-wk dose-limiting toxicity period. The initial dose was 1.13 MBq/kg, and subsequent cohorts received an incremental 30% dose increase until a tumor response or a dose-limiting toxicity was observed. The maximum total dose per subject was 296 MBq in the single-ascending-dose cohort and 888 MBq in the multiple-ascending-dose cohort. Treatment was well tolerated, with the most common adverse events being nausea, fatigue, and alopecia. No serious treatment-emergent adverse events were related to the study drug, and no subjects required treatment delay or a dose reduction. Of the 10 subjects who received all 4 cycles, 8 (80%) demonstrated an objective, long-lasting radiologic response by RECIST 1.1 (26).
225AC-PSMA-617 FOR THERAPY OF PROSTATE CANCER
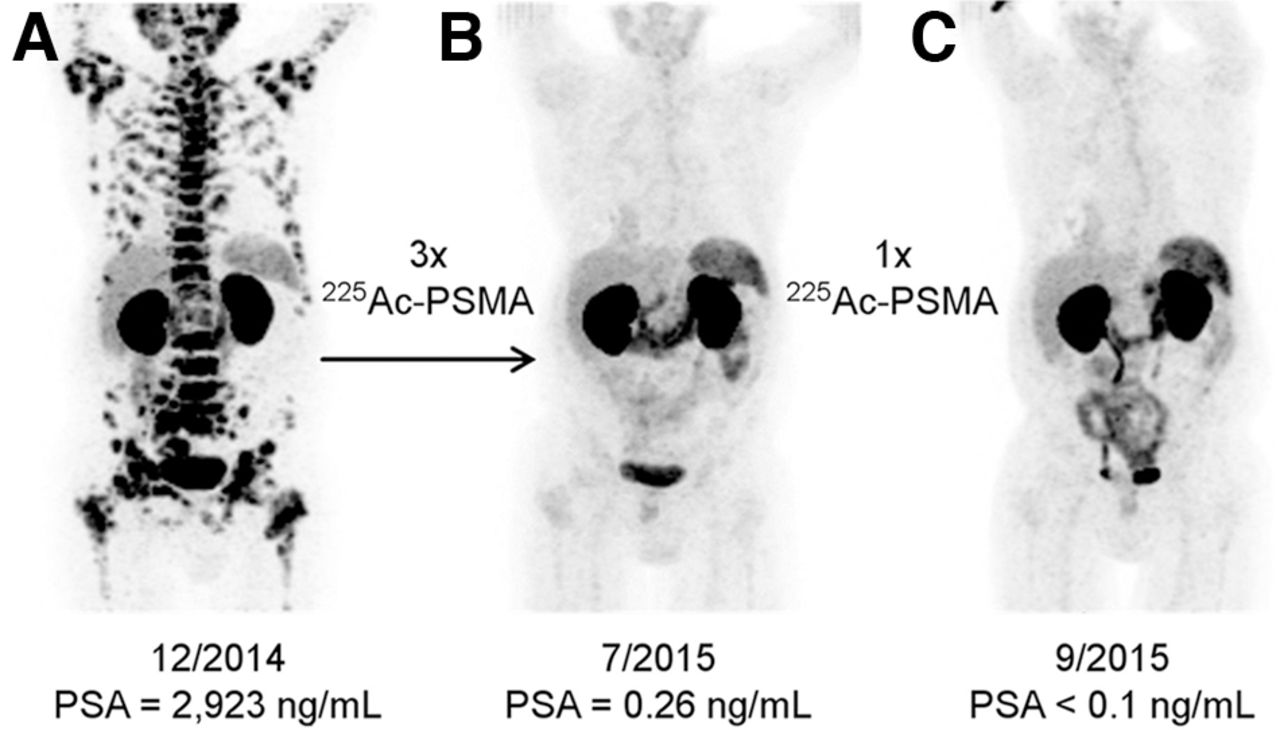
Treatment of metastatic prostate cancer has been complemented by the use of α-emitters in the past few years. 225Ac-PSMA-617 was first developed in vitro at the Joint Research Centre Karlsruhe in 2013 and 2014 (1). Kratochwil et al. first described a remarkable therapeutic effect of 225Ac-PSMA-617 in patients with late-stage prostate cancer (Fig. 3) (27). The initial report included 2 patients who showed complete remission after exhausting conventional therapies, including chemotherapy and advanced hormone treatment.
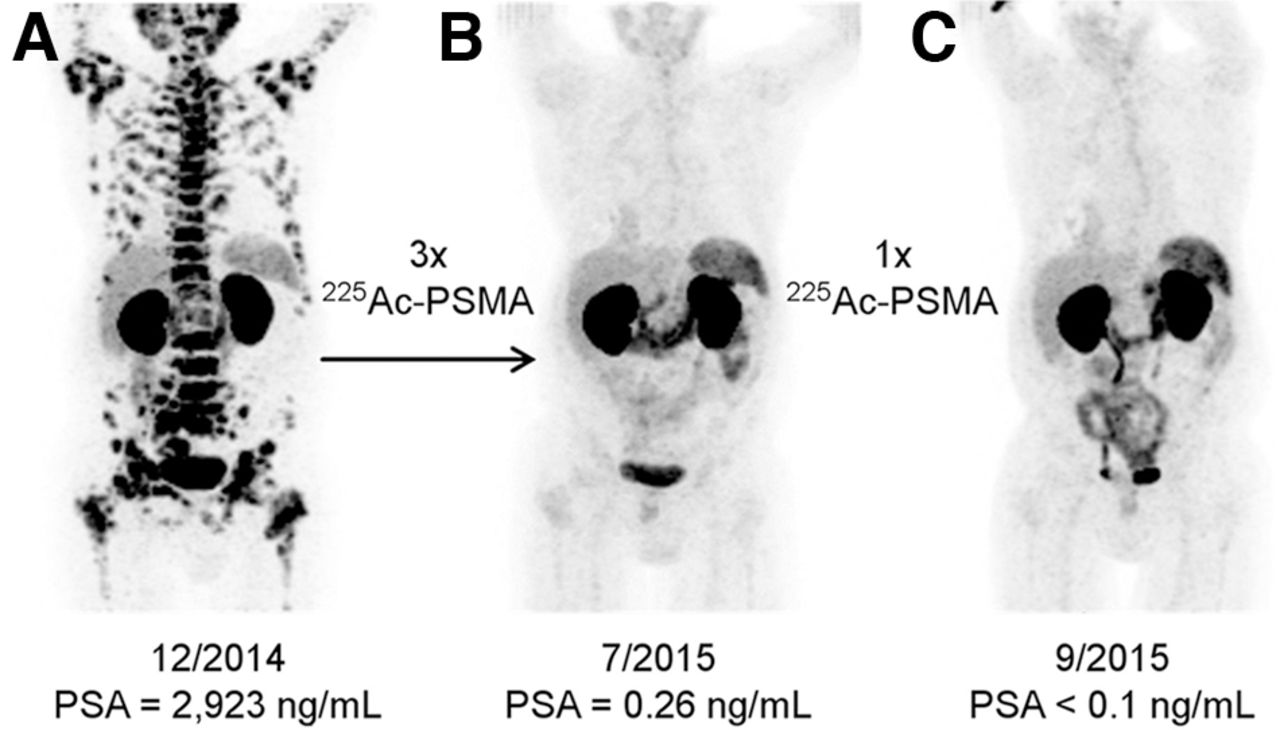
Initial promising results from Heidelberg group for patient with diffuse red bone marrow infiltration of mCRPC, which was considered contraindication for treatment with β-emitters. 68Ga-PSMA-11 PET/CT scans demonstrating pretherapeutic tumor spread (A), restaging 2 mo after third cycle of 225Ac-PSMA-617 (B), and restaging 2 mo after 1 additional consolidation therapy (C). (Reprinted from (27).)
The favorable pharmacologic properties and kinetics of PSMA-617 with fast tumor uptake, good internalization, long tumor retention, and rapid clearance of unbound ligand are desirable properties for the combination with an α-emitter (1). Because of the long half-life of several days and multiple α-emissions in the decay chain of 225Ac, these pharmacokinetic properties are highly relevant to reduce potential clinical side effects. Almost all available studies used 225Ac-PSMA-617 for treatment (Table 1). One study also investigated 225Ac-PSMA-I&T (28) and demonstrated similar biochemical response rates, with any prostate-specific antigen (PSA) decline in 79% of patients, comparable to PSMA-617 ranging between 79% and 94% (Table 1). However, no data on clinical progression-free survival (cPFS) and OS is currently available using 225Ac-PSMA-I&T.
Overview of Current Data on Compassionate Use of 225Ac-PSMA-617 RLT in Different Patient Cohorts: Effects of Treatment on PSA, Complete Remission in PET, and Median OS Are Summarized (34)
Dose Finding and Dosimetry Estimates
Subsequent studies focused on dose finding using between 50 and 200 kBq/kg activities of 225Ac-PSMA-617 with the aim of finding a reasonable trade-off between therapeutic efficacy and side effects. For 8-week intervals, a reasonable trade-off was found to be a 100 kBq/kg activity of 225Ac-PSMA-617 (29). At 50 kBq/kg, side effects were low but antitumor effects were insufficient, and at 200 kBq/kg, side effects increased significantly, with xerostomia as the dose-limiting toxicity (29). In this study, a dosimetry estimate showed that assuming a relative biological effectiveness of 5, 1 MBq of 225Ac-PSMA-617 leads to mean doses of 2.3 Sv for salivary glands, 0.7 Sv for kidneys, and 0.05 Sv for red marrow that are composed of 99.4% α, 0.5% β, and 0.1% photon radiation, respectively (29). However, dosimetry of α-emitters is challenging, in part because of the short range of α-emitters in tissue and the complex decay schemes with multiple α-, β-, and γ-daughters with a broad range of energies and half-lives of the emitted nuclides. To date, no standardized means of imaging α-emitters exists (30).
Surrogate Markers of Response
The number of patients experiencing any PSA decline is in the range of 79%–94% (31–33). A maximum PSA decline of at least 90% was achieved in only 12% of the late-stage mCRPC patients (34) but in 40%–82% of patients in earlier stages (Table 1). Data on complete remission on PET are not available from all studies but are in the range of 0%–65% (Table 1). Median cPFS was between 4.1 and 15.2 mo, and the median OS was 7.7–18 mo (31,33–35). cPFS and OS, 4.1 and 7.7 mo, respectively, appear to be shorter in patients with late-stage mCRPC (e.g., median of 6 treatment regimens before 225Ac-PSMA-617) compared with early- to intermediate-stage mCRPC patients (up to a median of 3 prior treatment regimens), with a cPFS of 7–15.2 mo and an OS of 12–18 mo, respectively.
Risk Factors
A study of advanced mCRPC patients, treated with 225Ac-PSMA-617, found that those with high baseline immunohistochemical PSMA expression or DNA damage repair alterations tended to have longer OS (36). In a cohort of 72 patients, a PSA decline of at least 50% was significantly associated with OS and PFS in multivariate analysis after 225Ac-PSMA-617 treatment (33). Additionally, previous 177Lu-PSMA treatment was negatively correlated with PFS (33). In a study of 26 late-stage mCRPC patients, it is reported that liver metastases are associated with significantly shorter PSA PFS (median, 1.9 vs. 4.0 mo), cPFS (median, 1.8 vs. 5.2 mo), and OS (median, 4.3 vs. 10.4 mo) (34).
Xerostomia
Xerostomia has been reported to be a relevant side effect of 225Ac-PSMA treatment. It seems to be more pronounced after intensive pretreatment. In late-stage mCRPC patients, xerostomia (grade 1/2) was reported in all patients, with 23% requesting stop of treatment to preserve quality of life (34). Kratochwil et al. (31) reported that 10% of patients discontinued treatment because of intolerable xerostomia. Sathekge et al. (33) reported grade 1/2 xerostomia in 85%–100% patients. Yadav et al. reported xerostomia of grade 1/2 in only 29% in an earlier-stage patient cohort (35). Khreish et al. reported that 20 patients who received a tandem therapy of 225Ac-PSMA-617/177Lu-PSMA-617 with 5.3 MBq (range, 1.5–7.9 MBq) of 225Ac-PSMA and 6.9 GBq (range, 5.0–11.6 GBq) of 177Lu-PSMA- 617, had grade 1 (very mild) xerostomia in 8 of 20 patients (40%) and grade 2 (mild) in 5 of 20 (25%) (Fig. 1) (37). Xerostomia was reported as grade 1 (very mild) in 8 of 20 patients (40%) and grade 2 (mild) in 5 of 20 (25%) (37). In an attempt to maintain salivary gland function, Rathke et al. performed sialendoscopy on patients undergoing 225Ac-PSMA-617 treatment (38). Despite sialendoscopy support with, for example, steroid injections, xerostomia was present after multiple cycles (38). Using monoclonal antibodies such as 225Ac-J591 might reduce salivary gland uptake and hence xerostomia as previously reported, with only 8 of 32 (25%) patients showing grade 1 xerostomia (39). However, these antibodies have less renal excretion and may increase bone marrow toxicity.
Hematologic Side Effects
Compared with 177Lu-PSMA, grade 3/4 hematologic toxicity seems to be higher for 225Ac-PSMA. In a series of patients with late-stage mCRPC, hematologic grade 3/4 toxicities were reported in 35%, 27%, and 19% for anemia, leukopenia, and thrombocytopenia, respectively (34). In patients treated with 177Lu-PSMA-radioligand therapy, the frequencies of grade 3/4 anemia, thrombocytopenia, and leukopenia were only 9%–10%, 2%–13%, and 3%–32%, respectively (40–43). However, the patients receiving 225Ac-PSMA had been treated at a substantially later stage with a median of 6 prior treatments (34). Notably, the frequency of these adverse hematologic events was similar to that of investigational agents (44) or, for example, carboplatin/etoposide (45). Yadav et al. reported no grade 3/4 thrombocytopenia or leukopenia, but 1 of 28 patients had grade 3 anemia (35). Khreish et al. reported at least one grade 3/4 hematotoxicity in 25% of patients: 5% with grade 3 anemia, 10% with grade 3 leukopenia, and 10% with grade 4 anemia and thrombocytopenia each using a tandem therapy of 225Ac-PSMA-617 and 177Lu-PSMA (37).
225Ac-PSMA-617/177Lu-PSMA-617 Tandem Therapy
Up to 30% of mCRPC patients do not respond to 177Lu-PSMA therapy (37). In patients with resistance to β-emitters, treatment with an α-emitter may still be effective. The combination of α- and β-emitters may allow for a lower dose of 225Ac, reducing salivary gland toxicity. In 13 of 20 patients who received one course of 225Ac-PSMA-617/177Lu-PSMA-617 tandem therapy, biochemical response (PSA decline > 50%) and grade 1/2 xerostomia was observed (37). Rosar et al. reported partial response in 5 of 17 patients and stable disease in 7 of 17 patients, treated with tandem therapy after progression with 177Lu-PSMA (46).
NEW TARGETS
In addition to hydroxyapatite, somatostatin receptors, and the prostate-specific membrane antigen, there are additional targets under investigation for their utility in TAT. One of the most prominent new targets is the fibroblast activation protein (FAP) that is overexpressed by cancer-associated fibroblasts in the stroma of several tumor entities as well as less frequently directly from tumor cells. FAP can be targeted with antibodies, peptides, and small molecules such as FAP inhibitors (FAPIs). FAP-targeted imaging has been explored in several malignancies, including glioma, nasopharyngeal carcinoma, gastric cancer, colorectal cancer, hepatocellular carcinoma, cholangiocarcinoma, soft-tissue sarcoma, pancreatic cancer, breast cancer, and prostate cancer (47). Research into the therapeutic application of FAPI radionuclides is still in the early stages. Breast cancer patients have been treated with 90Y-FAPI-04 (48) and 177Lu-DOTA.SA.FAPI (49), an ovarian cancer patient has been treated with 90Y-FAPI-46 (50), and sarcoma and pancreatic cancer patients have been treated with 90Y-FAPI-46 (50) and 177Lu-FAPI-46.
As for TAT, 225Ac-FAPI-46 has been used in pancreatic cancer mouse models and 153Sm-FAPI-46 was used to treat a patient with soft-tissue sarcoma metastatic to the lung. In one study, 34 kBq of 225Ac-FAPI-04 were injected into 6 PANC-1 xenograft mice, 3 wk after implantation with a tumor size of 0.98 ± 0.66 cm3 (51). Tumor size was compared with 6 control mice for up to 51 d. The mice who received 225Ac-FAPI-04 showed significant tumor growth suppression compared with the control mice, without a significant change in body weight, with the equivalent dose in the tumor estimated to be 5.68 ± 0.77 Gy/MBq. In another study, 3 kBq (n = 3), 10 kBq (n = 2), and 30 kBq (n = 6) of 225Ac-FAPI-04 were injected into PANC-1 xenograft mice and tumor size and weight were compared with 7 control mice (52). The tumor growth was suppressed immediately after treatment with 10 kBq and 30 kBq, whereas the tumor-suppressive effects in the 3-kBq group were very mild. The tumor size of the 30-kBq group was significantly smaller than that in the control group on days 5–9 and day 25. The body weight in all groups showed a trend to decrease in the first week but recovered in the 3- and 10-kBq groups after day 7. Lastly, in a patient with fibrous spindle cell soft-tissue sarcoma metastatic to the lung, 3 cycles of 20 GBq of 153Sm- and 8 GBq of 90Y-FAPI-46 were well tolerated and achieved stable disease for 8 mo (53). In the future, 212Pb-FAPI compounds will also be preclinically and potentially clinically explored.
Another target that is being investigated is human epidermal growth factor receptor type 2 (HER2), which is overexpressed in various cancers including breast, ovarian, bladder, pancreatic, and gastric. A preclinical study evaluated a HER2-targeting single-domain antibody labeled with 225Ac, called 225Ac-DOTA-2Rs15d, in mice with HER2-positive intraperitoneal ovarian cancer (54). Both a single dose of 86.84 ± 8.97 kBq of 225Ac-DOTA-2Rs15d 7 d after tumor inoculation and 3 consecutive administrations of 86.84 ± 8.97 kBq of 225Ac-DOTA-2Rs15d on days 7, 10, and 14 resulted in a significantly longer mean survival of 101 and 143 d, respectively, versus 56 d for mice receiving vehicle solution (P < 0.0001). Additionally, 3 consecutive doses of 225Ac-DOTA-2Rs15d increased the mean survival (143 d) compared with a group a receiving trastuzumab regimen (7.5 mg/kg loading dose, followed by 2 maintenance doses of 3.5 mg/kg) and a single dose of 225Ac-DOTA-2Rs15d (P < 0.0389). There was histopathologic evidence of kidney toxicity after repeated doses of 225Ac-DOTA-2Rs15d. The single-domain antibody 2Rs15d, also referred to as anti-HER2-VHH1, has also been labeled with 131I and studied in HER2-positive breast cancer patients (NCT02683083, NCT04467515) (55).
Other targets including type I insulin-like growth factor (NCT03746431) (56), a transmembrane protein that is overexpressed in non–small cell lung, prostate, and breast cancers; neurotensin receptor 1 (NCT05605522) (57), upregulated in colorectal and pancreatic cancers; and CD33 (NCT03867682) (58), found in myeloid tumor cells are being investigated in conjunction with 225Ac.
COMBINATION THERAPY
TAT may also be advantageous in combination with other cancer treatments such as chemotherapy, immunotherapy, DNA repair inhibitors, and other radionuclide treatments. These types of regimens are already being readily studied in prostate cancer patients in combination with 177Lu-PSMA treatment and can also be evaluated with TAT. For example, 225Ac-PSMA-617 could also be studied in combination with androgen receptor signaling inhibitors or chemotherapy. Other innovative concepts include the concomitant administration of 225Ac- and 177Lu-PSMA-617 in mCRPC patients (59). HER2-targeted therapy can be studied in conjunction with trastuzumab. Additionally, studies of a combination of 177Lu-DOTATATE and M3814 (an inhibitor of DNA-dependent protein kinase) for patients with pancreatic NETs (NCT04750954) and a combination of 223Ra-dichloride and M3814 for patients with mCRPC (NCT04071236) are already under way; these concepts can also be studied with TAT. Future investigations can focus on combination therapies to evaluate possible synergistic effects.
CONCLUSION
The field of TAT is currently one of the most promising in innovative targeted cancer therapy. The question as to whether these new TATs are merely an evolution or small improvement of currently used therapies versus a revolution leading to a complete paradigm shift remains to be answered. Whereas several early- and late-stage clinical trials on NETs and metastatic prostate cancer are already under way, there is also a significant interest (and investment) by multiple well-funded early-phase biotechnical companies (60) dedicated to the further development of novel TAT concepts. Despite the profound excitement and incredible clinical potential, it is also important to emphasize the need to understand short- and long-term toxicity of TAT and identification of suitable therapeutic combination partners.
DISCLOSURE
Clemens Kratochwil is coinventor of patents for radiolabeled PSMA and FAP ligands; owns shares of FAPi Holding AG; indirectly received research support from Telix Pharmaceuticals; worked as a scientific advisor for Novartis Radiopharmaceuticals, Hoffmann-La Roche, and AdvanCell; and received speaker fees from Novartis. Hojjat Ahmadzadehfar reports fees from Bayer (speaker), Novartis/AAA (speaker), and IPSEN (speaker) and is an unpaid consultant for Novartis/AAA. Matthias Eiber reports fees from Blue Earth Diagnostics Ltd. (consultant, research funding), Novartis/AAA (consultant, speaker), Telix (consultant), Bayer (consultant, research funding), RayzeBio (consultant), Point Biopharma (consultant), Eckert-Ziegler (speaker), Janssen Pharmaceuticals (consultant, speakers bureau), Parexel (image review), and Bioclinica (image review) outside the submitted work and a patent application for rhPSMA. Ken Herrmann reports personal fees from Bayer, Sofie Biosciences, SIRTEX, Adacap, Curium, Endocyte, BTG, IPSEN, Siemens Healthineers, GE Healthcare, Amgen, Novartis, ymabs, Aktis Oncology, Theragnostics, Pharma15, Debiopharm, AstraZeneca, and Janssen; nonfinancial support from ABX; grants from BTG; and other fees from Sofie Biosciences outside the submitted work. Kelsey Pomykala reports personal fees from ABX outside the submitted work. No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Apr. 13, 2023.
- © 2023 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication December 22, 2022.
- Revision received February 10, 2023.
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.