


Abstract
Discovery of biomarkers has been steadily increasing over the past decade. Although a plethora of biomarkers has been reported in the biomedical literature, few have been sufficiently validated for broader clinical applications. One particular challenge that may have hindered the adoption of biomarkers into practice is the lack of reproducible biomarker cut points. In this article, we attempt to identify some common statistical issues related to biomarker cut point identification and provide guidance on proper evaluation, interpretation, and validation of such cut points. First, we illustrate how discretization of a continuous biomarker using sample percentiles results in significant information loss and should be avoided. Second, we review the popular “minimal-P-value” approach for cut point identification and show that this method results in highly unstable P values and unduly increases the chance of significant findings when the biomarker is not associated with outcome. Third, we critically review a common analysis strategy by which the selected biomarker cut point is used to categorize patients into different risk categories and then the difference in survival curves among these risk groups in the same dataset is claimed as the evidence supporting the biomarker’s prognostic strength. We show that this method yields an exaggerated P value and overestimates the prognostic impact of the biomarker. We illustrate that the degree of the optimistic bias increases with the number of variables being considered in a risk model. Finally, we discuss methods to appropriately ascertain the additional prognostic contribution of the new biomarker in disease settings where standard prognostic factors already exist. Throughout the article, we use real examples in oncology to highlight relevant methodologic issues, and when appropriate, we use simulations to illustrate more abstract statistical concepts.
- statistics
- area under the ROC curve
- biomarker cut point
- biomarker discretization
- prognostic biomarker
- resubstitution statistics
Recent advances in biotechnologies have made it possible to perform extensive biologic characterizations of human diseases. These efforts have resulted in the discovery of a myriad of biomarkers and generated much excitement for their potential to guide patient care. Possible uses of biomarkers in research and clinical settings include individual risk stratification, disease monitoring, and guiding the use of specific treatment regimens. Despite the large volume of published articles in biomedical journals on newly identified biomarkers, very few of these have progressed to the point of being clinically actionable. Many biomarkers may appear promising in the initial research reports but fail to retain their utility in subsequent studies. One particular challenge that may have hindered the adoption of biomarkers into practice is the lack of reproducible biomarker cut points. To aid clinical decision making, medical practitioners are accustomed to discretizing a biomarker measured on a quantitative scale into different risk categories based on some partition of the scale, commonly called cut points. This practice is natural, as it is desirable to define patient groups sharing a similar expected prognosis (say, for treatment or surveillance), and an overly precise scale is not useful in this regard. However, research reports frequently lack sufficient details on how such cut points are identified. Moreover, naïve use of statistical methodology for cut point identification, invalid methods for analysis, and overconfidence in the reliability of cut point–defined risk groups have hampered the ability to compare results across different studies or to generalize the results to the larger disease population of interest in an unbiased fashion. Even in the same or a similar disease setting, the biomarker cut points reported are often inconsistent and irreproducible.
Our goal in this article is to highlight some common statistical issues that arise from biomarker cut point identification and to provide guidance on proper evaluation, interpretation, and validation of such cut points. First, we illustrate how discretization of a continuous biomarker using sample percentiles (e.g., sample median) results in significant information loss and should be avoided. Second, we review a popular method for cut point identification that entails testing a range of cut points and selecting the cut point that yields the smallest P value (i.e., the “minimal-P-value” approach). We show that this approach results in highly unstable P values and is associated with a severely inflated false-discovery rate (i.e., it unduly increases the chance of significant findings when the biomarker is not associated with the outcome) and estimates of the biomarker effect that are biased (suggesting a larger effect than is actually present). Some methods for correcting the P value and biomarker effect are referenced. Third, we critically review a common analysis strategy by which the selected biomarker cut point is used to categorize patients into different risk categories and then the difference in survival curves among these risk groups in the same dataset is claimed as the evidence supporting the biomarker’s prognostic strength. We show that this method yields exaggerated P values and overestimates the prognostic impact of the biomarker. We illustrate in a simulation study that the degree of the optimistic bias increases with the number of variables being considered in a risk model. We expand from that point to special considerations for biomarker cut points in disease settings where standard prognostic factors already exist. We discuss methods to appropriately ascertain whether there is any additional prognostic contribution from the new biomarker and the relevance of cut point determination in such a context.
Throughout the article, we use real examples in oncology to highlight relevant methodologic issues, and when appropriate, we use simulations to illustrate more abstract statistical concepts. Although the examples here pertain primarily to molecular biomarkers, these principles generally apply to other types of biomarkers (e.g., imaging biomarkers and blood biomarkers) so long as they are measured on a continuous scale. Similarly, these statistical principles can readily be adapted to other noncancer disciplines in biomedical research.
STATISTICAL PITFALLS IN BIOMARKER CUT POINT SEARCH AND ANALYSIS
Loss of Information Due to Discretization
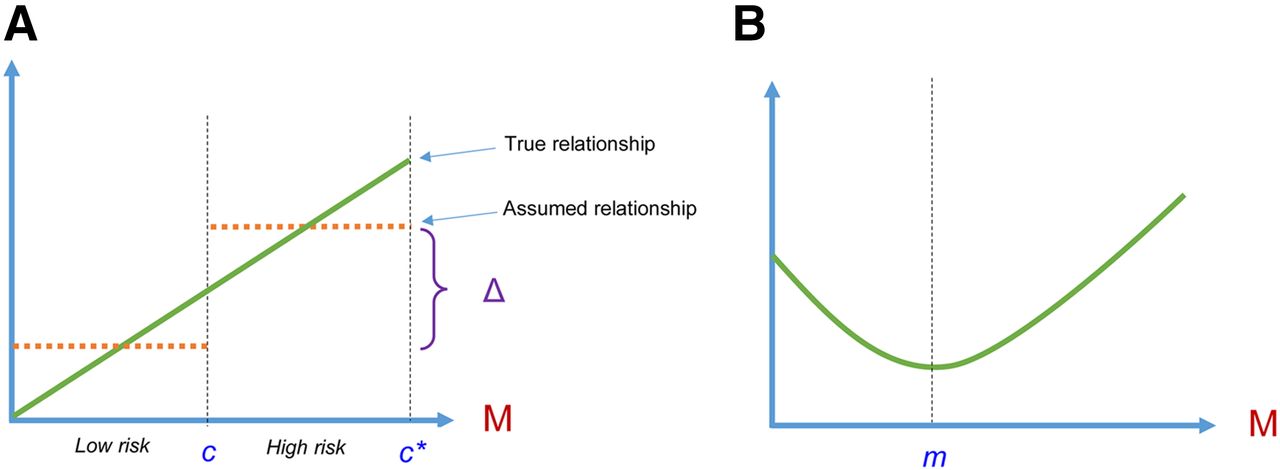
A popular strategy for handling continuous biomarkers is to convert them into discrete variables by grouping patients into distinct risk subgroups (e.g., groups based on sample percentiles of the biomarker values). This type of categorization avoids the need to make strong assumptions about the functional relationship between the biomarker and the outcome. In reality, however, the true relation between a continuous biomarker and outcome is almost always smooth. Such relations are seldom characterized by an abrupt jump at a given biomarker value. Figure 1 illustrates 2 true relationships between a biomarker and some continuous outcome of interest (e.g., patient survival)—one linear (the risk of death increases linearly with the biomarker value) and the other quadratic (the risk of death decreases up to a certain point but then increases linearly). Dichotomy of biomarkers into 2 patient groups assumes that a discontinuity in the risk occurs at some biomarker value and that the relationship between the biomarker and the outcome is flat for patients whose biomarker values are within the same intervals, as defined by the point of dichotomy. Such dichotomy presumes that there is a notable change in prognosis at the cut point in that patients whose biomarker values are below the cut point are conferred the same risk, which is lower than the risk conferred to patients whose biomarker values exceed the cut point. This risk stratification based on dichotomizing the biomarker clearly does not adequately reflect the true linear relationship between the biomarker and the outcome. In addition, categorizing a continuous biomarker causes considerable loss of valuable information, which may in turn increase the chance of missing a real association. For example, a patient whose true risk is highest in a high-risk subgroup is assumed to have the same prognosis as a patient whose true risk is lowest in the same risk category.

Hypothetical relationship between biomarker M and clinical outcome. (A) Green line depicts linear relationship between biomarker and outcome. Risk of outcome increases linearly with increasing biomarker values. Dashed lines illustrate effect of dichotomizing biomarker, assuming that discontinuity in risk occurs at cut point c (patients whose biomarker values are below cut point are conferred the same risk, which is lower by magnitude Δ than that conferred to patients whose biomarker values exceed cut point). c* represents the biomarker value of a patient whose true risk is highest in the high-risk subgroup. (B) Quadratic relationship between biomarker and outcome. Risk of outcome decreases with biomarker up to point m and increases linearly after m.
Consider the following example. In early-stage triple-negative breast cancer, an elevated neutrophil-to-lymphocyte ratio (NLR, a peripheral indicator of systematic inflammation) has been shown to be associated with poor outcomes in small retrospective patient cohorts (1–3). In a recent report, 605 patients were identified who underwent breast surgery for stage I–III breast cancer between 1985 and 2012 at the Mayo Clinic and met the criteria for the triple-negative breast cancer phenotype (4). Clinicopathologic factors and biomarkers (including NLR) were collected to assess their impact on clinical outcomes. In that study, the median NLR was 2.52. A common strategy for handling continuous biomarkers such as NLR is to dichotomize the biomarker at its sample median since this guarantees an equal sample size between the low- and high-risk groups. Figure 2A displays the relationship between NLR and patient survival using a restricted spline (5). Clearly, there is a nonlinear relationship between NLR and risk of death. If we apply a quadratic transformation to NLR (by including a continuous NLR term and its squared term in the regression model), there is a highly significant statistical association between NLR and risk of death (likelihood ratio test, 37.91; P < 0.0001). However, this association dissipates if NLR is dichotomized at its sample median (Fig. 2B; hazard ratio [HR], 1.16 [95% CI, 0.89–1.52]; log-rank P = 0.27). This example illustrates that arbitrary dichotomization of a continuous biomarker can distort its true relationship with outcome, resulting in significant information loss. The HR estimate of 1.16 suggests that an NLR above the sample median—that is, a high NLR—confers a 16% increase in the hazard of death, compared with a low NLR. In contrast, if NLR is modeled as a continuous variable in the Cox regression model, the resultant HR is 1.23, suggesting that a 1-unit increase in NLR is associated with a 23% increase in the hazard of death. When interpreting the prognostic effect of a continuous biomarker, it is important to pay attention to its range (in the Mayo triple-negative breast cancer dataset, NLR ranged from 0.14 to 10.50) since the magnitude of a 1-unit increase is relevant to the underlying scale of the biomarker.

Dichotomy of continuous biomarker (NLR example). (A) Nonlinear relationship between NLR and patient survival in Mayo Clinic triple-negative breast cancer dataset using restricted spline method. (B) Effect of dichotomizing NLR at its sample median. Association between NLR and survival is no longer significant (log-rank P = 0.27). AIC = Akaike’s information criterion; L.R. = likelihood ratio.
Because of the haphazard discretization of continuous biomarkers, the literature is plagued with biomarker cut points that are rarely reproducible, making comparison of biomarker effects across different studies impossible. For example, S-phase fraction, the percentage of tumor cells in the S phase obtained by cell cycle analysis, was of considerable scientific interest as a potential prognostic biomarker in breast cancer, but a review by Altman et al. found that a wide range of S-phase fraction cut points—from 2.6 to 15.0—has been reported as optimal in the literature, rendering the effect of S-phase fraction inconsistent among studies (6). Another example is the nuclear proliferation biomarker Ki-67. Ki-67 is of interest for various applications in research and clinical management of breast cancer. For instance, clinical decision making regarding treatment options for breast cancer often relies on the application of a Ki-67 cut point to classify patients into high-risk or low-risk groups. However, in a metaanalysis of 85 studies that included 32,825 patients with early breast cancer, Stuart-Harris et al. reported that Ki-67 cut points ranging from 0% to 28.6% have been investigated (7). This lack of consensus regarding the optimal cut point for Ki-67 in various settings has hindered its ability to facilitate clinical decision making or direct comparisons of Ki-67 results across laboratories and clinical trials (8).
In general, when the goal is to explore whether a biomarker is singly prognostic, it would be preferable not to categorize the biomarker at all. A preferred approach to characterizing the relationship between a continuous biomarker and time-to-event outcome is by modeling the biomarker as a continuous variable in a univariate Cox regression model without introducing any cut point. This method has the considerable advantage of retaining valuable information in the data and will improve the ability to directly compare results from different studies. When linearity assumption (i.e., the risk increases or decreases linearly as the biomarker increases) is called into question, modern statistical techniques such as regression splines or fractional polynomial models can be used to effectively model nonlinear relationships between values of the biomarker and risk (5,9). The relationship between biomarker values and risk is represented by the fitted regression function and its associated confidence bands. Cut points for the biomarker, if desired, can then be based on the nature of the relationship.
Cut Point Search via the Minimal-P-Value Approach
Another common approach for identifying biomarker cut point is to examine a range of biomarker values and select the cut point that yields the smallest P value. Altman et al. referred to this method as the minimal-P-value approach (6). Several authors have demonstrated that this naïve approach is associated with a considerable inflation of the type I error due to the well-known problem of multiple testing (6,9,10). Using the NLR example above, Figure 3A displays the log-rank P values (testing the association between dichotomized NLR and recurrence-free survival) based on a range of NLR cut points. We excluded the top and bottom 20% of NLR and used 200 cut points. The NLR cut point associated with the smallest P value is 3.95. It can be seen that the P values are highly unstable (range, 0–0.53) and that a minor change in the NRL cut point can lead to drastically different P values. As such, if P value were to be reported, some statistical adjustment for multiplicity is necessary. Altman described a formula to compute a corrected P value (6). When we apply this adjustment to the NLR example, the resulting P value is 4.7 × 10−5, substantially larger than the uncorrected P value, 0.14 × 10−6.

Minimal-P-value approach (NLR example). (A) Highly unstable P values of log-rank test as function of cut point used for NLR in Mayo Clinic triple-negative breast cancer dataset. Top and bottom 20% of NLR values were excluded, and 200 cut points were used. (B) Strong inverse correlation between estimated HRs and log-rank P values for NLR in Mayo Clinic triple-negative breast cancer dataset. Smallest P value corresponds to most extreme HR estimate.
We conducted simulation studies to investigate the severity of type I error inflation and how the type I error rate changes as a function of the number of cut points and sample size. Specifically, we simulated a continuous biomarker that follows a uniform distribution between 0 and 1 (the biomarker takes any value between 0 and 1 with equal probabilities) and a survival outcome that follows an exponential distribution with rate 0.0289 (translating to median survival of 24 mo) with no censoring. This data-generating mechanism ensures that the continuous biomarker and the survival outcome have no association. In each simulated dataset, we excluded 10% of the smallest and largest biomarker values as potential cut points, applied a fixed number of biomarker cut points, computed the 2-sided P value from the log-rank test associated with each cut point, and identified the cutoff that yields the minimal P value. We considered a variety of scenarios, varying the sample size (100, 300, 500) and number of biomarker cut points (50, 150, 300). For each sample size, 5,000 datasets were simulated as described above, and the type I error (the percentage of simulations for which the minimal P value is less than a nominal level of 5%) was recorded. The results of these simulations are shown in Figure 4. For a fixed number of cut points, the type I error hardly changed with the sample size. However, for a fixed sample size, the type I error increased with an increasing number of biomarker cut points. For example, for a sample size of 300, the type I error increased from 37.3% with 50 cut points to 43.3% with 300 cut points. Notably, in all scenarios considered, the type I errors exceeded 37%. These simulations confirmed that when a series of significance tests is performed on the same dataset, each with a prespecified nominal type I error rate of, for example 5%, the minimal-P-value approach leads to a global false-discovery rate that may be much higher than 5%. In particular, this approach may yield a “statistically significant” result (P < 0.05) with a probability greater than 37% for a biomarker that has no association with outcome at all when the number of attempted cut points exceeds 50.
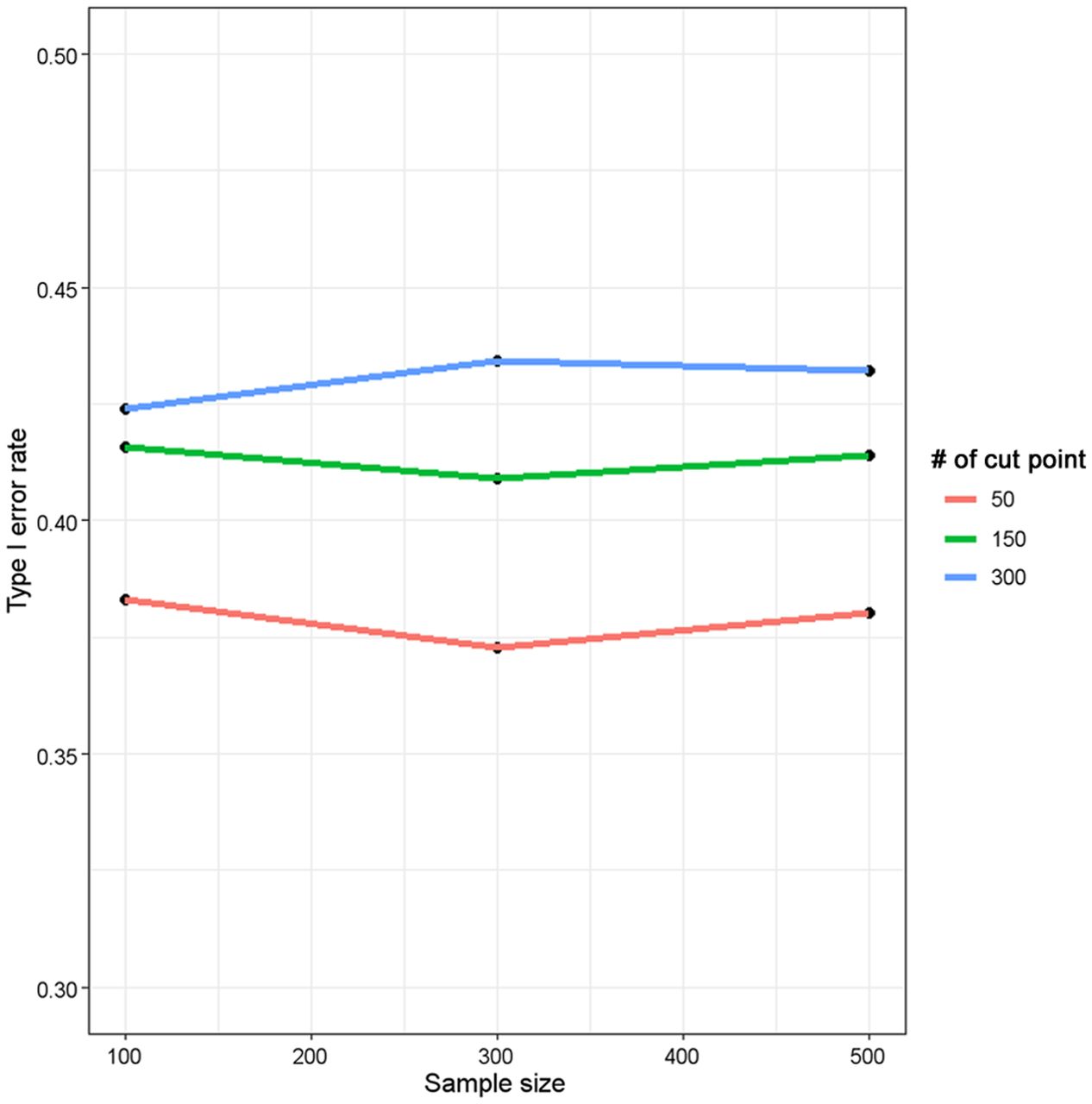
Type I error inflation as function of number of cut points and sample size using minimal-P-value approach. In each simulation, 10% of smallest and largest biomarker values were not considered as potential cut points. Two-sided P value from log-rank test was computed for each cut point applied. Each plotted point represents percentage of 5,000 simulations for which minimal P value is less than nominal 5% level based on assumption that there is no association between biomarker and time-to-event outcome (i.e., type I error). No censoring in outcome was assumed.
Another problem with the minimal-P-value approach concerns estimation of the biomarker effect. Specifically, this approach gives an exaggerated sense of association between the biomarker and the outcome because when there is an association between the continuous biomarker and outcome, the P values derived from the significance tests (e.g., log-rank) are associated with the effect estimates (e.g., HR). As such, the smallest P value would correspond to the most extreme HR estimate (e.g., positive association for HR < 1; negative association for HR > 1). Figure 3B illustrates the association between HR estimates and P values using the NLR example. The minimal P value corresponds to an HR estimate of 0.45 (i.e., NLRs above the cut point of 3.95 confer a 55% reduction in the hazard of death compared with NLRs below 3.95); this effect is overestimated. Several authors have proposed strategies to correct for overestimation of the effect of a biomarker using the same dataset (11,12). The best and clearly unbiased approach to estimating the biomarker effect is to apply the cut point identified from the current study to other independent datasets. This approach guarantees that no optimistic bias is introduced to the effect estimation by the data-derived cut point.
Comparison of Clinical Outcomes Using Data-Driven Cut Point
Other methods exist for identifying cut points of continuous biomarkers. In the radiology literature, for example, a common measure of discrimination for binary outcomes (e.g., alive vs. dead, cancer vs. noncancer) is the receiver-operating-characteristic (ROC) curve. Discrimination quantifies how well a biomarker differentiates subjects at higher risk of having an event from those at lower risk. More specifically, a biomarker with good discrimination would predict a higher probability of having an event among subjects who will develop the event. The ROC curve consists of plotting the pairs of sensitivity and (1 − specificity) (13), with a natural tradeoff between these 2 quantities. The area under the ROC curve (AUC) is a measure of discrimination, with values close to 0.5 indicating discrimination no better than chance alone (i.e., having equal probability of classifying to an event category those subjects with events vs. those without). AUCs close to 0 or 1 indicate that the biomarker almost always correctly predict the subject’s event status. Many methods are available for identifying a biomarker cut point that optimizes its discriminant performance. The index proposed by Youden (14), defined as ([sensitivity + specificity] – 1), is an example. This index, ranging from 0 to 1, gives equal weight to false-positive and false-negative values. Graphically, the Youden index represents the height above the 45° chance line (representing an AUC of 0.5). The biomarker value associated with the largest Youden index may be chosen as the optimal cut point. Other methods exist for identifying cut points from the ROC (15).
In some disease settings, a multitude of biomarkers or clinicopathologic variables may be of prognostic potential. It is sometimes useful to combine these prognostic factors via statistical modeling strategy (e.g., logistic regression model for binary endpoints, Cox proportional-hazards model for time-to-event endpoints) to form a risk system (also sometimes referred to as a prognostic signature). For individual patients, the composite risk score (or prognostic index) can be computed by adding up the weighted factors (with the weights being the estimated regression coefficients). The prognostic indices then represent a new variable combining the information from all prognostic factors that can be used for prognostication. For example, Haybittle et al. developed a prognostic index, the Nottingham Prognostic Index, from a Cox proportional-hazards model for patients with primary operable breast cancer. The prognostic index for each patient was expressed as a linear function, [0.17 × (tumor size in cm)] + [0.76 × (lymph node stage)] + [0.81 × (tumor grade)], where tumor grade = 1 or 2 or 3 and lymph-node stage = 1 or 2 or 3 (the lymph node stages are defined by Haybittle et al. in their publication (16)). The larger the value of the Nottingham Prognostic Index, the worse the patient prognosis. Three risk groups were then defined on the basis of the range of the Nottingham Prognostic Index. The choice of a cut point for the continuous prognostic index can be based on the ROC methodology as described above for a single continuous biomarker.
In practice, it is not uncommon for investigators to use the selected cut point of the model score to categorize patients and then compare the nonparametric survival curves of the 2 risk groups via the log-rank test using the same dataset. This approach tends to exaggerate the P value and overestimates the effect of the model. Optimizing a biomarker or risk model based on outcome and then claiming good discriminatory value based on the survival curves on that same dataset is a prevalent problem in the medical literature. Subramanian and Simon used the term resubstitution statistics to refer to a risk-model performance evaluation (e.g., discrimination) that is done using the same data utilized for some form of optimization (e.g., cut point selection or model development) (17). The separation between Kaplan–Meier curves for low- and high-risk patients as defined by the cut point derived from the same dataset is an example of resubstitution statistics. Subramanian and Simon maintain the importance of separating the data used for any aspect of optimization from the data used for performance assessment. Some complex statistical approaches (such as bootstrap, jackknife, and permutation tests) may be useful in providing a more unbiased assessment of the true utility of the dichotomized biomarker. These methods belong to a class of resampling methods (18). One simple form of resampling method is the sample split. With sample split, one portion of the dataset is used for cut point optimization or model development and the remaining (independent) data are used to evaluate the discriminatory power of the biomarker or model developed with the first portion (19). However, resampling methods represent interval validation and do not reflect many sources of variabilities present in broader practice settings. Therefore, large independent studies will still be required to confirm the results.
Consider the studies by Lin et al. (20) and Casasnovas et al. (21), both aiming to assess the prognostic value of early 18F-FDG PET using SUVs in patients with diffuse large B-cell lymphoma. A clinical endpoint of interest was event-free survival, defined by Lin et al. as months from study enrollment until first evidence of progression, relapse, or death due to any cause. To apply standard ROC methodology, the investigators first replaced the continuous event-free survival variable with a binary one (i.e., event vs. no event). The approach of using a binary outcome status (e.g., vital status = dead or alive) in place of a continuous outcome variable such as event-free survival has the drawback of information loss, because it ignores the varying length of follow-up among patients. For example, a patient who has survived for 5 y would have the same binary outcome status as another patient who has survived for 1 y (i.e., for both patients, the vital status would be “alive”). Statistical methods exist that extend the standard ROC methodology to accommodate time-to-event outcomes such as event-free survival (22). The investigators then applied the ROC methodology to identify an optimal cut point for SUV (65.7% for Lin et al. and 66% for Casasnovas et al.). Study subjects were then categorized into 2 risk groups based on the selected cut point, and the significant P values from log-rank testing (P = 0.028 for Lin et al. and P < 0.0001 for Casasnovas et al.) and notable separation in the Kaplan–Meier survival curves (Fig. 2B) in both studies were cited as strong evidence supporting the prognostic value of SUV. Again, because the cut point was preselected to distinguish outcome by some measure, the resultant estimated biomarker effect and P value obtained from the same dataset are optimistically biased and should not be interpreted as a confirmation of the prognostic utility of SUV.
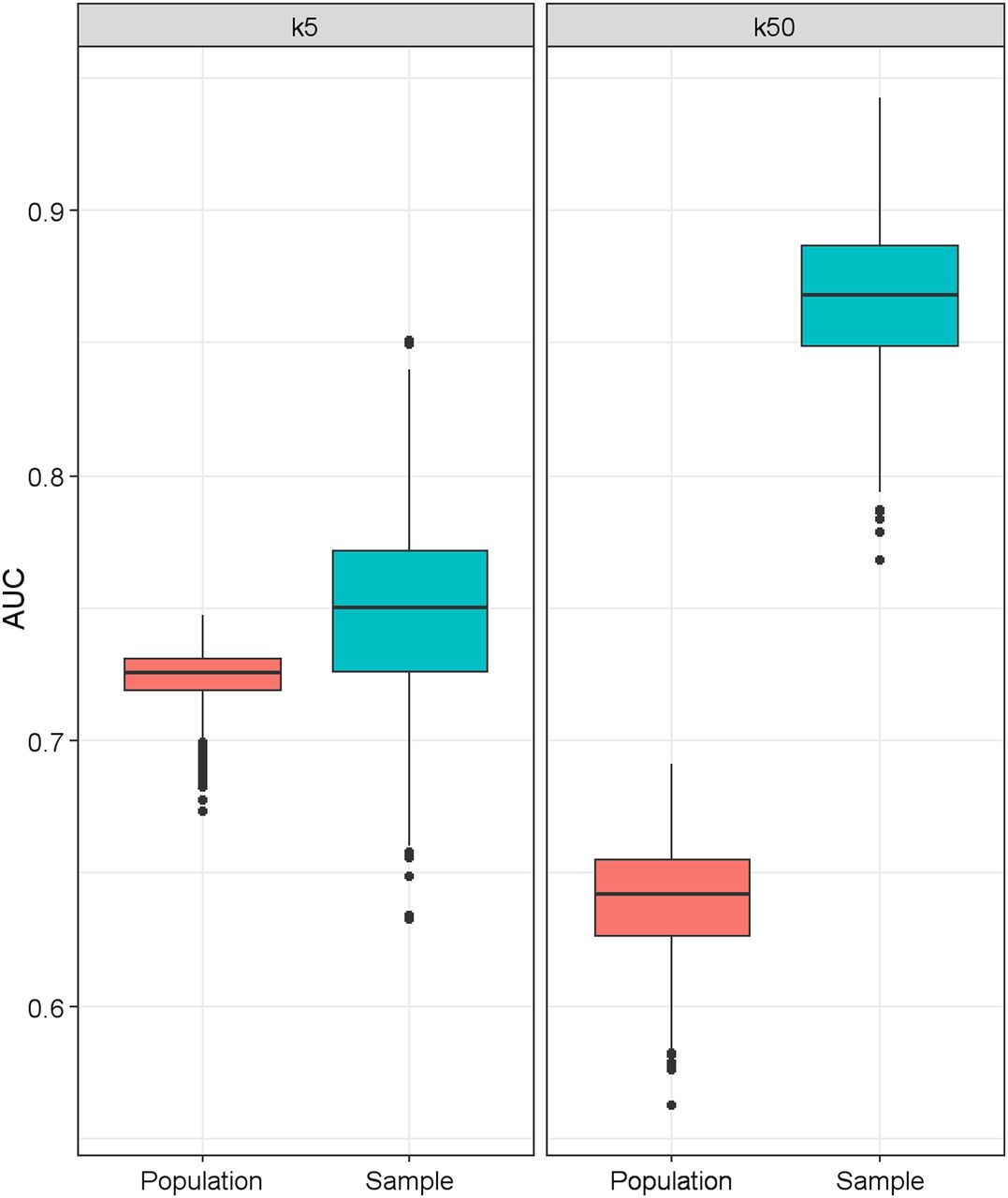
The magnitude of resubstitution bias is further exacerbated as the number of covariates in the risk model increases. This problem is known as overfitting, in that a complex statistical model containing a sufficiently large number of variables having no true association with clinical outcome can spuriously provide an excellent fit to a small dataset. We performed a simulation to illustrate this bias in estimated discrimination. We simulated a sample dataset with 200 patients and a population dataset with 10,000 patients. The latter represents the target population at large, and hence, the performance of the model evaluated in the population is regarded as the true value. In each simulated dataset, we randomly generated a set of k continuous variables, denoted as X = (X 1, X 2,…, X k), each following a standard normal distribution (with mean 0 and SD 1). We assumed that two of the k variables are associated with the binary endpoint Y. Specifically, the correlation between (X 1, X 2,…, X k) and Y is induced by a multivariable logistic regression with intercept 0 and regression coefficients B = (β1 = 1.2, β2 = 1.2, β3 = 0,…, βk = 0). Correspondingly, the association between (X 1, X 2) and Y is characterized by an odds ratio of exp(1.2) = 3.32 whereas the remaining (k-2) variables, (X 3, X 4, ..., X k), have no association with the outcome (i.e., odds ratio = 1). We considered 2 scenarios: k = 5 (small number of biomarkers) and k = 50 (large number of biomarkers). For each k, we generated 1,000 datasets as described above and compared the distributions of AUC between the sample datasets and the population datasets. To arrive at the AUC estimate in a sample dataset, we fit a multivariable regression model of X on Y and obtained k regression coefficient estimates. The prognostic scores for individual patients were calculated as the linear combination of the variables weighted by the regression coefficients. The AUC was then estimated from the ROC for the new continuous score variable. The regression model was constructed using only the sample dataset; the resultant regression coefficients were then fixed and applied to the population dataset to obtain individual prognostic scores and the true AUC (i.e., with no further model building or refinement).
Figure 5 displays side-by-side box plots of the distributions of AUCs from the simulated sample datasets and the population datasets for k = 5 and k = 50. When k = 5, AUCs were slightly biased upward in the sample distribution compared with the true population (median, 0.75 and 0.73 for samples and populations, respectively). The degree of optimistic bias increased drastically when the number of variables increased to 50 (median, 0.87 and 0.64 for samples and populations, respectively). This simulation exercise underscores the fact that the performance of a risk model is overestimated when the evaluation is performed using the same dataset as used to construct the model, and the degree of the optimistic bias increases with the number of variables in the model. These results highlight the importance of evaluating the performance of a risk model using a dataset that is independent from that used for model development.

Effect of number of covariates (k) in risk model on resubstitution bias in AUC. Population box plot represents true AUC distribution in interested population at large. Sample box plot represents distribution of AUC derived from sample dataset used to construct risk model. Each box plot was based on 1,000 simulations. When k = 5, there is slight upward (optimistic) bias in sample AUC distribution compared with true population. Degree of optimistic bias increases drastically when k increases to 50.
BIOMARKER CUT POINT IN THE PRESENCE OF ESTABLISHED PROGNOSTIC FACTORS
For many cancers, certain prognostic factors are known and well established. For example, tumor size and the number of positive lymph nodes are well-known prognostic factors in breast cancer. For patients with advanced non-Hodgkin lymphoma, the International Prognostic Index was a risk system developed to predict patient survival (23). The components of the International Prognostic Index were based on clinical features—including age, tumor stage, serum lactate dehydrogenase concentration, performance status, and number of extranodal disease sites—that are easy to measure and prognostically important. In these settings, it is more pertinent to determine whether a new biomarker adds prognostic information to that already provided by standard prognostic factors alone. Statistical models such as the Cox proportional-hazards regression model are often used to study the joint prognostic influence of multiple factors. To assess the independent prognostic influence of the new biomarker above and beyond recognized factors, 1 reduced multivariable model can be fitted containing only the standard factors and 1 full multivariable model can be simultaneously fitted, with both models containing the new biomarker and standard factors. The difference in how well the 2 nested models fit the data provides a measure of the statistical significance of whether the new factor contains additional prognostic information (e.g., via the likelihood ratio test) (24). If there are multiple new factors, this approach accounts for the number of new variables in the calculation of statistical significance. For example, Cheang et al. studied the additional prognostic information of a 5-biomarker panel (estrogen receptor, progesterone receptor, human epidermal growth factor receptor 2, epidermal growth factor receptor, and cytokeratin 5/6) above and beyond a 3-biomarker panel (estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2) in the presence of standard clinical variables for predicting breast cancer death-specific survival (25). To test the statistical significance of the 2 additional biomarkers, 2 Cox regression models were fitted, and a likelihood ratio test of the difference between the 2 models was used to evaluate the additional prognostic contribution of epidermal growth factor receptor and cytokeratin 5/6.
When the cut point of a biomarker is preselected on the basis of clinical outcome (e.g., via the minimal-P-value approach or the ROC methodology), the corresponding dichotomized biomarker will impart an inflated effect on the multivariable regression model, thus diminishing the relative importance of other known prognostic factors. Kaplan–Meier curves showing the difference in survival between risk groups correspond to univariate statistical tests (e.g., log-rank) and thus do not indicate the effect of the biomarker after accounting for the other variables that may influence survival. In fact, in the presence of existing prognostic factors, determination of a cut point for the new biomarker alone is not as relevant. Instead, a more holistic approach would be to develop a prognostic model incorporating both known prognostic factors and the new biomarker. Prognostic categories can then be based on the model-predicted prognostic indices of individual patients. For example, Paik et al. developed the Oncotype Dx assay, a 21-gene recurrence score, to quantify the likelihood of distant recurrence in women with node-negative, estrogen-receptor–positive breast cancer who have been treated with tamoxifen (26). The cut points were determined on the basis of the results of National Surgical Adjuvant Breast and Bowel Project trial B-20 and were validated using data from trial B-14. The cut points classified patients into 3 risk categories based on predicted 10-y distant recurrence rate: low risk (recurrence score < 18), intermediate risk (18 ≤ recurrence score < 31), and high risk (≥31). The authors also demonstrated that the model based on age, tumor size, and recurrence score provided significantly independent prognostic information compared with the model including age and tumor size alone (P < 0.001 by the likelihood ratio test).
CONCLUSION
Discovery of biomarkers has been steadily increasing over the past decade. Although a plethora of biomarkers and associated cut points has been reported in the biomedical literature, few have been sufficiently validated for broader clinical applications. In contrast to the abundance of classic clinical-trial principles for guiding the design, conduct, analysis, and reporting of studies, relatively fewer guidelines exist for biomarker research (27,28). In this article, we have attempted to identify some common methodologic issues related to biomarker cut point identification and evaluation. We strongly advocate that discretization of continuous biomarkers be avoided. If cut point identification is performed, it should be handled with statistical care. Biased resubstitution should either not be reported or be clearly noted as an unreliable representation of the true discriminant value of the biomarker. When feasible, large independent datasets are ideal for confirmation of the prognostic value of the biomarker and its cut point. A schema for the consideration of biomarker analysis and cut point evaluation is proposed in Figure 6. We hope that the discussions here will draw attention to critical statistical issues associated with development and evaluation of biomarker cut points and will, in turn, help improve methodologic rigor in this line of research.
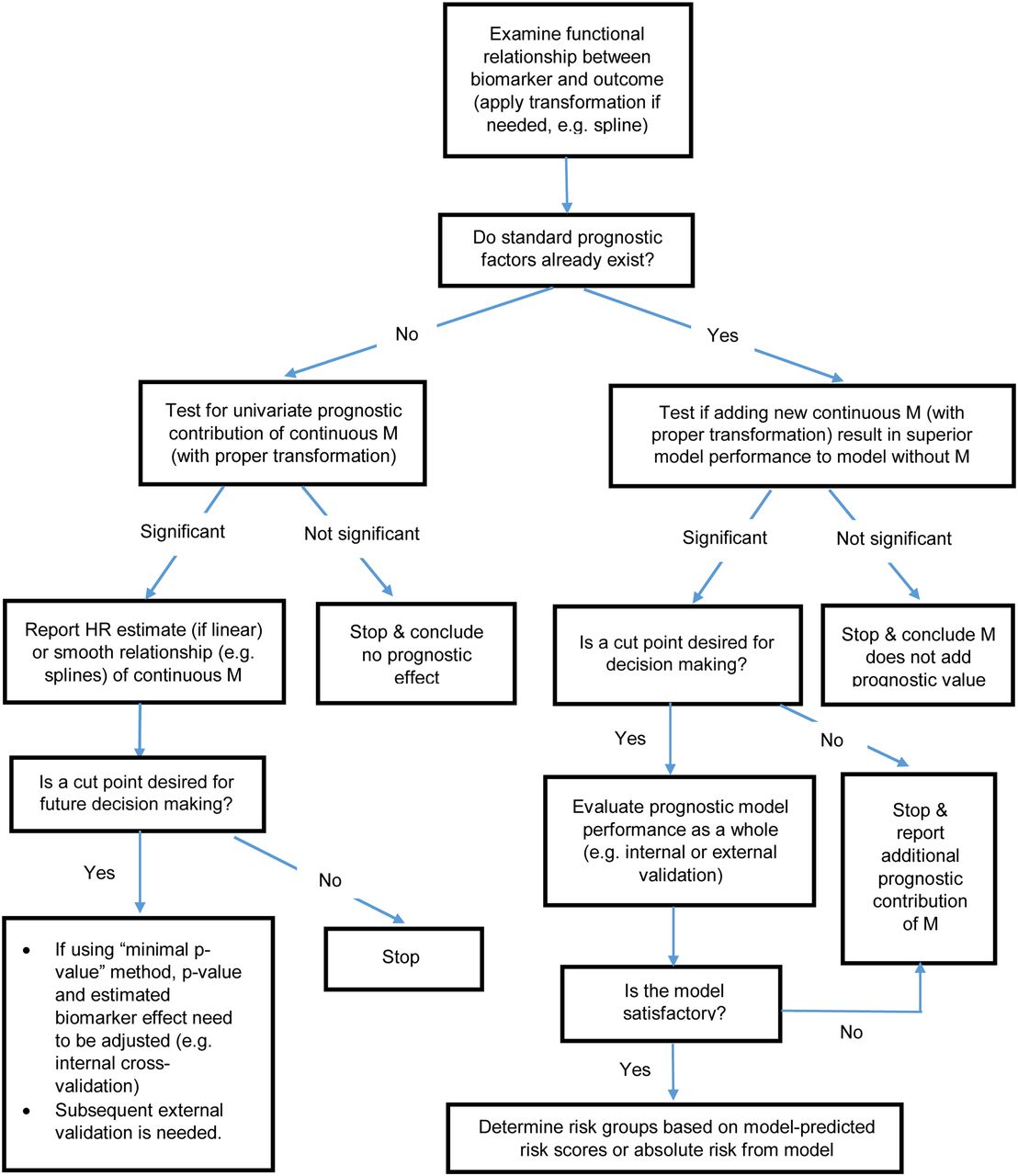
Schema for biomarker cut point analysis and evaluation.
Footnotes
Published online Feb. 12, 2021
Learning Objectives: On successful completion of this activity, participants should be able to (1) identify common statistical pitfalls in the identification of biomarker cut points; (2) describe statistical principles in the evaluation of biomarker cut points; and (3) recommend logical steps in the appraisal of prognostic biomarkers and associated cut points.
Financial Disclosure: This work was supported by the National Cancer Institute of the National Institutes of Health under awards U10 CA180822 (Statistics and Data Management Center, NRG Oncology) and P50 CA116201 (Mayo Clinic Breast Cancer Specialized Program of Research Excellence). The authors of this article have indicated no other relevant relationships that could be perceived as a real or apparent conflict of interest.
CME Credit: SNMMI is accredited by the Accreditation Council for Continuing Medical Education (ACCME) to sponsor continuing education for physicians. SNMMI designates each JNM continuing education article for a maximum of 2.0 AMA PRA Category 1 Credits. Physicians should claim only credit commensurate with the extent of their participation in the activity. For CE credit, SAM, and other credit types, participants can access this activity through the SNMMI website (http://www.snmmilearningcenter.org) through May 2024.
- © 2021 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication October 5, 2020.
- Accepted for publication January 19, 2021.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}