


Abstract
Although abnormally folded tau protein has been found to self-propagate from neuron to connected neuron, similar propagation through human brain networks has not been fully documented. We studied tau propagation in the left hemispheric syntactic network, which comprises an anterior frontal node and a posterior temporal node connected by the white matter of the left arcuate fasciculus. This network is affected in the nonfluent variant of primary progressive aphasia, a neurodegenerative disorder with tau accumulation. Methods: Eight patients with the nonfluent variant of primary progressive aphasia (age, 67.0 ± 7.4 y; 4 women) and 8 healthy controls (age, 69.6 ± 7.0 y; 4 women) were scanned with 18F-AV-1451 tau PET to determine tau deposition in the brain and with MRI to determine the fractional anisotropy of the arcuate fasciculus. Normal syntactic network characteristics were confirmed with structural MRI diffusion imaging in our healthy controls and with blood oxygenation level–dependent functional imaging in 35 healthy participants from the Alzheimer Disease Neuroimaging Initiative database. Results: Language scores in patients indicated dysfunction of the anterior node. 18F-AV-1451 deposition was greatest in the 2 nodes of the syntactic network. The left arcuate fasciculus had decreased fractional anisotropy, particularly near the anterior node. Normal MRI structural connectivity from an area similar to the one containing tau in the anterior frontal node projected to an area similar to the one containing tau in the patients in the posterior temporal node. Conclusion: Tau accumulation likely started in the more affected anterior node and, at the disease stage at which we studied these patients, appeared as well in the brain region (in the temporal lobe) spatially separate from but most connected with it. The arcuate fasciculus, connecting both of them, was most severely affected anteriorly, as would correspond to a loss of axons from the anterior node. These findings are suggestive of tau propagation from node to connected node in a natural human brain network and support the idea that neurons that wire together die together.
In Alzheimer disease (AD) and frontotemporal dementia (FTD), damage to the brain starts from a single cortical region and, over months or years, spreads to areas functionally and structurally connected to the one involved initially (1). Several hypotheses have been put forth to explain the network pattern of spreading. Neurotrophic factors normally secreted by neurons in one node may fail, inducing neuron loss in the connected node (2). Alternatively, dying neurons in the epicenter node may release substances that damage the neurons in the downstream node. Among these substances, prime candidates are the misfolded proteins typically found in these neurodegenerative disorders (3). Misfolded tau, characteristic of AD and several variants of FTD, acquires a β-sheet–rich structure that polymerizes into amyloid fibrils. These fibrils may behave as prions (3,4). Like other prions, tau prions would multiply by self-propagation, in which the β-sheet is a template for nascent prion formation (3,4). Tau prions would build up in impaired epicenter node neurons and propagate from sick neuron to healthy neuron connected through natural brain networks, thereby spreading damage across the brain. Evidence supporting this postulate comes from tissue culture experiments (5,6); from mouse models of tau spreading (7–11); and, with the shortcoming of partial brain sampling (12), from the pattern of tau deposition in postmortem brain (13,14). PET tracers for aggregated tau are now available to quantify in vivo tau in humans (15). Using these tracers, tau has been found to be associated with network brain characteristics in AD (16–18). However, the complexity of the default network affected in AD (19), and the confounding overlap in AD of tau with another misfolded protein, amyloid-β, which also alters functional connectivity (19,20), have hindered the conclusive demonstration of network tau propagation in humans.
We addressed this issue by studying tau spreading in a simpler model, the syntactic language network (Fig. 1) (21). This network is affected in the nonfluent variant of primary progressive aphasia (nfvPPA), an unusual variant of FTD associated with tau pathology in more than 80% of cases (22). We hypothesized that abnormal tau would preferentially localize in the 2 nodes of the network, an anterior node (Broca area) devoted to language production and a posterior node in the middle temporal gyrus devoted to syntactic processing during comprehension (Fig. 1; Supplemental Fig. 1 [supplemental materials are available at http://jnm.snmjournals.org]). These nodes are widely separated in the cortex but are connected by the arcuate fasciculus white matter pathway (21). As the disease progresses, neurons containing tau die but tau remains in the tissue (23). Thus, if the tau prion propagation hypothesis is true, tau would accumulate in the anterior node, which is affected first, and in the posterior node, infected by tau prions from neurons in the anterior node (Supplemental Fig. 1). Additionally, neuronal death would cause axonal loss in the arcuate fasciculus (Supplemental Fig. 1), which would be abnormal, with decreased fractional anisotropy (FA), a marker of axonal integrity, in the affected left hemisphere.
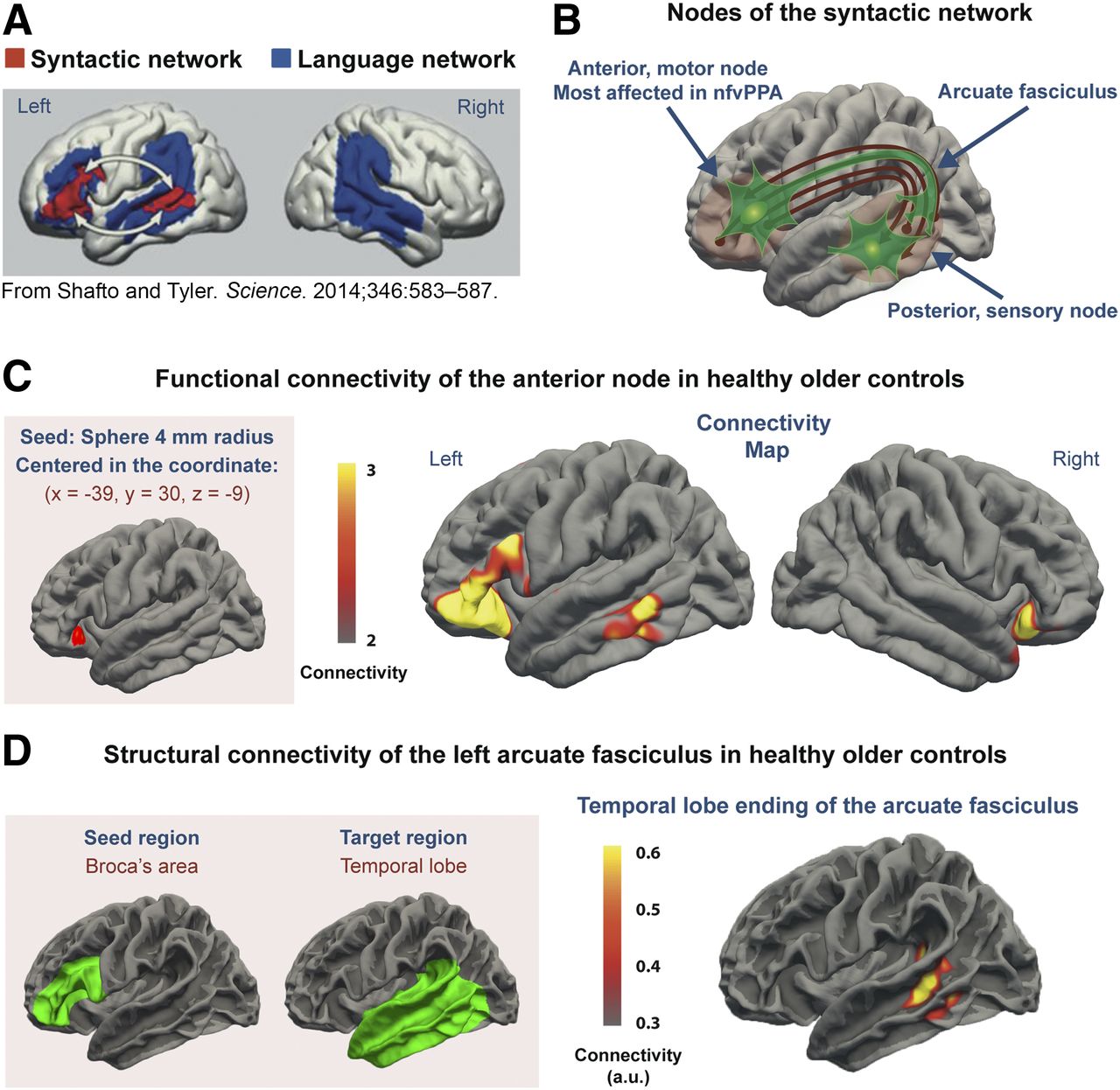
Map of syntactic network. (A) Syntactic network is located in left hemisphere and is a component of larger language network. (Reprinted with permission of (21).) (B) Anterior node is involved in syntax production and posterior node in syntax decoding. The 2 nodes are connected by arcuate fasciculus, a white matter tract. (C) Functional MRI connectivity from seed in anterior node showed anterior node to be most strongly connected to region in posterior portion of left middle temporal gyrus, well known to correspond to posterior node of syntactic network. Color bar represents normalized z scores. Only positive z scores are plotted, ranging from ≥2 (P ≤ 0.02) to ≤3 (P ≤ 0.001). (D) DTI structural connectivity. Seed in Broca area and group-averaged image of ending of arcuate fasciculus in lateral temporal lobe are displayed on FreeSurfer average gray–white matter boundary surface. For each subject, significance cluster was defined by 90th percentile of arcuate fasciculus density at gray–white matter boundary. Color bar indicates in arbitrary units (a.u.) for how many patients a given voxel was significant.
MATERIALS AND METHODS
Subjects
Participants were recruited from 2015 to 2017 at the Houston Methodist Nantz National Alzheimer Center and consisted of 8 patients meeting the diagnostic criteria (24) for nfvPPA and 8 healthy controls matched to the patient group by age and sex (Table 1). Patients were diagnosed by board-certified neurologists and underwent a thorough clinical history and a neurologic, cognitive, and neuropsychiatric evaluation, together with a detailed speech and language evaluation (25). To facilitate network specificity and avoid the amyloid-β confounder, we excluded patients with AD-related language deficits by means of the clinical phenotype and a negative amyloid-β PET scan with either 11C-Pittsburgh compound B or 18F-florbetapir (24,26). Healthy volunteers had clinical and neuropsychologic screening, normal MRI results, and absence of neurologic, psychiatric, or other major medical illnesses. Procedures were approved by the Methodist Hospital Research Institute Committee on Human Research, and written informed consent was obtained from all participants.
Demographics and Neuropsychologic Test Performance
To determine the normal functional connectivity of the syntactic network (Fig. 1C) in a sample similar to our nfvPPA patients, resting state functional MRI data from 35 healthy controls (average age, 74.1 ± 5.1 y; 20 women) were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database. ADNI was launched in 2003 as a public–private partnership led by Principal Investigator Michael W. Weiner (27). The primary goal of ADNI has been to test whether serial MRI, PET, other biologic markers, and clinical and neuropsychologic assessment can be combined to measure the progression of mild cognitive impairment and early AD. MRI data from ADNI were preprocessed using a standard method (28). With MATLAB software (version 2018a; MathWorks), a 4-mm-radius spheric seed was drawn in the left inferior frontal gyrus, centered on the voxel (coordinates: x, −39; y, 30; z, −9) where peak values for syntactic-task activation and syntactic disruption by lesions has been described in the literature (Fig. 1C) (21). The average blood oxygenation level–dependent time-series signal within the seed was extracted for each subject. Functional connectivity with the rest of the brain was calculated as previously described (28).
Neuropsychologic, Speech, and Language Evaluation
Our patients and controls were studied with the Mini-Mental State Examination (29), DemTect (30), Clock Drawing Test (31), and the naming component of the Addenbrooke Cognitive Examination (Table 1) (32). Language was further evaluated with the Progressive Aphasia Severity Scale (25), which rates the presence and severity of impairment across 10 domains (Fig. 2A; Supplemental Table 1). Impairment in each domain was assessed by integrating information from patient test performance in the office and a structured questionnaire that was completed by the primary caregiver.
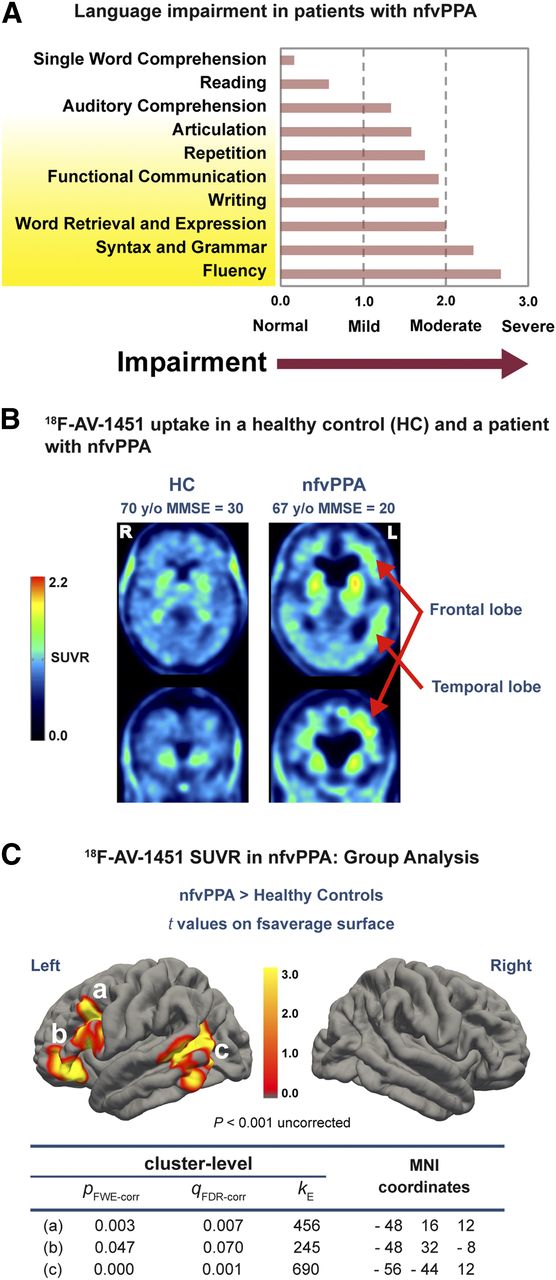
Language and brain tau in nfvPPA. (A) Mean scores of nfvPPA patients on Progressive Aphasia Severity Scale (25); fluency and syntax, mediated by anterior node of syntactic network, were more impaired than syntactic auditory comprehension, mediated by posterior node. (B) Uptake of tau tracer 18F-AV-1451 was higher in this nfvPPA patient than in healthy control and higher in left hemisphere than right. (C) In group analysis, nfvPPA patients had higher 18F-AV-1451 SUV ratios in both anterior (A and B) and posterior (C) nodes of syntactic network but nowhere else in cerebral hemispheres. Location of tau PET signal matches location of nodes of syntactic network, as determined by MRI connectivity (Figs. 1C and 1D; Supplemental Fig. 2). FDR-corr = false discovery rate correction; fsaverage = FreeSurfer average; FWE-corr = familywise error correction; κ = number of voxels in cluster; MMSE = Mini-Mental State Examination; SUVR = SUV ratio.
Tau PET Acquisition and Processing
Eighty minutes after the injection of about 370 MBq of 18F-AV-1451, participants (Table 1) were positioned in a Philips Gemini TF 64-DS PET/CT scanner, with their head firmly secured using a thermoplastic face mask. After a CT scan for attenuation correction, PET emission data were acquired for 20 min in four 5-min frames. The 4 frames were motion-corrected, averaged, and smoothed via a gaussian filter of 6 mm in full width at half maximum (version 3.906; PMOD Technologies LLC). Then, each subject’s PET image was coregistered to the corresponding T1-weighted structural MR images with SPM12 software. After segmentation of the T1-weighted MR images with FreeSurfer software (version 6.0.0; Athinoula A. Martinos Center for Biomedical Imaging), the average value of the cerebellar gray matter was used as a normalization factor to produce SUV ratio parametric images. The SUV ratio map was then transferred to the standard Montreal Neurological Institute space using the transformation parameters of the MR image.
MRI Acquisition for PET Coregistration and Diffusion Tensor Imaging (DTI) of Arcuate Fasciculus
All subjects enrolled at our institution were imaged with the same 3.0-T Ingenia scanner (Philips). The protocol included a 3-dimensional T1-weighted turbo field echo sequence for PET anatomic coregistration and DTI echo planar imaging (EPI) processing. Because enrollment had been through different institutional review board–approved protocols, the parameters of the 3-dimensional T1-weighted structural MRI for the patients differed slightly from the parameters for the controls (patients: 1 × 1 × 1.2 mm voxels, 7.4-ms repetition time, 3.4-ms echo time, 8° flip angle; controls: 1 × 1 × 1 mm voxels, 8.2-ms repetition time, 3.7-ms echo time, 8° flip angle). However, structural images were used only for PET coregistration and DTI EPI processing and were not compared across groups. Diffusion imaging was acquired for all subjects using a DTI EPI sequence with a 7,633-ms repetition time, 82-ms echo time, 2.5 × 2.5 × 2.5 mm voxels isotopically distributed along 32 directions, 2 b values (0 and 800 s/mm2), and a 90° flip angle with no cardiac gating. One patient declined to undergo MRI and was excluded from the DTI analysis (a previously obtained 3-dimensional fast spoiled gradient echo T1-weighted structural image was used for PET preprocessing). Only 4 controls underwent both tau PET and DTI. Another 3 controls matched in sex and age to the patients and studied with the same DTI EPI sequence were used to complete the analyses.
DTI Processing
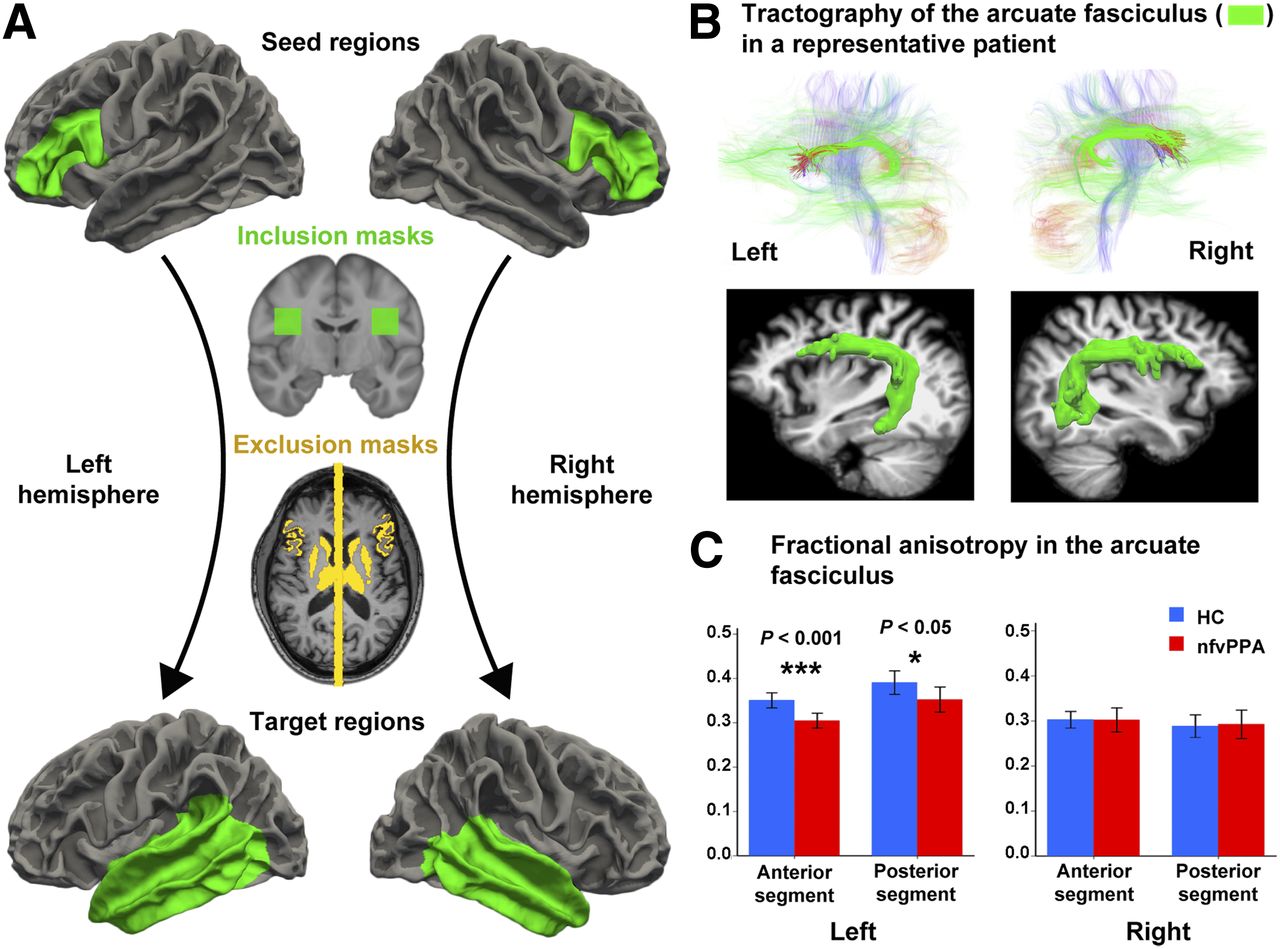
With some minor modifications, we used the method described by Takaya et al. (33) to isolate the arcuate fasciculus in each subject and compare FA across groups. To perform tractography of the arcuate fasciculus, the seed region in the Broca area (anterior node of the syntactic network) was defined by the white–gray matter boundary of the pars opercularis and the pars triangularis of the inferior frontal gyrus (Fig. 3A) (33). For a separate analysis, we used a seed region, largely coincident with the previous one, that colocalized with the area of significantly increased tau in the patients (Fig. 2C; Supplemental Fig. 2). The target region was defined liberally as the white–gray matter boundary of the entire temporal lobe, including the banks of the superior temporal sulcus (Fig. 3A). For the right hemisphere, we selected the homologous seed and target regions.
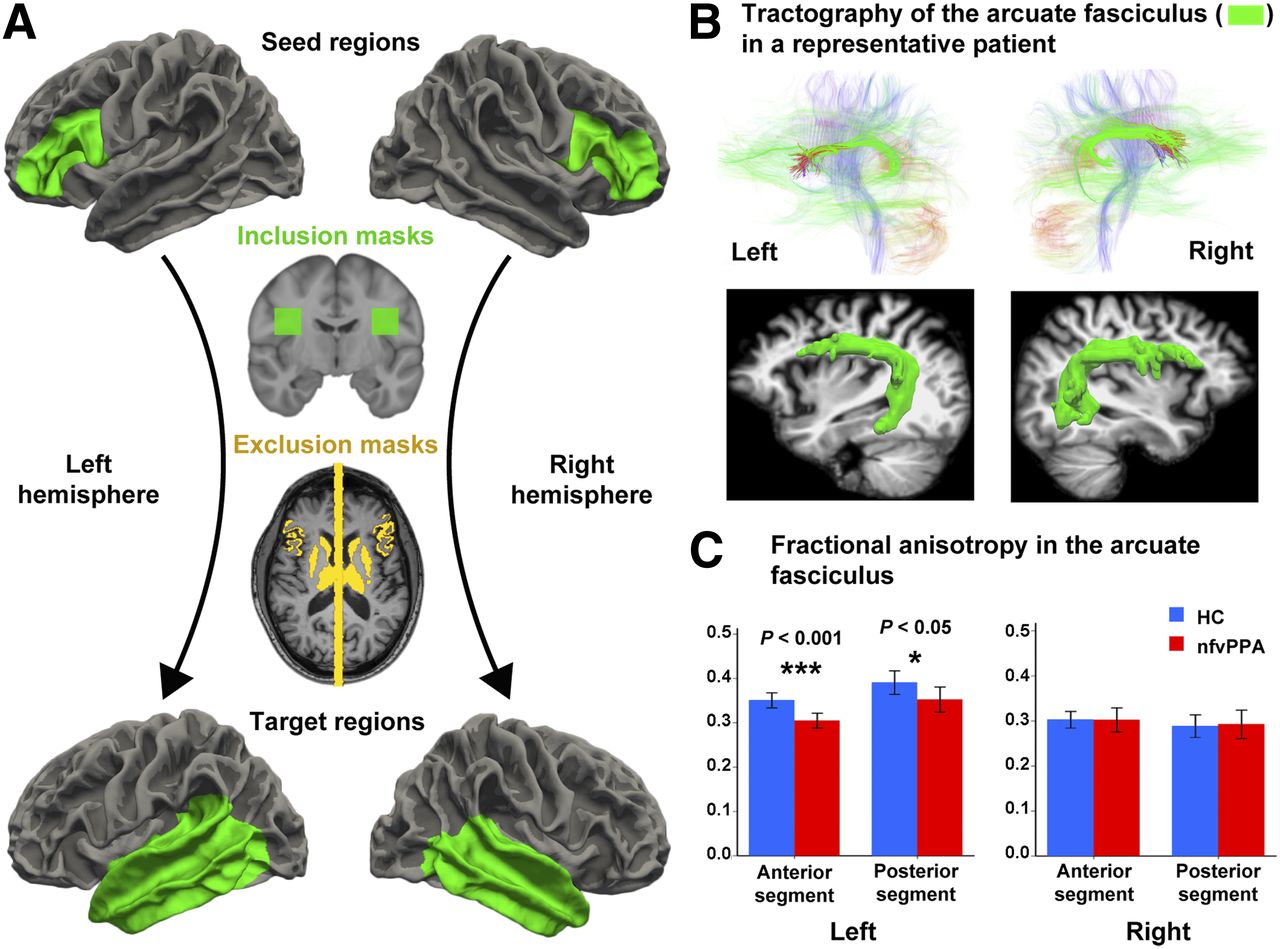
Arcuate fasciculus measurements. (A) Seed and target regions, as well as inclusion and exclusion masks for arcuate fasciculus tractography and FA analyses. Using probabilistic tractography, we seeded tracts from gray–white matter boundary surface of anterior node in left hemisphere or its right homolog (green areas, top row) and terminated them at boundary surface of temporal lobes (green areas, bottom row). We included only those tracts that passed through white matter inclusion mask in each hemisphere (green areas, second row). We used as exclusion masks subcortical gray nuclei and midline sagittal plane, as well as cortex volumes attaching gray–white matter boundary surface of seed regions (yellow areas, third row). (B) Tractography of main white matter tracts, including arcuate fasciculus (green areas, top row) in both hemispheres of representative patient with nfvPPA, and 3-dimensional reconstruction of fasciculus over structural MRI (green areas, bottom row) using technique in Figure 2A. Left, normally thicker than right, is thinner in nfvPPA. (C) Compared with healthy controls, nfvPPA patients had lower FA in both anterior and posterior segments of left arcuate fasciculus, effect size (r) being larger in anterior segment, but right arcuate fasciculus was similar between both groups.
To increase sensitivity and specificity, the gray matter associated with the seed regions was used as an exclusion mask to exclude tracks that propagated exclusively toward the gray matter. Similarly, the gray matter of the target regions was used as a stop mask to avoid tracks that terminate in the target through the gray matter as well as tracts merging within the gray matter and being extended by the tracking algorithm. To further isolate the arcuate fasciculus, we used as exclusion masks 2 subcortical structures that border the arcuate fasciculus, namely the thalamus and striatum, and a midline mask to exclude interhemispheric tracks (Fig. 3A). As a final step, to restrict tractography to the known anatomic distribution of the arcuate fasciculus (33), in Montreal Neurological Institute space in each hemisphere we defined an inclusion mask through which all fibers had to pass, the coordinates of which are given below:
As all DTI analysis was performed in each subject’s native DTI space, masks defined in Montreal Neurological Institute space were transformed to the anatomic space of each individual subject using the SPM12 coregistration algorithm. The structural MRI was then coregistered (again via SPM12) to the first frame of each subject’s diffusion series, which was acquired without any diffusion weighting (B0), and all masks were transformed from the anatomic space to diffusion space using the resulting transformation matrix. The rest of the procedure followed the method described by Takaya et al. (33). To further restrict our FA analysis to voxels most likely containing arcuate fasciculus fibers, we included only voxels that contained at least 3.08 × 10−5 percent of the total streamlines sent out from the ROI masks used to trace that tract (34). The average value of FA (also produced via the software library of the Oxford Centre for Functional Magnetic Resonance Imaging of the Brain) was then extracted for each subject using the entire arcuate fasciculus mask. In addition, to determine differences in FA between the anterior and posterior arcuate fasciculus, we divided the arcuate fasciculus into 2 segments in Montreal Neurological Institute space. The anterior segment was defined by all coronal slices anterior to slice y = −24 (y > −24), and the posterior segment was defined by slice y = −24 and all coronal slices posterior to it (y ≤ −24). Both regions were transformed first into each subject’s anatomic space and then, using the resulting transformation matrix, into each subject’s diffusion space. Then, the average value for FA along the anterior and posterior segment of the arcuate fasciculus was computed.
To generate the projections of the arcuate fasciculus in the temporal lobe of healthy controls, each subject’s probabilistic tractography results were intersected with the target mask of the temporal lobe white–gray boundary, thresholded to consider only the top 10% of endpoints and binarized. The resulting images were coregistered via each subject’s B0 image to the anatomic MRI and then normalized to Montreal Neurological Institute space with SPM12. A group average over all controls was then computed and projected onto the FreeSurfer average white matter surface for display.
Statistical Analysis
Tau PET Analysis
18F-AV-1451 tau PET data were obtained in SUV ratio units for each brain voxel. Statistical analysis of SUV ratio parametric images across patients and healthy controls was performed at the voxel level using SPM12 with a 2-sample, 2-tailed t test with unequal variance. For screening and display, we used a cluster threshold of P < 0.001 uncorrected and a 150-voxel extent threshold. Results were then analyzed on the cluster level within SPM12, using familywise error correction for multiple comparisons from random-field theory implemented in SPM12.
DTI Analysis
Wilcoxon–Mann–Whitney tests, implemented in SPSS (version 20.0; IBM SPSS Statistics for Mac [Apple]), were used to compare the FA of the anterior and posterior portions of the arcuate fasciculus between nfvPPA patients and healthy controls. The effect size (r) for the statistic was also calculated (35).
RESULTS
Cognitive and Language Impairment in nfvPPA
Both the clinical evaluation and formal language testing showed that the nfvPPA patients uniformly had impaired verbal production but largely spared comprehension (Fig. 2A; Supplemental Table 1). Verbal fluency, syntax/grammar, and word retrieval/expression scored in the moderately to severely impaired range on the Progressive Aphasia Severity Scale (25). By contrast, single-word comprehension, reading, and auditory comprehension were normal or only mildly impaired (Fig. 2A).
Brain Tau Load in nfvPPA
18F-AV-1451 PET showed increased tau in nfvPPA compared with controls (Fig. 2B), and 3 clusters survived multiple-comparison correction: 2 contiguous clusters were in the anterior node and the other one was centered on the posterior node of the syntactic network (Fig. 2C). The location of the tau clusters was strikingly similar to the nodes of the syntactic network identified in healthy subjects by functional and structural connectivity MRI (Figs. 1C and 1D; Supplemental Fig. 2) (21,36).
Loss of Integrity of Left Arcuate Fasciculus
FA, an index of white matter integrity, did not differ between patients and controls for the right arcuate fasciculus, but in the left, particularly in the anterior portion, nfvPPA patients had lower FA than controls (Fig. 3C). The effect size was large but was larger for the anterior (r = 0.84) than for the posterior segment (r = 0.70). Additionally, there was a trend toward increased tau levels in the anterior node to correlate with lower FA in the anterior segment of the arcuate fasciculus; a similar correlation was not found for the posterior node and the posterior segment of the arcuate fasciculus (Supplemental Fig. 3).
DISCUSSION
Our finding of an abnormal tau load in the anterior and posterior nodes of the syntactic network suggests that network arrangement determines tau deposition in nfvPPA. The location of the posterior node, known from blood oxygenation level–dependent MRI activation studies (Fig. 1A) (21), coincided with the temporal lobe area where the arcuate fasciculus projects most heavily (Fig. 1D) (33) and with the area most structurally connected to the frontal lobe area—the anterior node—where our nonfluent PPA patients had increased tau (Supplemental Fig. 2). Because the disease begins with impairment of functions such as verbal fluency (Fig. 2A), mediated by the anterior node, it is logical to postulate that the anterior node is the epicenter of damage (37,38) and that tau buildup starts there. Clinically detectable impairment, as we found in our patients, begins when anterior node neurons become dysfunctional or die. By contrast, although there was tau buildup in the posterior node, functions mediated by this node were less impaired (Fig. 2A), suggesting that tau-containing neurons in the posterior node were still functional and therefore likely to have been affected later than those in the anterior node. As predicted by the tau-as-prion hypothesis, the arcuate fasciculus, connecting both nodes, was affected on the left hemisphere and more so anteriorly, where there was a trend for FA to correlate with the amount of tau in the anterior node (Fig. 3; Supplemental Fig. 3). These findings suggests that the axons of anterior node neurons were the most heavily affected (Supplemental Figs. 1 and 3).
Explanations other than tau propagation might be posited for our findings. Neuronal loss and dysfunction in the anterior node may decrease postsynaptic neuroprotection, provided by trophic factors and other constituents required by posterior node neurons to function normally. Abnormal function may then induce the production of tau in posterior node neurons. However, this is unlikely, because aggregated tau, which is the type that 18F-AV-1451 binds (39), has not been reported after cortical infarction causing extensive necrosis, even when the brain was examined years after a stroke (40,41). A more nuanced form of the same explanation may postulate that a loss of neuroprotection, combined with a milieu prone to tau production by neurons caused by genetic and other factors, may yield the findings we obtained. Still, tau propagation explains our findings most parsimoniously.
Although in our nfvPPA sample we did not find significant tau deposition in other regions of the brain connected with the anterior node of the syntactic network, such as the supplementary motor area or anterior cingulate area, which is connected to the anterior node through the aslant tract (37), these other regions could show abnormal deposition with lower thresholds or in other samples. In nfvPPA, another group has found increased tau deposition in the supplementary motor area, but the sample showed heterogeneous findings (42), which suggests that this unusual and still poorly understood syndrome may have various underlying neuroanatomic substrates loading differently in different patient samples.
The uptake of 18F-AV-1451 is known to be considerably lower in FTD than in AD (39). However, 18F-AV-1451 is an adequate tracer to study tau deposition in nfvPPA, a FTD variant most often associated with the 4-repeat tau isoform (22). 18F-AV-1451 uptake correlated well with quantitatively measured 4-repeat tau burden, as determined post-mortem in the brain of a patient presenting with nfvPPA (43). Furthermore, unlike other tau PET tracers, 18F-AV-1451 is not appreciably blocked by monoamine oxidase B inhibitors (44), ruling out the possibility that the increased 18F-AV-1451 signal may simply correspond to an increase in monoamine oxidase B induced by the pathologic process (45). In FTD, small but detectable and accurate 18F-AV-1451 binding has been found in most studies (39,43), and its regional distribution corresponds well to the clinical syndromes (46). The lower 18F-AV-1451 signal in FTD than in AD could be related to lower 18F-AV-1451 binding to FTD tau but also reflects the lower tau load in FTD, which is about 10% of that in AD (47).
Our cross-sectional imaging study provided only a snapshot of the neurodegenerative process. It would have been ideal to image first the first stage of the disease, likely with tau only in the anterior node (Supplemental Fig. 1A), and then, sometime later, the stage documented in this paper. However desirable that may be, it may not be realistically possible: our patients were imaged shortly after presentation, when the functions of the anterior node faltered (Fig. 2A), causing them to seek medical attention. At the other temporal end of the process, imaging these patients again later could yield additional useful information by documenting increasing tau in the posterior node. However, as the disease progresses, local spreading through shorter pathways may actually obscure the network effect (48) detected at this stage in the evolution of nfvPPA.
CONCLUSION
Our data, obtained in vivo from the entire human brain, support a mechanistic model in which abnormal tau buildup starts focally and spreads via a neuron-to-connected-neuron pattern in natural brain networks. This model could facilitate important aspects of treatment discovery, such as monitoring disease progression by tau brain imaging and targeting therapies to tau propagation.
DISCLOSURE
Meixiang Yu and Joseph Masdeu received research funding from Eli Lilly, parent company of Avid Radiopharmaceuticals, manufacturer of 18F-AV-1451. Paolo Zanotti-Fregonara and Joseph Masdeu are on a speaker bureau for Eli Lilly. The study was funded by the Chao, Graham, Harrison, and Nantz Funds from the Houston Methodist Foundation. Avid Radiopharmaceuticals provided the 18F-AV-1451 precursor free of charge. The ADNI data used in this study was collected and shared through a project, ADNI, funded by the National Institutes of Health (grant U01 AG024904) and DOD (Department of Defense award W81XWH-12-2-0012). The ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research provides funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (https://fnih.org/). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Does abnormal tau propagate through natural brain networks in dementia?
PERTINENT FINDINGS: In a cross-sectional study of nfvPPA, the anterior and posterior nodes of the syntactic network, separated from each other in the cortex, had increased tau as measured with 18F-AV-1451 PET. The white matter pathway (the left arcuate fasciculus) connecting these 2 nodes had decreased FA, particularly near the anterior node, which is affected first in this disease.
IMPLICATIONS FOR PATIENT CARE: Our findings support tau spreading from neuron to connected neuron through natural brain networks, a concept that is important for monitoring disease progression by tau brain imaging and may facilitate targeting therapies to tau propagation.
Acknowledgments
Dr. Henry J. Pownall provided helpful comments. Some data used in this article were obtained from the ADNI database (http://adni.loni.usc.edu/). As such, the investigators within the ADNI contributed to the design and implementation of the ADNI or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at http://adni.loni.usc.edu/wp-content/themes/freshnews-dev-v2/documents/policy/ADNI_Acknowledgement_List%205-29-18.pdf.
Footnotes
Published online Jul. 26, 2019.
- © 2020 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication December 29, 2019.
- Accepted for publication July 6, 2019.





{kind=link}
{kind=link}
{kind=link}