


Abstract
In recent years, targeted radionuclide therapy (TRT) has emerged as a promising strategy for cancer treatment. In contrast to conventional radiotherapy, TRT delivers ionizing radiation to tumors in a targeted manner, reducing the dose that healthy tissues are exposed to. Existing TRT strategies include the use of 177Lu-DOTATATE, 131I-metaiodobenzylguanidine, Bexxar, and Zevalin, clinically approved agents for the treatment of neuroendocrine tumors, neuroblastoma, and non-Hodgkin lymphoma, respectively. Although promising results have been obtained with these agents, clinical evidence acquired to date suggests that only a small percentage of patients achieves complete response. Consequently, there have been attempts to improve TRT outcomes through combinations with other therapeutic agents; such strategies include administering concurrent TRT and chemotherapy, and the use of TRT with known or putative radiosensitizers such as poly(adenosine diphosphate ribose) polymerase and mammalian-target-of-rapamycin inhibitors. In addition to potentially achieving greater therapeutic effects than the respective monotherapies, these strategies may lead to lower dosages or numbers of cycles required and, in turn, reduce unwanted toxicities. As of now, several clinical trials have been conducted to assess the benefits of TRT-based combination therapies, sometimes despite limited preclinical evidence being available in the public domain to support their use. Although some clinical trials have yielded promising results, others have shown no clear survival benefit from particular combination treatments. Here, we present a comprehensive review of combination strategies with TRT reported in the literature to date and evaluate their therapeutic potential.
NOTEWORTHY
TRT is growing in popularity for cancer treatment.
Efforts to improve TRT have led to increasing numbers of combination strategies being attempted.
Increasing our understanding of the radiobiology of TRT will help inform the successful implementation of combination strategies.
Targeted radionuclide therapy (TRT) involves the use of radiopharmaceuticals designed to specifically target cancer cells. These radiopharmaceuticals consist of β, α, or Auger electron–emitting radionuclides coupled to a tumor-targeting vector, such as a monoclonal antibody or peptide. In recent years, TRT has rapidly grown in popularity, with agents such as 177Lu-DOTATATE,131I-metaiodobenzylguanidine, (Bexxar; GlaxoSmithKline), and (Zevalin; Acrotech Biopharma) now clinically approved for the treatment of low-grade neuroendocrine tumors (NETs), neuroblastoma, and non-Hodgkin lymphoma, respectively. 177Lu-PSMA, which targets prostate-specific membrane antigen (PSMA), is also emerging as an attractive strategy for metastatic castration-resistant prostate cancer, with large late-phase clinical trials under way. Because of its highly targeted approach, the application of TRT could result in fewer side effects than conventional external-beam radiotherapy (EBRT) and allow for more effective treatment of disseminated cancers (1). Promising results have been obtained with several TRTs in clinical trials, but there remains room for improvement; for instance, whereas the phase III NETTER-1 trial showed that 177Lu-DOTATATE can be a life-extending treatment, with significant improvement in the median progression-free survival being reported, objective response was observed in just 18% of patients (2). To further improve TRT outcomes, administering TRT at earlier disease stages, as is being investigated in the UpFrontPSMA trial (NCT04343885), and the use of combination therapies is being attempted.
Combination strategies investigated thus far include the use of agents to improve tumor perfusion to allow better distribution of the radiopharmaceutical (3), upregulation of the target receptor to increase cellular uptake (4), the combination of TRT with other DNA-damaging drugs (5–7), radiosensitization through inhibiting essential processes such as DNA damage repair (8–10), and the use of immune checkpoint inhibitors (11–13). Several of these strategies have shown promise in preclinical studies and are currently in clinical trials, but there remains a lack of research into TRT radiobiology, making it difficult to predict which of these strategies will prove most effective. Attempts to enhance tumor perfusion and cellular uptake have shown promise, but we do not currently know the exact absorbed radiation dose required for a complete response, making the optimization of this strategy challenging (3,4). Efforts toward modeling the necessary dose required for tumor response after 177Lu-PSMA therapy are under way, but this modeling is difficult to achieve given the heterogeneity of PSMA expression (14). Additionally, although there are a wide variety of drugs being combined with TRT in an attempt to achieve effective radiosensitization, the mechanisms by which these combination therapies work and their effects on downstream biologic pathways are not necessarily fully understood. Many of these combination strategies make use of known radiosensitizers of EBRT, but because of differences in the dose rates, duration of radiation exposure, and radiobiologic effects between EBRT and TRT, our understanding of EBRT radiobiology cannot simply be extrapolated to TRT (1,15–17). Radiosensitizers of EBRT may therefore not always be effective radiosensitizers of TRT, and vice versa. Here, we present a comprehensive review of TRT-based combination therapies tested to date, focusing primarily on those involving β-emitting TRTs because several of these combinations are being evaluated clinically (Fig. 1; Supplemental Tables 1–8 [supplemental materials are available at http://jnm.snmjournals.org]). We also discuss possible limitations of such studies and suggest further research that is needed if these combination strategies are to be effectively translated to patients.
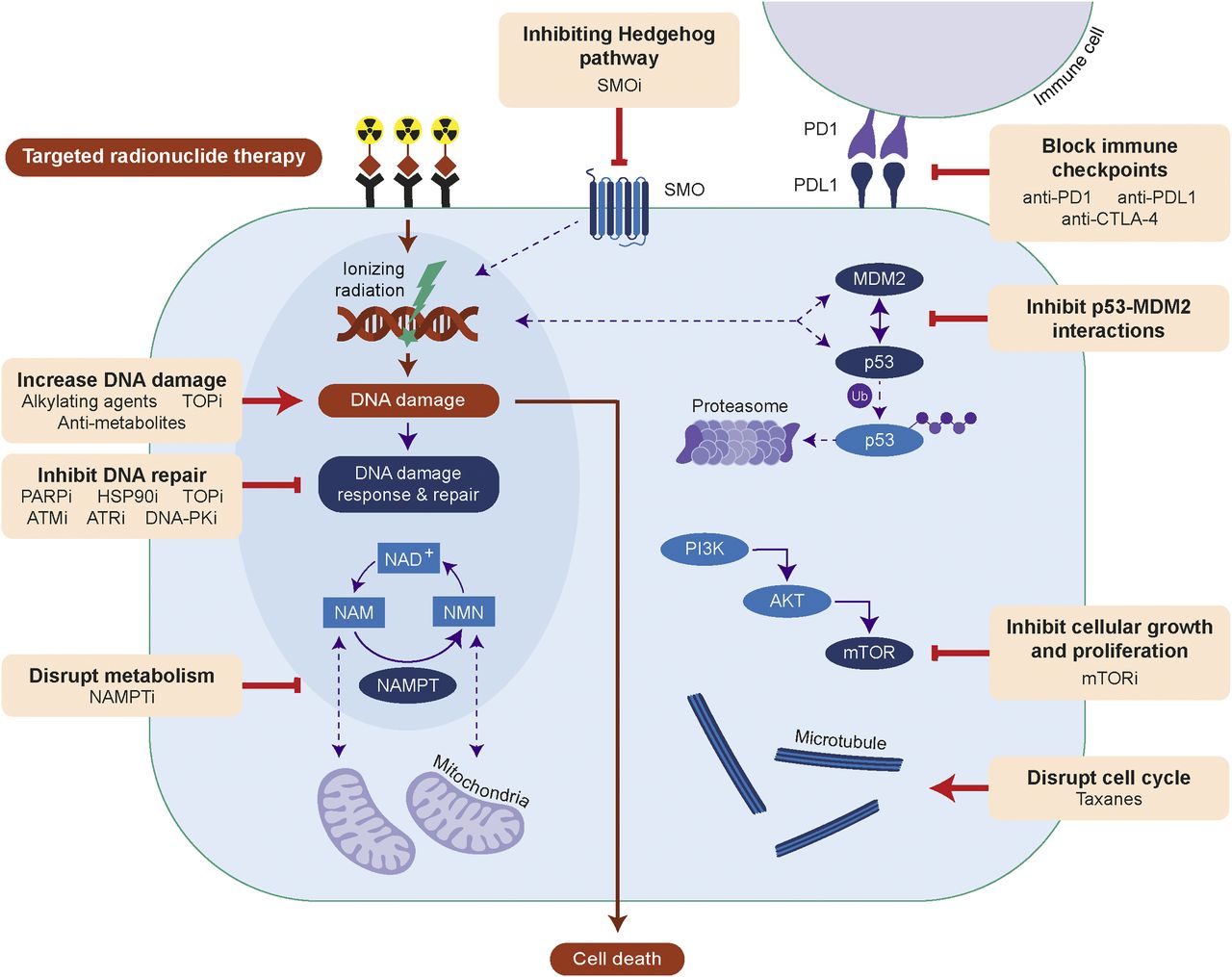
Efficacy of TRT can be improved by increasing DNA damage, inhibiting DNA repair, disrupting metabolism or cell cycle, inhibiting signaling of Hedgehog, phosphoinositide-3-kinase (PI3K)/protein kinase B (AKT)/mTOR or p53-MDM2, or blocking immune checkpoints. AKT = protein kinase B; ATMi = ataxia telangiectasia mutated inhibitor; ATRi = ataxia telangiectasia and Rad3-related inhibitor; CTLA-4 = cytotoxic T-lymphocyte antigen 4; HSP90i = HSP90 inhibitor; mTORi = mTOR inhibitor; NAM = nicotinamide; NAMPTi = NAMPT inhibitor; NMN = nicotinamide mononucleotide; PI3K = phosphoinositide-3-kinase; PARPi = PARP inhibitor; PD1 = programmed death 1; PDL1 = programmed death 1 ligand; PKi = protein kinase inhibitor; SMOi = smoothened inhibitor; SMO = smoothened; TOPi = topoisomerase inhibitor; Ub = ubiquitin.
INCREASING DNA DAMAGE WITH TRADITIONAL CHEMOTHERAPIES
As TRT primarily acts by inducing DNA damage, the efficacy of TRT can be increased by creating additional DNA damage through combining TRT with conventional chemotherapy or radiotherapy. Prominent examples include the combination of 177Lu-DOTATATE with the antimetabolite capecitabine, the alkylating agent temozolomide, or both (CAPTEM) for the treatment of NETs (Supplemental Table 1). These are all standard chemotherapy options for advanced NETs, making their combination with 177Lu-DOTATATE easily translatable if effective. To date, promising tumor response rates have been observed with 177Lu-DOTATATE + capecitabine, 177Lu-DOTATATE + temozolomide, and/or 177Lu-DOTATATE + capecitabine + temozolomide (CAPTEM) (6,18,19), and phase II clinical trials are currently under way (NCT02736500 and NCT02358356). Initial results from the phase I/II CAPTEM trial suggest that although the objective tumor response rate in the cohort treated with 177Lu-DOTATATE + CAPTEM was higher than in patients treated with 177Lu-DOTATATE alone, more treatment-related adverse effects were observed (20). Though rare, both 177Lu-DOTATATE and CAPTEM have been associated with hematologic toxicities when used as monotherapies (2,21); thus, the increase in adverse events observed in the combination group may be due to overlapping toxicities. The final outcomes of the phase I/II CAPTEM trial should provide further insight.
To our knowledge, there exists just one preclinical study attempting to optimize 177Lu-DOTATATE + capecitabine + temozolomide, 177Lu-DOTATATE + temozolomide chemotherapy (3). In mice bearing human small cell lung cancer H69 tumors, Bison et al. noted that although temozolomide outperformed 177Lu-DOTATATE as a monotherapy, the combination of 177Lu-DOTATATE + temozolomide led to additive effects (Fig. 2) (3). Moreover, administering temozolomide 14 d before 177Lu-DOTATATE treatment was found to be significantly more effective than vice versa, which was attributed to enhanced tumor perfusion, radiosensitivity, and tumor oxygenation (3). In clinical studies involving 177Lu-DOTATATE + temozolomide, 177Lu-DOTATATE + capecitabine + temozolomide therapy, chemotherapy is typically initiated before or concomitantly with 177Lu-DOTATATE (5,7,19). Further studies are necessary to optimize the dosing schedules to produce the greatest survival benefits, particularly for CAPTEM protocols, for which the relative timings of capecitabine and temozolomide administration are already known to affect synergism (22).
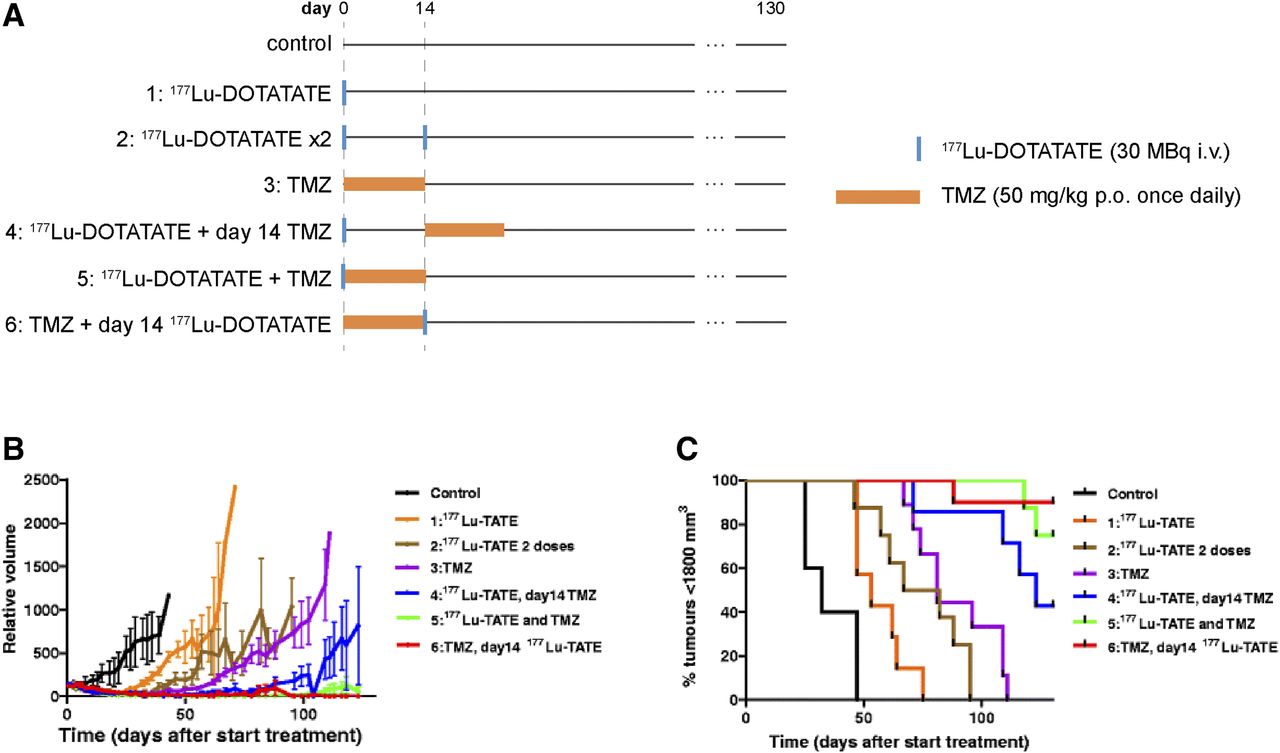
Evaluation of different 177Lu-DOTATATE + temozolomide (TMZ) treatments in H69 tumor–bearing mice. (A) Study timeline. (B and C) Average tumor volume and percentage of mice with tumors smaller than 1,800 mm2 (8–10 mice per group). i.v. = intravenously; p.o. = orally. (Reprinted with permission of (3).)
Aside from 177Lu-DOTATATE–based combination therapies, there are several examples of other TRTs being combined with chemotherapy, but there is currently insufficient evidence to support the use of these combinations over their respective monotherapies (Supplemental Table 1). Recent reports have, however, alluded to significantly longer overall survival times in chemotherapy-naïve patients treated with TRT than in patients with a history of chemotherapy (23–25). Until the precise reason behind these observations is understood, TRT + chemotherapy combinations should be carefully monitored if implemented.
RADIOSENSITIZATION THROUGH INHIBITING DNA REPAIR
Inhibiting key proteins involved in the DNA damage response (DDR) could lead to radiosensitization of TRT. To date, there are several examples of this strategy being used: most involve poly(adenosine diphosphate ribose) polymerase (PARP) inhibitors, but inhibitors of other DDR proteins, heat shock protein 90 (HSP90), and checkpoint kinase 1 have also been used (Supplemental Table 2). Inhibition of DNA topoisomerases I and II has also been suggested to induce radiosensitization by disrupting DNA repair (15,16). It should be noted, however, that many DDR-targeting agents are associated with bone marrow and gastrointestinal toxicities, which could lead to increased adverse side effects when combined with TRT (2,26,27). Careful monitoring of normal-tissue toxicity is therefore warranted for these combinations.
DDR Inhibitors
PARP enzymes constitute a family of proteins that are vital for the maintenance of cellular homeostasis and form an essential part of DDR. PARP proteins—in particular, PARP-1—have been implicated in the repair of both single-stranded and double-stranded breaks (28). Because the main mechanism by which ionizing radiation causes cell death is through the induction of DNA damage, reducing the capacity for DNA repair with PARP inhibitors is a potential radiosensitization strategy (29,30). Several preclinical studies investigated whether PARP inhibitors can also potentiate TRT with 177Lu-DOTATATE: in vitro with the human cancer cell lines osteosarcoma U2OS expressing somatostatin receptor 2, gastroenteropancreatic BON-1, and bronchopulmonary NCI-H727, and in vivo in AR42J tumor–bearing mice (8–10). The combination of PARP inhibitors with other TRTs, such as 131I-MIP-1095, 177Lu-RM2, and 227Th-HER2, has also been tested, with promising results (Supplemental Table 2).
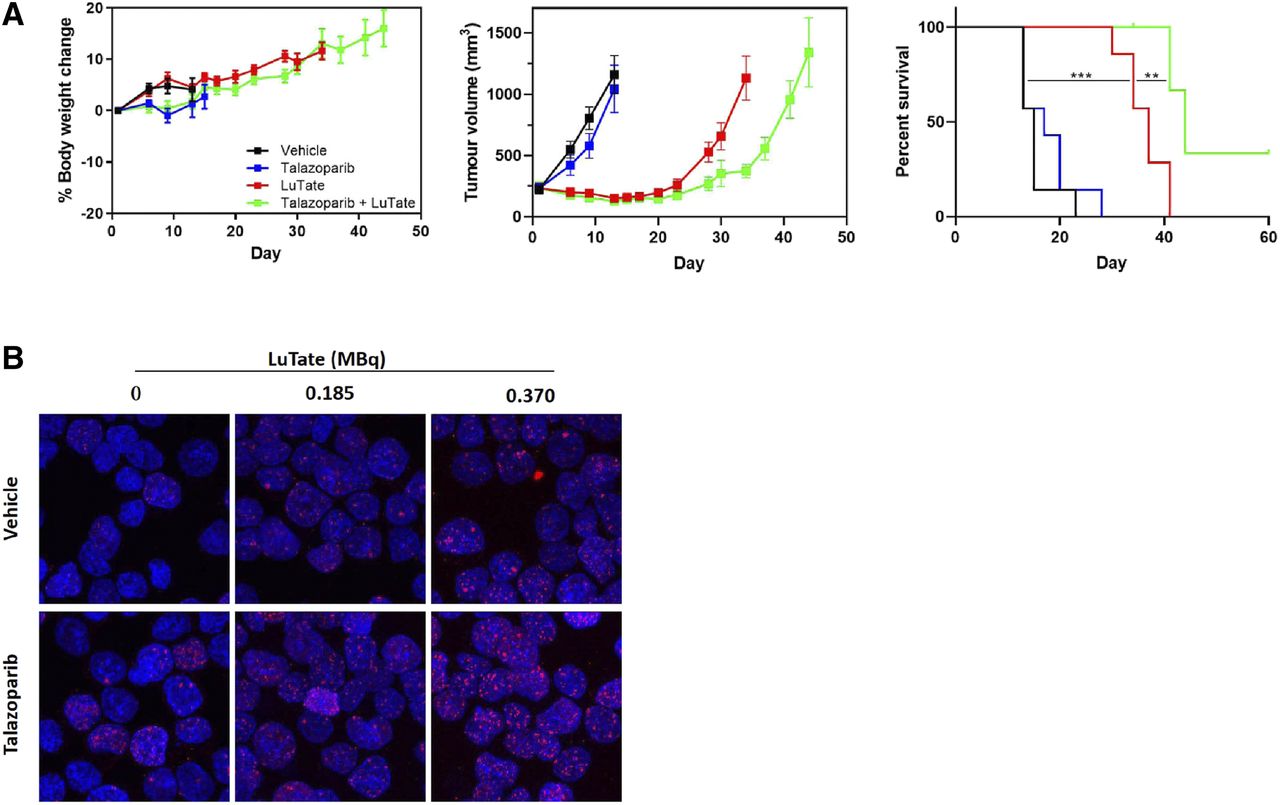
Although few studies have aimed to elucidate the mechanism behind PARP inhibitor–induced radiosensitization of TRT, 177Lu-DOTATATE + PARP inhibitor treatment has been associated with an increased number and persistence of double-stranded breaks; increased levels of DNA damage markers, such as γH2AX, phosphorylated p53, and 53BP1; and increased cell cycle arrest (Fig. 3) (8–10). Therefore, it has been proposed that PARP inhibition leads to an inability to repair the single-stranded breaks caused by 177Lu-DOTATATE, resulting in the formation of additional double-stranded breaks on cellular replication and, ultimately, cell death (8–10). Several clinical trials assessing the safety and efficacy of TRT + PARP inhibitors are under way (NCT04086485, NCT04375267, and NCT0387484), and the field is looking forward to their outcome. In the future, it would be of interest to compare the effects of TRT + PARP inhibitors in patients with deficient homologous recombination (e.g., with BRCA1/2 mutations) versus patients with functioning homologous recombination, as mutations in homologous recombination repair proteins are known to induce PARP inhibitor sensitivity (31).

Evaluation of TRT + talazoparib treatment in exocrine pancreatic AR42J model. (A) Body weight, tumor volume, and percentage survival in AR42J tumor–bearing mice treated with 30 MBq of 177Lu-DOTATATE (day 1) with or without 0.25 mg/kg dose of talazoparib twice daily (days 1–5). (B) ɣH2AX staining of AR42J cells treated with 177Lu-DOTATATE with or without talazoparib for 2 h. (Reprinted with permission of (10).)
In addition to PARP inhibitors, inhibitors of ataxia telangiectasia mutated, ataxia telangiectasia and Rad3-related, and DNA-dependent protein kinase, key effector proteins involved in DDR, have also been investigated as radiosensitizers. These combination studies are mainly with EBRT (32–34), but there are a few examples showing their ability to also radiosensitize TRT (35,36); however, it remains to be seen how these TRT combinations perform clinically. Combining TRT with inhibitors of epidermal growth factor receptor, which modulates DNA repair through activation of DNA-dependent protein kinase, has also been attempted in a few preclinical studies (37,38).
HSP90 Inhibitors
177Lu-DOTATATE has been combined with the HSP90 inhibitors onalespib and ganetespib in vivo, with promising results and a favorable toxicity profile reported for NET xenografts (39,40). HSP90 is a molecular chaperone that participates in key processes such as protein degradation, folding, and intracellular transport. Clients for HSP90 include kinases involved in cell growth, such as epidermal growth factor receptor and protein kinase B, and DDR proteins, such as ataxia telangiectasia and Rad3-related and checkpoint kinase 1 (41,42). HSP90 inhibitors have been reported to induce radiosensitization when combined with EBRT, but because of the multiple clients associated with HSP90, the exact mechanism of HSP90 inhibitor–induced radiosensitization is unknown. However, it is known to negatively affect DNA repair signaling and the activation of cell cycle checkpoints, potentiating radiation-induced damage (43,44). To date, no study has sought to elucidate the mechanism of HSP90 inhibitor–induced radiosensitization in the context of TRT. Moreover, although several HSP90 inhibitors have been tested in clinical trials as potential monotherapies, a lack of efficacy and unacceptable toxicity have prevented any from being clinically approved, making this strategy challenging to translate to patients (45,46).
Topoisomerase Inhibitors
Topoisomerases 1 and 2 are nuclear enzymes that are essential for maintaining the correct topological state of DNA, which is crucial for RNA transcription, chromatin remodelling, and DNA replication. Through the formation of a topoisomerase DNA covalent intermediate—the topoisomerase cleavable complex—the main role of topoisomerases 1 and 2 is to catalyze the relaxation of positive or negative DNA supercoiling by nicking DNA strands to allow controlled rotation, followed by religation (47). Many inhibitors of topoisomerases 1 and 2 work by stabilizing the topoisomerase cleavable complex, leading to the accumulation of single- and double-stranded breaks, respectively (47). In addition to creating DNA damage, numerous studies have shown that topoisomerase inhibitors can also act as radiosensitizers of EBRT (48–50); however, the precise radiosensitization mechanism remains unelucidated. Shih et al. suggested that topoisomerase inhibitor–mediated cytotoxicity and radiosensitization occur via different pathways, as the former is Ku80-independent whereas the latter is Ku80-dependent (51). Ku80 is required for nonhomologous end joining, suggesting that topoisomerase inhibitors can potentiate radiation-induced DNA damage through interfering with DDR. The schedule of topoisomerase inhibitor administration is also thought to be crucial for effective radiosensitization of EBRT, with varying degrees of radiosensitization observed depending on the dosing frequency (49,50).
Several studies investigated the use of topoisomerase 1 inhibitors with TRT, with the most frequently tested combination being 131I-metaiodobenzylguanidine + topotecan for the treatment of advanced neuroblastoma. In vitro and in vivo studies using the neuroblastoma SK-N-BE(2c) and glioma UVW/NAT models have shown that 131I-metaiodobenzylguanidine + topotecan treatment leads to superadditive DNA damage and reduced efficiency of DNA repair (52,53). As with EBRT + topoisomerase inhibitor combinations, the benefits of TRT + topoisomerase inhibitor treatment appears to depend on the timing of topoisomerase inhibitor administration: McCluskey et al. reported greater tumor growth delay in SK-N-BE(2C) and UVW/NAT tumor–bearing mice when topotecan was administered simultaneously with 131I-metaiodobenzylguanidine than when administered 24 h beforehand or afterward (52,53). This observation has been linked to differences in cell cycle distribution induced by the different schedules; however, further studies are needed for confirmation (53). Several clinical studies have been conducted with 131I-metaiodobenzylguanidine + topotecan in neuroblastoma patients, but it remains unclear whether the addition of topotecan leads to any significant benefit over 131I-metaiodobenzylguanidine monotherapy (Supplemental Table 2).
RADIOSENSITIZATION THROUGH INHIBITING PHOSPHOINOSITIDE-3-KINASE/PROTEIN KINASE B/MAMMALIAN TARGET OF RAPAMYCIN (MTOR) SIGNALING
mTOR is a serine/threonine kinase that forms 2 complexes, mTOR complex 1 and mTOR complex 2, which differ in their subcellular location, structure, and function: mTOR complex 1 regulates cell growth and metabolism, and mTOR complex 2 regulates cell proliferation and survival (54). mTOR activity is regulated primarily by the phosphoinositide-3-kinase/protein kinase B/mTOR signaling pathway. In many human cancers, mTOR signaling is hyperactivated, leading to increased tumorigenesis, increased tumor progression, and decreased survival (55,56). mTOR is also known to influence DDR; for example, mTOR can positively and negatively regulate ataxia telangiectasia mutated (57).
Activation of the phosphoinositide-3-kinase/protein kinase B/mTOR pathway has been linked to the development of radioresistance, and multiple studies have investigated mTOR inhibitors for their potential radiosensitizing effects with EBRT (58–60). Multiple preclinical studies have demonstrated that EBRT + mTOR inhibitors leads to increased radiosensitivity through decreased expression of nonhomologous end joining and homologous recombination repair pathway proteins and increased expression of apoptosis pathway proteins (58–60). However, the use of mTOR inhibitors as radiosensitizers for TRT remains controversial, as preclinical studies investigating the efficacy of 177Lu-DOTATATE + mTOR inhibitors have reported contrasting results: Johnbeck et al. observed greater antitumor effects with 177Lu-DOTATATE + everolimus treatment than with 177Lu-DOTATATE alone in the mouse H727 non–small cell lung carcinoma model, Zellmer et al. found that the combination treatment was as effective as 177Lu-DOTATATE alone in the athymic mouse AR42J pancreatic model, and Pool et al. reported reduced antitumor effects with the combination treatment in the immunocompetent rat CA20948 pancreatic tumor model (61–64). Pool et al. also noted the development of distant metastases in more than 70% of the CA20948 rats in the everolimus-treated groups, whereas no metastases were observed in the control or 177Lu-DOTATATE–only groups (61,62). This increased propensity for metastasis formation was not observed in athymic AR42J tumor–bearing mice or in the phase I NETTLE trial evaluating the maximum tolerated dose of everolimus when administered with 177Lu-DOTATATE (64,65); thus, it is probable that the increased metastases observed are specific to the genotype of the CA20948 model. Alternatively, there is evidence that mTOR inhibition can influence endothelial–mesenchymal transition, with both the promotion and the suppression of endothelial–mesenchymal transition being observed when using mTOR inhibitors under different conditions (66,67). Endothelial–mesenchymal transition is known to contribute toward tumor progression and metastasis, and thus, the precise role of mTOR inhibitors on endothelial–mesenchymal transition when combined with TRT merits close investigation (68). Understanding the source of these increased metastases is crucial, particularly as a phase I/II trial aimed at assessing 177Lu-DOTATATE + everolimus treatment in NET patients is under way (NCT03629847).
In addition to its role in tumorigenesis, mTOR plays an essential part in immune system regulation—a fact that should be considered when designing TRT + mTOR inhibitor therapies. Everolimus is clinically used as an immunosuppressant because of its ability to promote the expansion of regulatory T cells (69). A study testing 177Lu-DOTATATE + everolimus in non–tumor-bearing rats saw no significant increases in renal or hematologic toxicities compared with 177Lu-DOTATATE treatment, but decreases in the white blood cell count were noted, in line with the immunosuppressive effects of everolimus (70). Although immunosuppression is typically undesirable in cancer treatment because it decreases immune surveillance, there is little evidence suggesting that using mTOR inhibitors as a monotherapy could promote tumor growth (71). Nevertheless, in LEW/SsNHsd Lewis rats—a substrain that shows an enhanced autoimmune response—bearing CA20948 tumors, Bison et al. observed complete tumor regression in 50% of the rats in the control group, compared with 12.5% in the group treated with everolimus alone (62). No cases of complete tumor regression were seen in control LEW/HanHsd rats, which possess a less active immune response (62). Both findings indicate immune system involvement and suggest that any immunosuppressive effects should be considered when developing treatment combinations with TRT.
RADIOSENSITIZATION THROUGH INHIBITING HEDGEHOG SIGNALING
The Hedgehog signaling pathway is involved in embryonic development and is abnormally activated in many cancers. Inhibitors of the transmembrane protein smoothened, a key component of the Hedgehog pathway, have been investigated as potential anticancer drugs, with several of these inhibitors now approved by the U.S. Food and Drug Administration. Spetz et al. showed that combining 177Lu-DOTATATE with the smoothened antagonist sonidegib in GOT1 tumor–bearing mice led to an increased time to progression compared with the respective monotherapies (72). Moreover, pathway analysis predicted that this combination therapy impacted cancer related pathways, such as Wnt/β-catenin, Notch, and nuclear factor κ-light-chain enhancer, differently from either sonidegib or 177Lu-DOTATATE alone (72). Although experimental validation is required for these predictions, this finding suggests that radiosensitization of 177Lu-DOTATATE by sonidegib could involve multiple biological pathways and may therefore be challenging to optimize.
RADIOSENSITIZATION THROUGH INHIBITING P53–MURINE DOUBLE MINUTE 2 (MDM2) INTERACTIONS
The tumor suppressor protein p53 regulates cell cycle progression, DNA repair, and apoptosis and is negatively regulated by MDM2. MDM2 is overexpressed in many human tumors, leading to decreased p53 activity; thus, disruption of the p53–MDM2 interaction is a promising therapeutic strategy. Several studies have shown that ionizing radiation induces p53-dependent MDM2 expression, and p53/MDM2 inhibitors may therefore act as radiosensitizers by promoting p53-dependent apoptosis (73,74). Despite the fact that p53–MDM2 inhibitors are being investigated as radiosensitizers for EBRT in multiple studies, there are few examples of them being used with TRT to date and further studies are needed to validate this approach (Supplemental Table 5). However, this strategy depends on the presence of wild-type p53 and may therefore not be as effective in cancers in which TP53 mutations are common, such as metastatic castration-resistant prostate cancer (75,76).
RADIOSENSITIZATION THROUGH DISRUPTING CELL CYCLE
Microtubules, composed of heterodimers of α- and β-tubulin, are essential for cell signaling, division, and mitosis and for maintaining cellular structure (77). Taxane drugs stabilize microtubules by binding to β-tubulin, promoting microtubule polymerization and, ultimately, G2/M cell cycle arrest and apoptosis (77). The cell cycle plays an important role in radiosensitivity, with cells being most sensitive in the G2/M phases and most resistant in the S phase (78). Because of their effect on the cell cycle, there are many examples of the use of taxanes as radiosensitizers for EBRT, with encouraging outcomes observed (79). However, only a few TRT + taxane combination therapies have been investigated; although promising preclinical results have been reported for 177Lu- and 188Re-based TRTs, Kessel et al. reported that the benefit of 177Lu-PSMA was decreased in patients with a history of second-line cabazitaxel therapy, suggesting that further investigation into the mechanism of TRT + taxane combinations is warranted (Supplemental Table 6) (80). In particular, the sequencing of the therapies is likely to be important, as ionizing radiation itself influences the cell cycle (81); Liebmann et al. noted that irradiating human breast MCF-7 and lung A549 adenocarcinoma cells with EBRT before or concurrently with paclitaxel antagonized paclitaxel cytotoxicity (82). Optimizing this sequencing with TRT is likely to be challenging as, in contrast to EBRT, TRT delivers radiation heterogeneously over an extended period with less temporal control over the absorbed radiation dose (1).
RADIOSENSITIZATION THROUGH DISRUPTING NICOTINAMIDE ADENINE DINUCLEOTIDE (NAD+) METABOLISM
Nicotinamide phosphoribosyl transferase (NAMPT) is an enzyme that is essential for NAD+ metabolism. NAD+ is required for many cellular processes, including the activation of PARP-1, and is regenerated via a salvage pathway involving NAMPT. NAMPT inhibitors have been proposed to work as radiosensitizers of TRT by preventing NAD+ regeneration; on PARP-1 activation due to TRT-induced DNA damage, NAD+ is consumed and cannot be regenerated, leading it to drop to lethally low levels (83,84). To date, there has been just one investigation of TRT + NAMPT inhibitors: Elf et al. showed that the combination of 177Lu-DOTATATE and the experimental NAMPT inhibitor GMX1778 led to reduced tumor volumes and prolonged antitumor response in GOT1 tumor–bearing mice (83). However, more studies are required to assess the efficacy of this strategy and determine the exact radiosensitization mechanism. Moreover, since NAD+ is involved in many other processes, this approach could lead to off-target effects.
RADIOSENSITIZATION THROUGH BLOCKING IMMUNE CHECKPOINTS
Irradiation in the form of EBRT is known to have several immunomodulatory effects; for instance, EBRT can enhance tumor immunogenicity by inducing immunogenic cell death and promoting the release of tumor-associated antigens while simultaneously reducing tumor immunogenicity by upregulating programmed death ligand 1 expression (85). Combining EBRT with immune checkpoint blockade antibodies targeting programmed death 1, programmed death ligand 1, and cytotoxic T-lymphocyte antigen 4 represents an attractive strategy to potentiate radiation-induced antitumor immunity and has produced encouraging results (86,87). Although little is currently known about the effects of TRT on tumor immunogenicity, preclinical studies have shown that TRT also leads to upregulation of programmed death ligand 1 expression and that combinations of TRT with immune checkpoint blockade can lead to improved survival (11–13). Several clinical trials combining TRT + programmed death 1 inhibitors are currently under way (e.g., NCT03325816 and NCT03658447), though it is likely that further optimization of when immunotherapy should be administered during TRT treatment will be required; as with EBRT, Chen et al. noted in their preclinical studies that the benefits of combining TRT + anti-programmed death ligand 1 varies depending on whether the two treatments are given concurrently or sequentially (12,86).
OUTLOOK
The rising interest in TRT has led to the evaluation of numerous combination strategies, with promising results being reported and increasing numbers of clinical trials being conducted. In addition to the combination strategies discussed here, alternative strategies that have been proposed include those aimed at increasing the cellular uptake of radiopharmaceuticals and the rates of tumor perfusion (3,4). Multiple studies have also investigated the combination of TRT with EBRT, as well as with other β- or α-emitting TRTs (88–90). Growing numbers of triple-combination therapies are also being evaluated, such as TRT + CAPTEM (NCT02358356) and TRT + vincristine + irinotecan (NCT01313936), though it remains to be seen whether these strategies are effective in increasing the therapeutic index.
Despite the growing number of studies being conducted in this field, there remain few key outcomes. Increasing our understanding of TRT radiobiology is critical if we are to fully capitalize on the potential benefits of these combination therapies. For instance, the synergistic effects of taxanes and topoisomerase inhibitors in combination with TRT are believed to be cell cycle–dependent, and the effects of both the drugs and TRT on the cell cycle must be understood before we can optimize the dosing schedule (53,81,82). Greater characterization of the tested combinations over a range of concentrations is also necessary to identify combinations that are superadditive, as opposed to simply additive or even antagonistic. Moreover, many of the current combination strategies being used for TRT involve known radiosensitizers for EBRT, despite its markedly different radiobiology (1,15,16). Although these strategies may identify combinations that are effective with both forms of ionizing radiation, it is likely that there exist effective radiosensitizers for TRT that are ineffective when combined with EBRT and vice versa. In addition, although this review focused on β-emitting TRTs, there is growing interest in the use of α-and Auger emitters (91–93). However, because of differences in radiobiology and therefore biologically effective dose, effective radiosensitizers of β-emitting TRTs may not be effective radiosensitizers of α- and Auger-emitting TRTs. Differences may also exist between different β-emitters due to differences in dose rates and dose deposition profiles.
Furthermore, many of the commonly used preclinical models thus far are not entirely clinically relevant; for example, despite the use of U2OS cells expressing somatostatin receptor 2 and BON-1 cells to evaluate 177Lu-DOTATATE combination therapies, U2OS cells expressing somatostatin receptor 2 is a non-NET cell line and BON-1 cells express much lower levels of somatostatin receptor than are found in human NET tumors (4,8). BON-1 cells also show mutations of key DDR genes such as TP53—mutations that are rarely seen in G1 and G2 NETs (94,95). Preclinical studies on a greater variety of models are necessary, as well as identification of more clinically relevant model systems. Most preclinical studies to date have been on immune-compromised mice, and possible influences of the immune system on combination therapies and vice versa have not been explored in detail.
Moving forward, an increasing mechanistic understanding of TRT-based combination therapies is crucial if these strategies are to be effectively—and safely—translated to patients, particularly given the outcome of the phase III ERA223 trial, in which abiraterone acetate + prednisone/prednisolone + 223Ra resulted in no improvement in survival compared with abiraterone acetate + prednisone/prednisolone alone but was associated with an increased frequency of skeletal fractures (96). Similarly, initial results from the phase I/II CAPTEM trial (NCT02358356) show that although 177Lu-DOTATATE + CAPTEM treatment led to higher objective tumor-response rates than 177Lu-DOTATATE alone, the former was associated with more treatment-related adverse effects (20). Recent evidence has also alluded to better survival for chemotherapy-naïve patients treated with TRT than for those with a prior history of chemotherapy, possibly due to the acquisition of resistance mechanisms as has been suggested for second and subsequent lines of chemotherapy (25,97). The use of TRT combination therapies as first-line treatments may therefore produce greater survival benefits and should be explored further. In addition to potentially increasing therapeutic efficacy and minimizing the occurrence of toxicities, greater understanding of the radiobiology behind these combination strategies may also allow us to stratify patients and tailor combination therapies to the grade and mutational landscape of each patient’s cancer.
DISCLOSURE
Funding was provided to Tiffany Chan by Prostate Cancer Research Centre (U.K.), to Edward O’Neill by UKRI/MRC, and to Bart Cornelissen by UKRI/MRC and Cancer Research U.K. through the Oxford Institute for Radiation Biology. Bart Cornelissen serves as a consultant for Theragnostics Ltd. No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Oct. 9, 2020.
- © 2020 by the Society of Nuclear Medicine and Molecular Imaging.
Immediate Open Access: Creative Commons Attribution 4.0 International License (CC BY) allows users to share and adapt with attribution, excluding materials credited to previous publications. License: https://creativecommons.org/licenses/by/4.0/. Details: http://jnm.snmjournals.org/site/misc/permission.xhtml.
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.
- 60.↵
- 61.↵
- 62.↵
- 63.
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.
- 90.↵
- 91.↵
- 92.
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- Received for publication July 12, 2020.
- Accepted for publication September 9, 2020.





{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- INCREASING DNA DAMAGE WITH TRADITIONAL CHEMOTHERAPIES
- RADIOSENSITIZATION THROUGH INHIBITING DNA REPAIR
- RADIOSENSITIZATION THROUGH INHIBITING PHOSPHOINOSITIDE-3-KINASE/PROTEIN KINASE B/MAMMALIAN TARGET OF RAPAMYCIN (MTOR) SIGNALING
- RADIOSENSITIZATION THROUGH INHIBITING HEDGEHOG SIGNALING
- RADIOSENSITIZATION THROUGH INHIBITING P53–MURINE DOUBLE MINUTE 2 (MDM2) INTERACTIONS
- RADIOSENSITIZATION THROUGH DISRUPTING CELL CYCLE
- RADIOSENSITIZATION THROUGH DISRUPTING NICOTINAMIDE ADENINE DINUCLEOTIDE (NAD+) METABOLISM
- RADIOSENSITIZATION THROUGH BLOCKING IMMUNE CHECKPOINTS
- OUTLOOK
- DISCLOSURE
- Footnotes
- REFERENCES
- Figures & Data
- Info & Metrics
Related Articles
Cited By...
- No citing articles found.