


Abstract
The P2X7 receptor (P2X7R) is an adenosine triphosphate–gated ion channel that is predominantly expressed on microglial cells in the central nervous system. We report the clinical qualification of P2X7-specific PET ligand 18F-JNJ-64413739 in healthy volunteers, including dosimetry, kinetic modeling, test-retest variability, and blocking by the P2X7 antagonist JNJ-54175446. Methods: Whole-body dosimetry was performed in 3 healthy male subjects by consecutive whole-body PET/CT scanning, estimation of the normalized cumulated activity, and calculation of the effective dose using OLINDA (v1.1). Next, 5 healthy male subjects underwent a 120-min dynamic 18F-JNJ-64413739 PET/MRI scan with arterial blood sampling to determine the appropriate kinetic model. For this purpose, 1- and 2-tissue compartment models and Logan graphic analysis (LGA) were evaluated for estimating regional volumes of distribution (VT). PET/MRI scanning was repeated in 4 of these subjects to evaluate medium-term test-retest variability (interscan interval, 26–97 d). For the single-dose occupancy study, 8 healthy male subjects underwent baseline and postdose 18F-JNJ-64413739 PET/MRI scans 4–6 h after the administration of a single oral dose of JNJ-54175446 (dose range, 5–300 mg). P2X7 occupancies were estimated using a Lassen plot and regional baseline and postdose VT. Results: The average (mean ± SD) effective dose was 22.0 ± 1.0 μSv/MBq. The 2-tissue compartment model was the most appropriate kinetic model, with LGA showing very similar results. Regional 2-tissue compartment model VT values were about 3 and were rather homogeneous across all brain regions, with slightly higher estimates for the thalamus, striatum, and brain stem. Between-subject VT variability was relatively high, with cortical VT showing an approximate 3-fold range across subjects. As for time stability, the acquisition time could be reduced to 90 min. The average regional test-retest variability values were 10.7% ± 2.2% for 2-tissue compartment model VT and 11.9% ± 2.2% for LGA VT. P2X7 occupancy approached saturation for single doses of JNJ-54175446 higher than 50 mg, and no reference region could be identified. Conclusion: 18F-JNJ-64413739 is a suitable PET ligand for the quantification of P2X7R expression in the human brain. It can be used to provide insight into P2X7R expression in health and disease, to evaluate target engagement by P2X7 antagonists, and to guide dose selection.
The homomeric P2X7 receptor (P2X7R), a member of the purinergic family of receptors, is an adenosine triphosphate–gated ion channel expressed predominantly on macrophages and monocytes in the periphery and on microglia and astrocytes in the central nervous system (CNS). It has received particular attention because of its widespread involvement in inflammatory diseases as a key regulatory element of the inflammasome complex, with initiation and sustenance of the inflammatory cascade, and it plays an important role in CNS pathology (1). Activation of the P2X7R promotes nucleotide-binding domain–like receptor protein 3 inflammasome assembly and the release of neuroactive cytokines, such as interleukin 1β (IL-1β) and IL-18 (2). Within the CNS, IL-1β is thought to contribute to a neuroinflammatory diathesis that conceivably contributes to the pathophysiology of neuropsychiatric and neurodegenerative disorders (1,3). Although P2X7-mediated signaling has been most extensively studied in terms of its role in IL-1β regulation in the periphery, recent evidence indicates that CNS P2X7 activation also contributes to microglial activation, astrogliosis, and neuroinflammation. In addition to IL-1β, P2X7 antagonism in the brain may also dampen glutamate and chemokine release; this property may differentiate this mechanism from stand-alone IL-1β therapy (4).
Studies with genetic deletion and pharmacologic blockade of P2X7R have shown altered responsiveness in animal models for stroke (5), trauma (6), epilepsy (7), and multiple sclerosis and neurodegeneration (8) (e.g., amyotrophic lateral sclerosis (9,10), Alzheimer disease (11), Parkinson disease (12), and Huntington disease (13)) as well as mood disorders (14). All of these studies have stirred high interest in P2X7Rs as drug targets (15,16), and CNS-penetrating P2X7 antagonists appear to constitute a valid, novel therapeutic approach for treating CNS disorders (16).
In this context, a PET ligand for the P2X7R could improve understanding of the underlying pathophysiology in these CNS disorders, provide evidence of drug target engagement, and assist in clinical dose selection for novel drug therapies. Recently, several PET and SPECT ligands, including 11C-GSK1482160 (17), 11C-JNJ-54173717 (18), 18F-EFB (19), and 123I-TZ6019 and 11C-SMW139 (20,21), were evaluated preclinically. To date, no clinical studies using P2X7R radiotracer imaging have been reported.
We report the clinical qualification of 18F-JNJ-64413739, a selective P2X7 antagonist that was previously extensively evaluated in preclinical models as a candidate PET ligand for P2X7, showing high affinity and selectivity (22). This study includes the dosimetry of 18F-JNJ-64413739, tracer kinetic modeling optimization with test-retest variability (TRV) , and in vivo CNS blocking by pharmacologic doses of the P2X7 antagonist JNJ-54175446 (23,24).
MATERIALS AND METHODS
Details on the characteristics of 18F-JNJ-64413739 and JNJ-54175446 and on the synthesis and radiolabeling of 18F-JNJ-64413739 are given in the supplemental materials (supplemental materials are available at http://jnm.snmjournals.org). The safety, pharmacology, pharmacokinetics, metabolism, and toxicology of JNJ-54175446 were established without relevant concerns. Before the current study, previous phase 1 single-dose and multiple (ascending)-dose studies had been performed, without observation of severe or serious adverse events (25).
Study Design and Subjects
The 3 primary objectives of this open-label phase 1 study were to measure the whole-body (WB) distribution and radiation dosimetry of 18F-JNJ-64413739; to model the tissue-specific kinetics of 18F-JNJ-64413739 in the brain of healthy subjects with the appropriate metabolite-corrected arterial input function and to measure TRV by comparing PET scans obtained at least 1 wk apart; and, after the administration of a single oral dose of JNJ-54175446, to measure the blocking of 18F-JNJ-64413739 uptake in the brain at the time of maximum plasma concentration (Tmax) of JNJ-54175446 and to model the relationship between exposure and receptor occupancy for JNJ-54175446.
In total, 16 healthy subjects were included. All subjects were male, as the effects of sex and menstrual cycle on the expression of P2X7 are not known. Inclusion criteria and preparation of subjects before scanning are detailed in the supplemental materials. Corresponding demographic data and tracer information are summarized in Supplemental Table 1. Overall, the age of the subjects was 27 ± 7 y (mean ± SD) (range, 20–41 y), and the weight was 75.7 ± 11.9 kg (range, 53.0–96.2 kg). In terms of PET tracer dose, subjects received a dose of 145.2 ± 25.0 MBq (range, 87.5–196.1 MBq), with a corresponding injected mass of 1.56 ± 0.83 μg (range, 0.17–4.16 μg) and a specific activity of 54.36 ± 67.08 GBq/μmol (range, 14.16–421.64 GBq/μmol). The study was approved by the local ethics committee and was performed in accordance with the World Medical Association Declaration of Helsinki. All subjects signed an informed consent form before the study.
Biodistribution and WB Dosimetry
Three healthy male subjects (age, 21–25 y) underwent 10 consecutive WB PET/CT scans covering the WB tracer distribution for approximately 5 h after tracer administration. WB PET scans were acquired from the midfemoral position to the head in 3 segments (8 sequential WB scans from tracer injection to 70 min, a ninth scan at 150 min, and a 10th WB PET scan 270 min after tracer injection). Data acquisition and calculation of the normalized cumulated activities (NCA) or residence times are detailed in the supplemental materials. The NCA for the upper large intestine, lower large intestine, and small intestine were calculated using the International Commission on Radiological Protection (ICRP) 30 gastrointestinal model as incorporated in OLINDA v1.1 (26), with the fraction entering the small intestine as the input. This value was set to equal the decay-corrected plateau fraction of injected activity encompassed by the intestinal volume of interest (22%, 24%, and 15% for each of the 3 subjects, respectively).
On the basis of the NCA, absorbed doses were calculated using OLINDA v1.1 (26) according to current ICRP 60 definitions and using the MIRD scheme of a 73.7-kg adult male for each subject. The effective dose was calculated from the individual organ doses on the basis of predefined organ weighting factors, as specified by ICRP 60 in 1991 (27).
Tracer Kinetic Modeling and TRV
Five healthy male subjects (age, 20–38 y) underwent a dynamic PET/MRI scan combined with arterial blood sampling and tracer radiometabolite analysis. Retest scans were acquired for 4 of the 5 subjects (interscan interval, 26–97 d). Details of the image acquisition and reconstruction parameters for the GE Healthcare Signa PET/MRI data are given in the supplemental materials. Regional total distribution volume (VT) estimates were determined by applying a 1-tissue compartment model (1TCM) and a 2-tissue compartment model (2TCM) with a fixed blood volume of 5% and using the composite cortical time–activity curve to estimate potential small time shifts between the PET time–activity curve and the arterial blood and the plasma input functions. The Akaike information criterion was used to select the most appropriate model for estimation.
Next, a reduction in the acquisition time of dynamic PET scanning was evaluated on the basis of the most suitable kinetic model. Finally, the TRV and reliability of VT values determined with the preferred kinetic model and the shortest possible acquisition time interval were evaluated and compared with VT estimates using Logan graphic analysis (LGA) (28). The TRV and absolute TRV (aTRV) were assessed over the 4 test-retest datasets (supplemental materials). Next, the between-subject variability was assessed using the coefficient of variation of the average test-retest VT for each brain region across the 4 subjects, and reliability was evaluated using the intraclass correlation coefficient (ICC) (supplemental materials).
Single-Dose Occupancy Study
To determine the dose occupancy of JNJ-54175446, 8 healthy male subjects (age, 21–41 y) underwent baseline and postdose 18F-JNJ-64413739 PETMR scans. Postdose scans were performed at Tmax of 4–6 h after the administration of different oral doses of JNJ-54175446 (Supplemental Table 1) at least 7 d apart. The initial doses of JNJ-54175446 were selected on the basis of translational pharmacokinetic (PK)/pharmacodynamic modeling using PK and receptor occupancy data from PET studies in a rhesus macaque and translating the data to a human using a human population PK model (29) and adjusting for differences in plasma protein binding between a monkey and a human. During the study, the doses were adapted on the basis of the interim human scan data, resulting in a range of 5- to 300-mg single doses of JNJ-54175446 and the inclusion of 3 subjects per dose.
The same dynamic PET/MRI imaging protocol as that described earlier was used with a reduced acquisition time of 90 min and manual blood sampling (to avoid any clotting problems with the arterial line at dosing). Global P2X7 occupancy was estimated by analyzing baseline and postdose VT values using a Lassen plot, and the most suitable kinetic model was used to determine regional VT values. The corresponding dose occupancy curve was determined by nonlinear fitting of a 4-parameter log(dose) versus occupancy curve with a variable slope. Details of the fitted dose occupancy curve are given in the supplemental materials. For the PK analysis of JNJ54175446, blood samples were drawn just before the scan, at midscan, and at the end of the scan. The mean concentration of these 3 samples was then plotted against the P2X7 occupancy, and a sigmoid asymptotic maximum effect model was fitted through the data.
In addition to regional baseline and postdose VT values for dose and exposure occupancies, we also compared regional baseline and postdose K1 values to determine potential drug-induced effects on the rate of tracer uptake from arterial plasma to brain tissue. Finally, we compared the baseline and postdose VT values of the centrum semiovale to evaluate subcortical white matter as a potential reference region.
RESULTS
Biodistribution and WB Dosimetry
Figure 1 shows a representative coronal series of emission scans over time (subject 1 from Supplemental Table 1); the WB biodistribution and routes of excretion of the tracer and its radiometabolites indicate both hepatobiliary and urinary excretion. Table 1 shows the calculated NCA for all source organs with activity above the background, and Table 2 summarizes the individual organ doses of 18F-JNJ-64413739 for all subjects. Example fits for the time–activity curves are shown in Figure 2. In short, the gallbladder wall showed the highest organ dose (257 μGy/MBq); next were the urinary bladder (109 μGy/MBq), upper large intestine (89 μGy/MBq), and small intestine (78 μGy/MBq). The average effective dose was 22.0 ± 1.0 μSv/MBq, with a range of 20.9–22.6 μSv/MBq.
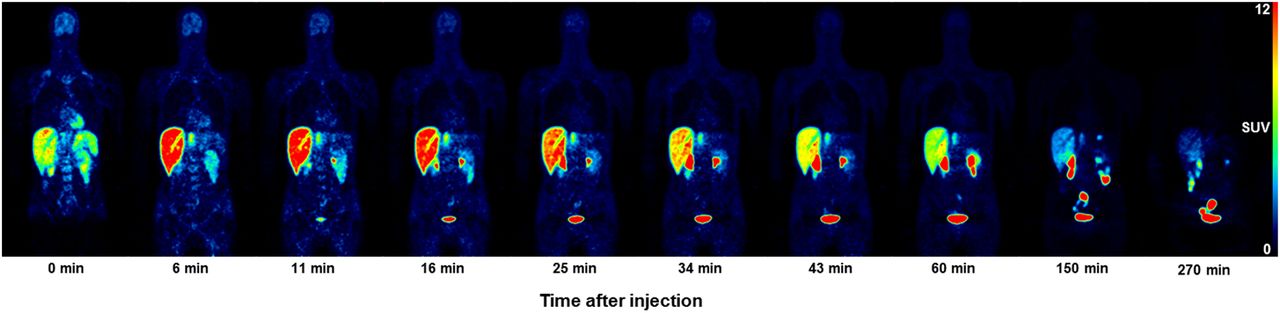
Whole-body time–activity distribution of 18F-JNJ-64413739 as function of the start time of the WB scan after tracer injection for representative coronal slices. PET data are presented as SUV. Lower rowindicates start of WB scan after tracer injection.
NCA of 18F-JNJ-64413739 for 12 Source Organs and Remainder (n = 3)
Radiation Absorbed Dose Estimates for 18F-JNJ-64413739 in 25 Organs (n = 3)*

Mean fractional activities for kidneys, urinary bladder, liver, and a gastrointestinal tract for all 3 subjects of part A (dosimetry study). %ID = percentage injected dose.
Tracer Kinetic Modeling and TRV
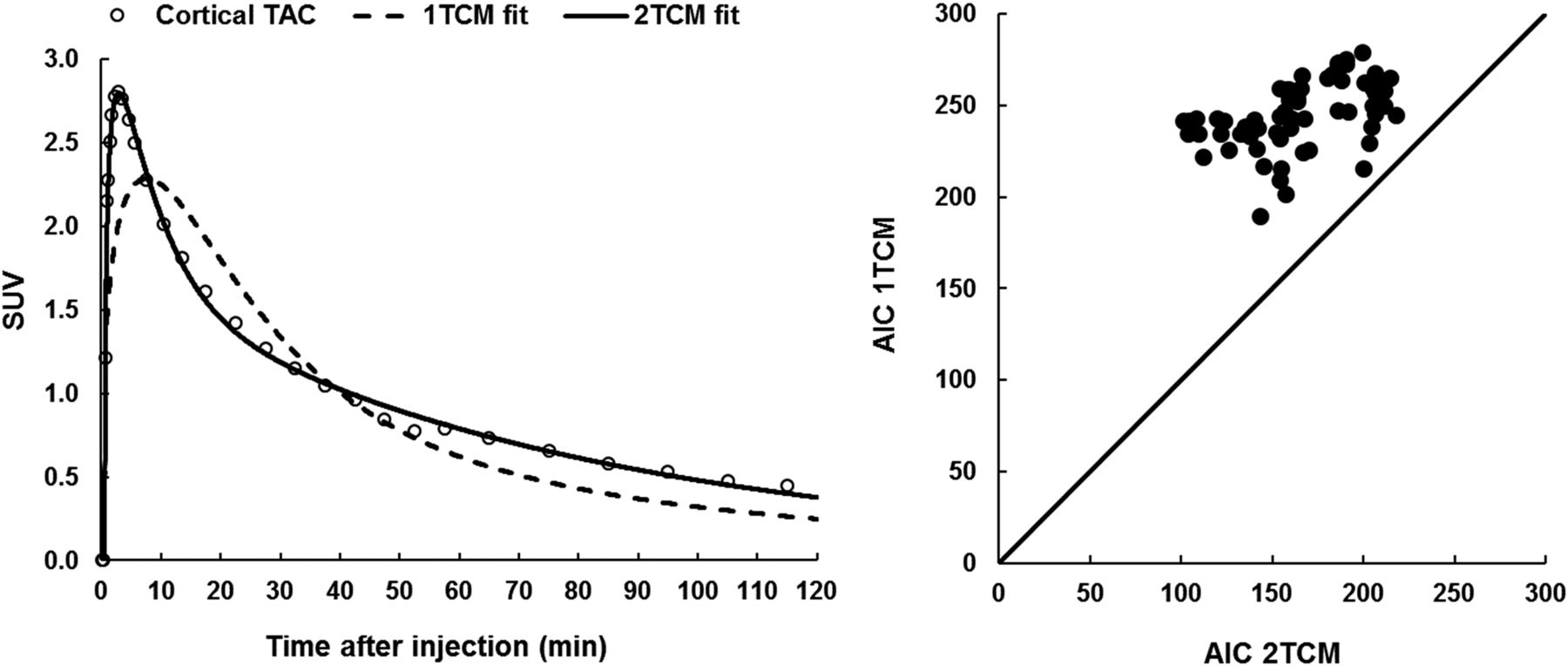
In terms of tracer kinetic modeling of 18F-JNJ-64413739 brain uptake, Akaike information criterion values were consistently lower for 2TCM than for 1TCM in all brain regions and all subjects, clearly demonstrating that 2TCM is the preferred model over 1TCM (Fig. 3). Representative 1TCM and 2TCM fits are shown in Figure 3 for the cortical time–activity curve. Therefore, we reported only 2TCM VT values in Table 3. Overall, 2TCM VT values were about 3, with a 2.5-fold range in VT values across subjects. Interregional variability was low across cortical and subcortical regions, with slightly higher values for the striatum, thalamus, and brain stem.

(Left) Representative 1TCM and 2TCM fits for baseline composite cortical time–activity curve (TAC) and corresponding arterial blood or plasma input function. (Right) 1TCM and 2TCM Akaike information criterion values for model fitting to baseline time–activity curves for different brain regions in 5 subjects.
Baseline 2TCM VT Values for Acquisition Times of 120, 90, and 60 Minutes and Baseline LGA VT Values for a 90-Minute Acquisition Time Interval (n = 5) for Different Brain Regions
In terms of time stability, we assessed the impact of using 90- and 60-min acquisition times instead of the full 120-min acquisition time. The results for 2TCM VT values (Table 3) showed a small average negative bias of −2.4% ± 2.6% (range, −9.3% to 0.5%) when the 90-min acquisition time interval was used and an average negative bias of −8.3% ± 5.8% (range, −25.2% to 2.3%) when the 60-min time interval was used (Supplemental Fig. 2). With the 90-min acquisition time interval, LGA VT values showed an average negative bias of −3.4% ± 2.4% (range, −10.8% to 0.9%) (Supplemental Fig. 2). Therefore, a 90-min acquisition time interval was considered acceptable for both 2TCM and LGA. A further reduction to 60 min resulted in a bias that was too high to be considered for clinical applicability.
The TRV and reliability of 18F-JNJ-64413739 brain PET imaging were assessed for 2TCM VT and LGA VT with a 90-min acquisition time interval. The findings (Table 4) for different brain regions indicated excellent reliability for both 2TCM VT and LGA VT, with ICCs being consistently much higher than 0.75. In terms of variability, an average aTRV of 12.1% ± 1.2% and an average TVR of 10.7% ± 2.2% were found for 2TCM VT across different brain regions, whereas for LGA VT, the average aTVR and TRV across brain regions were 14.2% ± 1.1% and 11.9% ± 2.2%, respectively. The between-subject variability values for 2TCM VT and LGA VT (percentage coefficient of variation) were 33.5% ± 2.2% and 32.7% ± 2.2%, respectively (n = 4).
Percentage TRV and aTRV, Between-Subject Variability, and Intraclass Correlation Coefficients for Regional VT Values of Test-Retest Datasets of 4 Subjects
Single-Dose Occupancy Study
On the basis of our findings so far, 2TCM VT values obtained with a 90-min acquisition time interval starting at tracer injection were used to estimate single-dose occupancies with a Lassen plot. Baseline 2TCM VT values are summarized in Supplemental Table 2 for comparison with baseline 2TCM VT values of the kinetic modeling and test-retest portion of the study. The 5- to 300-mg range of single doses of JNJ-54175446 resulted in occupancies of 12.5%–93.9% at Tmax (Supplemental Table 1). The corresponding dose occupancy and exposure occupancy curves are shown in Figure 4. The effective dose ED50 was 14.52 mg, and the half-maximal effective concentration EC50 was 44.3 ng/mL. There were no treatment-related effects on vital signs, electrocardiogram, or clinical chemistries in any of the 3 portions of the study. All adverse events were mild to moderate, did not require any intervention, and were most commonly related to study procedures.
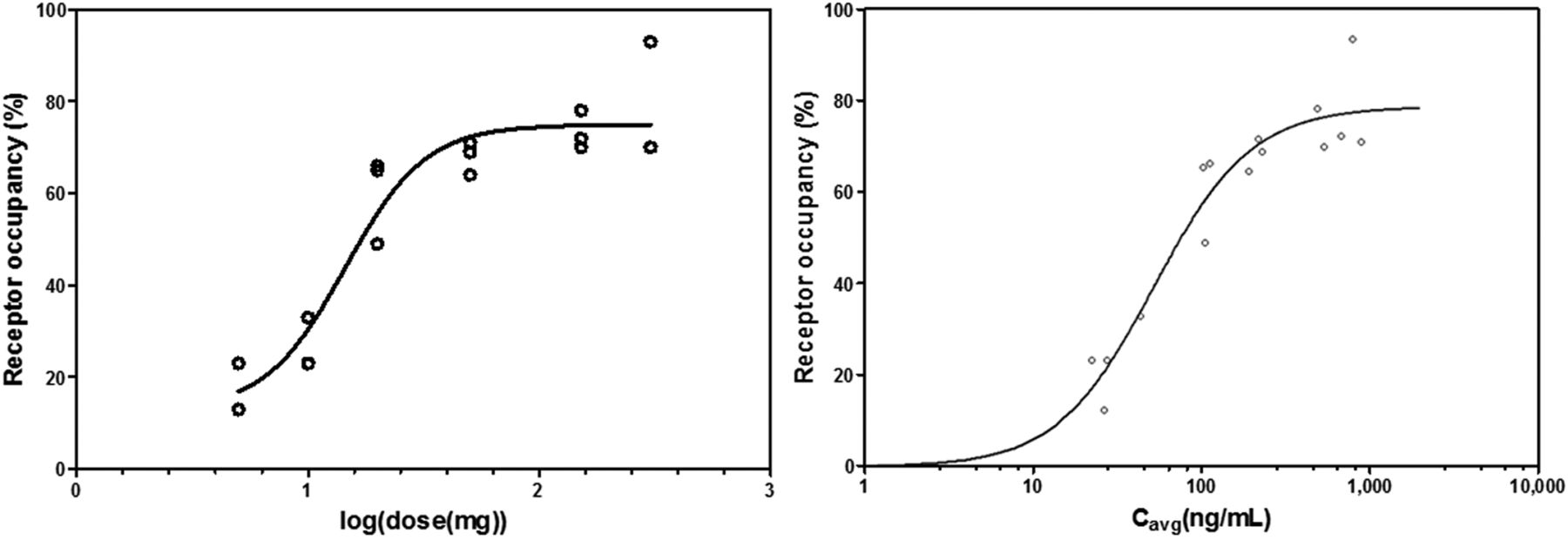
Dose occupancy and exposure occupancy curves based on baseline and postdose 18F-JNJ-64413739 2TCM VT values obtained with 90-min acquisition time interval and 5- to 300-mg single doses of JNJ-54175446. Occupancy values were estimated using Lassen plots. (Left) Dose occupancy curve determined by nonlinear fitting of 4-parameter log(dose) vs. response curve with variable slope. (Right) Exposure vs. occupancy curve representing P2X7R occupancy estimates as function of mean concentration (Cavg) for 3 plasma samples taken just before scan, at midscan, and at end of scan. Sigmoid asymptotic maximum effect model was used to fit curve through data.
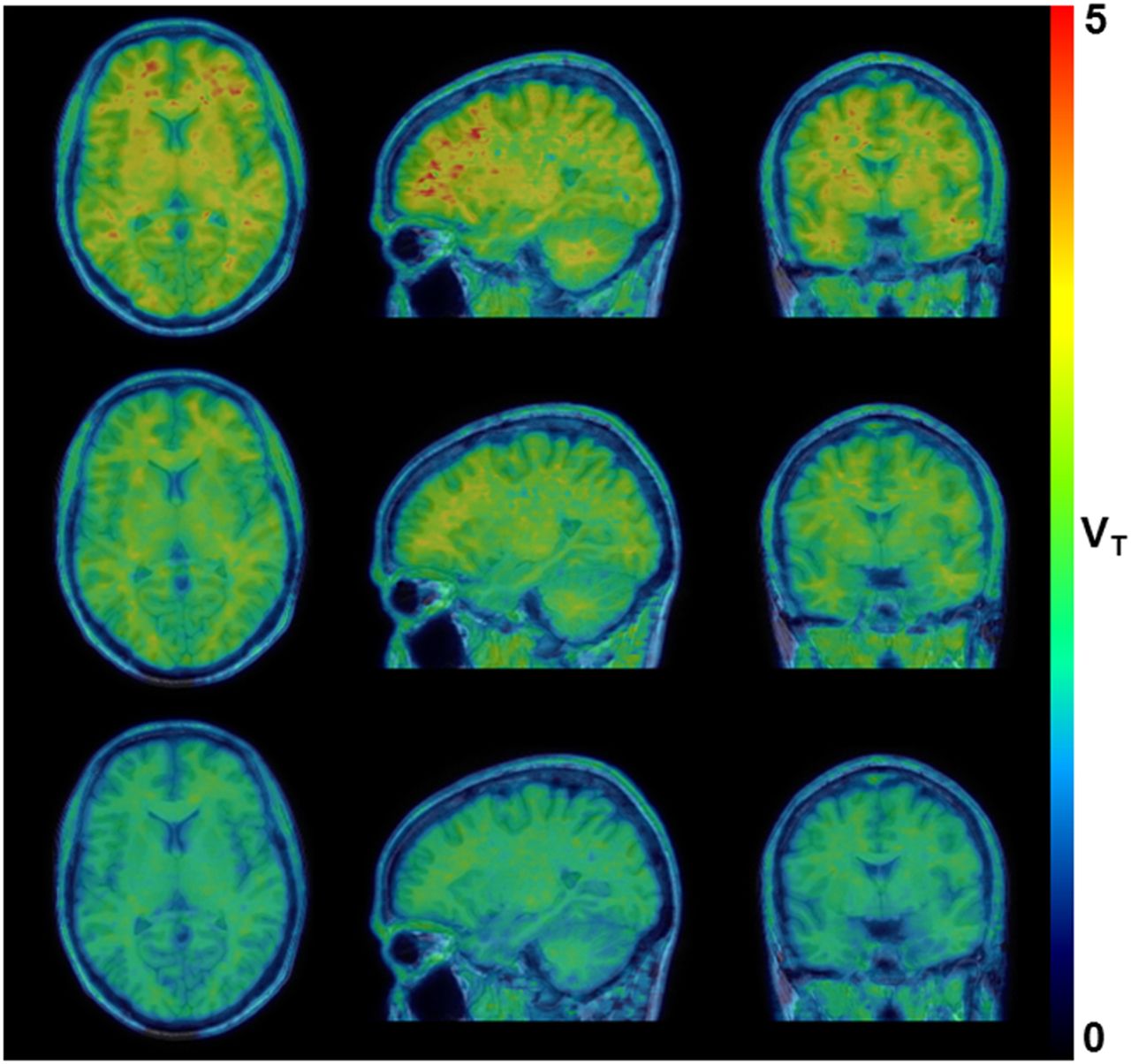
Figure 5 shows the parametric LGA VT datasets generated for a baseline 18F-JNJ-64413739 PET scan and the corresponding postdose 18F-JNJ-64413739 PET scans obtained 4 h after the administration of single doses of 20 and 50 mg of JNJ-54175446. These data clearly demonstrated a higher displacement of 18F-JNJ-64413739 binding with a corresponding higher dose of JNJ-54175446 in all brain regions, including white matter, as the baseline VT (mean ± SD, 5.34 ± 1.41; range, 3.74–7.42) and postdose VT (mean ± SD, 3.23 ± 1.29; range, 2.11–6.85) values for the centrum semiovale were significantly different (Wilcoxon signed rank test; P < 0.05). Regional baseline K1 (mean ± SD, 0.24 ± 0.04; range, 0.15–0.35) and postdose K1 (mean ± SD, 0.23 ± 0.04; range, 0.13–0.38) values were analyzed using a 2-way repeated-measures ANOVA to assess a possible drug effect. No significant interaction was observed between brain region and dosing, and Bonferroni post hoc tests did not show a significant difference between regional baseline and postdose K1 values (P > 0.05).
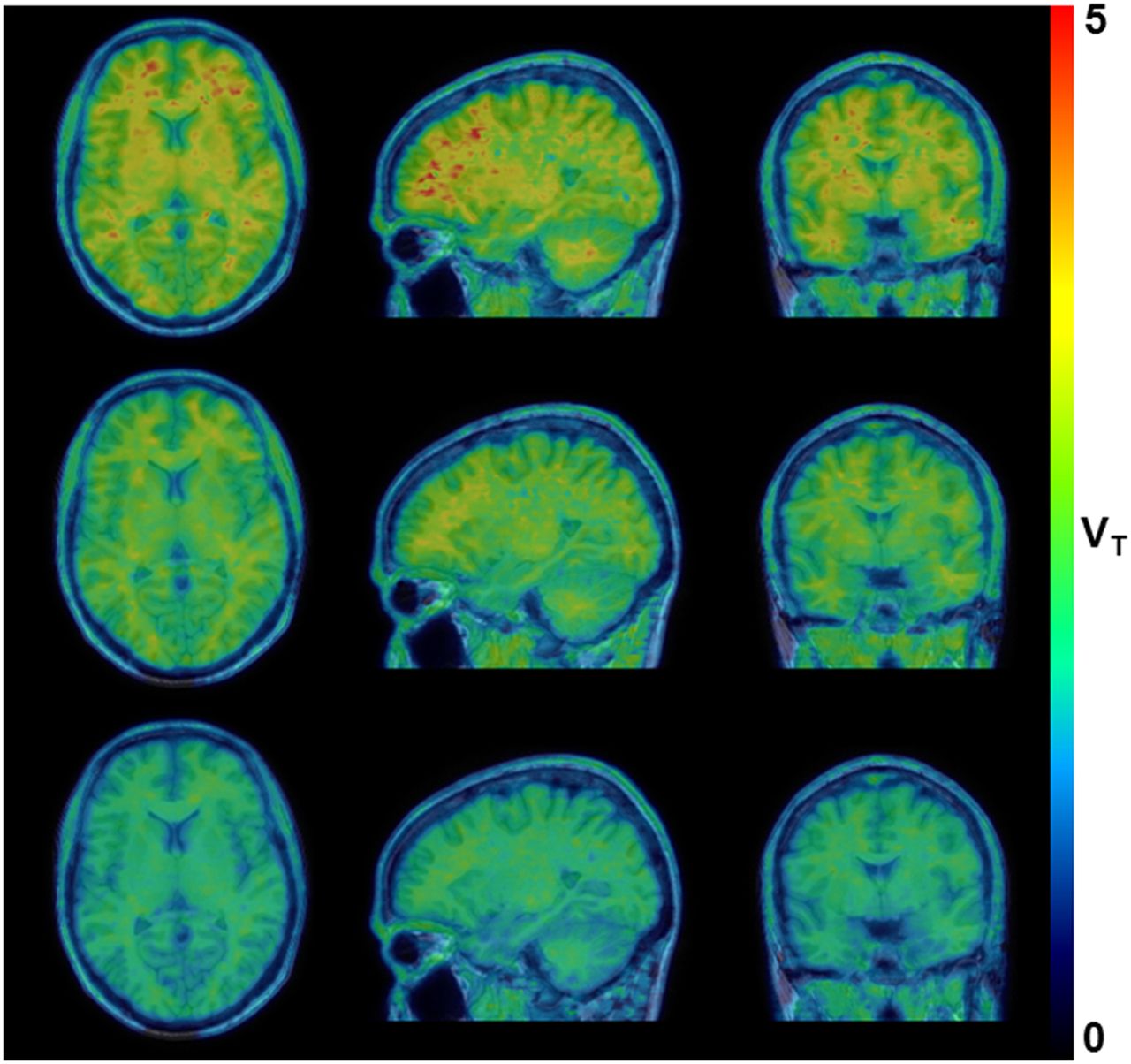
Representative parametric LGA VT dataset for baseline 18F-JNJ-64413739 PET scan (top row) and postdose 18F-JNJ-64413739 PET scan obtained 4 h after dosing with 20-mg (middle row) and 50-mg (bottom row) single doses of JNJ-54175446.
DISCUSSION
To our knowledge, the present study is the first biodistribution, brain kinetic modeling, and dose occupancy study in humans with P2X7 as a target. The biodistribution of 18F-JNJ-64413739 showed an excretion pattern of combined hepatobiliary clearance and renal clearance. No organs were found to have abnormally high specific uptake. The effective dose corresponded to 22.0 ± 1.0 μSv/MBq, which is within the typical range of 15–30 μSv/MBq for 18F-labeled radiopharmaceuticals (30). Thus, subjects participating in the dosimetry portion of the study received a total effective dose of 4.1 ± 0.3 mSv for the PET scan. The gallbladder was the organ with the highest absorbed dose, receiving a maximal dose of approximately 55 mGy.
The kinetic modeling results clearly identified 2TCM as the preferred model for 18F-JNJ-64413739 brain PET quantification and demonstrated that acquisition time could be reduced to 90 min with only a limited impact on VT bias and variability. Although differences were very limited, LGA VT values were slightly lower than 2TCM VT values, in line with literature data reporting a noise-induced underestimation of VT values by a Logan plot. However, these results supported LGA as a valid approach for generating parametric 18F-JNJ-64413739 VT maps. The regional distribution of VT values was very similar for cortical and subcortical regions. For white matter, tracer uptake should be attributed partially to specific binding, as postdose white matter VT values were significantly lower than baseline values. There is indeed direct immunohistochemical evidence that P2X7Rs also are present on white matter astrocytes, oligodendrocytes, and axonal myelin sheets, where they are involved in Ca2+ signaling (31,32). This macroscopic finding of significant displaceable binding excluded white matter as a candidate reference tissue for noninvasive 18F-JNJ-64413739 brain PET quantification.
TRV values were in line with previously published test-retest data for translocator protein (TSPO)–specific PET ligands and other PET ligands that do not allow for a reference tissue approach but require arterial blood sampling for accurate quantification (33,34). On the other hand, the between-subject variability of baseline VT values was relatively high, with a coefficient of variation of more than 30%. This variability of baseline VT values in the kinetic modeling and TRV portion of the study was driven mainly by the low VT values for 1 subject and was confirmed by the retest scan. Meanwhile, the variability of baseline VT values in the single-dose occupancy study was lower by a factor of 2 in comparison to the kinetic modeling and test-retest portion of the study.
So far, the influence of genetic polymorphisms on receptor expression levels or tracer binding properties are unknown. Interestingly, several human genetic studies have associated the highly polymorphic P2X7R gene with mood disorders (35–37), and some of these mutations have been linked to the modulation of P2X7 channel function in vitro (38). Therefore, whether P2X7R polymorphisms also may account for part of the variability of baseline P2X7 expression and therefore may render between-group comparisons of baseline P2X7 availability more challenging needs to be further investigated. However, the reliability of 18F-JNJ-64413739 VT quantification was very good, as ICCs of ≥0.75 are considered indicators of good reliability. On the other hand, the number of test-retest scans was limited, and the high between-subject variability compared with the intrasubject variability contributed to the high ICCs.
The dose occupancy study demonstrated a clear dose-response relationship for JNJ-54175446, in line with preclinical monkey and ex vivo P2X7R occupancy studies with this compound (29).
CONCLUSION
The novel radioligand 18F-JNJ-64413739 has appropriate properties for clinical PET imaging of the P2X7R. Its biodistribution shows sufficient brain uptake and favorable dosimetry. Full kinetic modeling identified 2TCM as the most suitable model for 18F-JNJ-64413739 brain PET quantification, although no reference region could be identified. A dynamic acquisition of 90 min provided limited bias, good TRV, and excellent reliability. Therefore, 18F-JNJ-64413739 proved to be a suitable PET ligand for the quantification of P2X7Rs in the human brain; for study of the pathophysiologic aspects of P2X7R involvement in neuroinflammation, neurodegeneration, and mood disorders; or for assessment of the drug occupancy of brain-permeating P2X7 antagonists that are currently used in clinical trials. Future studies are needed to determine whether P2X7 polymorphisms account for the high interindividual signal variability across subjects.
DISCLOSURE
This phase 1 PET study was sponsored by Janssen Pharmaceutica. Koen Van Laere is Senior Clinical Investigator of the Fund for Scientific Research, Flanders, Belgium (FWO). Mark E. Schmidt, Anja Hijzen, Paulien Ravenstijn, Marc Ceusters, Wei Zhang, Luc Van Nueten, and Hartmuth Kolb are employees of Janssen Research and Development. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Janssen Research and Development scientists Anindya Bhattacharya and Peter De Boer for guidance and suggestions regarding the biology of P2X7R and clinical applications and Xin Miao for translational PK/pharmacodynamic modeling used to support dose selection for the study. We explicitly want to thank Kwinten Porters and Jef Van Loock for contributions to scanning and data handling; the PET radiopharmacy and the medical physics group for skilled contributions and support; Jolien Laenen for contributions in recruitment, practical organization, and data handling; and Els Ampe for contributions in the pharmacy aspects of JNJ-54175446.
Footnotes
Published online Sep. 27, 2018.
- © 2019 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication June 28, 2018.
- Accepted for publication September 18, 2018.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}