


Abstract
Peptide receptor radionuclide therapy (PRRT) is an established treatment for nonoperable or metastatic neuroendocrine neoplasms that highly and frequently express somatostatin receptors. More generally, PRRT is an attractive therapy option for delivering cytotoxic radiation to tumor cells through specific binding of a radiolabeled peptide to a molecular target. The development of imaging companions gave rise to the concept of radiotheranostics, important for in vivo tumor detection, characterization, and staging but also, and more importantly, for individual patient selection and treatment. The success of somatostatin receptor targeting paved the way for the clinical translation of other peptide-based radiopharmaceuticals targeting, for example, the receptors cholecystokinin 2, gastrin-releasing peptide, neurokinin-1, and C-X-C motif chemokine 4. Although historically the Auger emitter 111In and the high-energy β− emitter 90Y were used, most PRRT are currently performed with the medium-energy β− emitter 177Lu, whereas α emitters are increasingly studied in various clinical applications.
- targeted radionuclide therapy
- theranostics
- G-protein coupled receptors
- beta radiation therapy
- alpha radiation therapy
Peptide receptor radionuclide therapy (PRRT) is part of the wider concept of targeted radionuclide therapy. PRRT delivers destructive radiation to cancer cells via radiolabeled peptides able to bind specifically to peptide receptors expressed in higher density on the tumor cell membrane than in nontumor tissues. This is the case for many G-protein–coupled receptors, whose overexpression is linked to numerous human malignancies. Regulatory peptides targeting G-protein–coupled receptors are a rich source of vectors (1) that can be chemically tuned to transport radioactivity while preserving their receptor affinity. PRRT is used in the treatment of metastatic or unresectable cancers through systemic or, more occasionally, locoregional administration.
Three types of radiation are used in PRRT: β− particles (electrons), α particles, and Auger electrons (2), with a strong focus on β− emitters (e.g., 177Lu and 90Y). Because β particles have a long range in tissues (0.05–12 mm), neighboring cells around the targeted cell are also irradiated (cross-fire effect). This effect is considered ideal for targeting large tumors with a heterogeneous target distribution. In contrast, α particles (e.g., 213Bi and 225Ac) have a very short range in tissues (20–100 μm), irradiating volumes with cellular dimensions and therefore sparing normal surrounding tissues from cytotoxic radiation. Their linear energy transfer is much higher than the one of β− particles (50–230 vs. 0.2 keV/μm), making α radiation far more cytotoxic. Finally, Auger emitters (e.g., 111In), having a very short range in tissue (<20 μm, subcellular dimensions) and intermediate linear energy transfer (4–25 keV/μm), may be better suited for targeting microscopic disease (i.e., micrometastases), such as in the adjuvant setting. All above-mentioned radionuclides are radiometals (Table 1).
Radionuclides Used for PRRT in Patients
Peptide-based radiotherapeutics require a chelator that stably chelates the radiometal in vivo (3), in order to exclude deposition of free radiometal in normal tissues. The conjugation of a universal chelator, such as DOTA, allows radiolabeling with different radiometals sharing similar chemical properties, such as the trivalent radiometals 90Y, 177Lu, 225Ac, or 68Ga (used in PET). This versatility enabled the development of radiotheranostics, which are essential for selecting patients who are suitable for PRRT. Such an approach may provide more efficient and personalized patient care, because properties of the vector, type of radionuclide, and dose of radioactivity can be tailored to address specific clinical needs.
Radiolabeled somatostatin analogs have been archetypal for the development of PRRT, since neuroendocrine neoplasms (NENs) strongly and frequently overexpress somatostatin receptor (sstr). This article discusses new approaches to overcoming current limitations, improving treatment outcome—considering that the objective response is still limited and few patients can be cured—and extending the application of PRRT beyond NENs. Also, recent developments in PRRT targeting the receptors cholecystokinin 2, gastrin-releasing peptide (GRPR), neurokinin-1, and C-X-C motif chemokine 4 (CXCR4) are presented. In addition, targeting of the prostate-specific membrane antigen (PSMA) with radiolabeled PSMA inhibitors has been recently a major breakthrough in targeted radionuclide therapy of prostate cancer. Strictly speaking, PSMA ligands are not peptides but peptidomimetics and PSMA is not a receptor but a type II transmembrane protein; therefore, PSMA-targeted radionuclide therapy is not discussed here.
SOMATOSTATIN IN NEN AND BEYOND
More than 20 y after the pioneering work in isolated centers in Europe such as Rotterdam, Basel, and Milan, PRRT with radiolabeled sstr agonists (e.g., DOTATOC or DOTATATE, Table 2) is part of the standard of care for NENs (4). Recently, the randomized phase III study NETTER-1 showed an improved objective response, an improved quality of life, improved progression-free survival, and a clear trend toward an overall survival benefit from PRRT with 4 cycles of 177Lu-DOTATATE (plus long-acting-release single-dose octreotide) compared with long-acting-release double-dose octreotide alone (5,6). Consequently, 177Lu-DOTATATE (177Lu-oxodotreotide) received marketing authorization for patients with metastatic and progressive midgut NEN. It is expected that approval of 177Lu-DOTATOC (177Lu-edotreotide) will follow completion of the COMPETE-NCT03049189 phase III trial comparing 177Lu-DOTATOC with a mammalian target of rapamycin inhibitor, everolimus, in patients with inoperable, progressive gastroenteropancreatic NEN. These and other trials (e.g., OCCLURANDOM-NCT02230176) should more precisely determine the position of PRRT in the current clinical algorithm with regard to other systemic therapies, such as everolimus and sunitinib.
Peptide Analogs Used for PRRT in Patients
Routes other than intravenous administration may be interesting for enhancing the therapeutic and safety window of PRRT. NENs and liver metastases are often highly perfused, and the intraarterial route can exploit the first-pass effect to more efficiently treat liver-dominant disease. Such an approach can also be used for inoperable primary tumors to downstage the disease in the neoadjuvant setting (7,8). However, large comparative prospective trials, to support its wider use, are lacking.
New Somatostatin Analogs
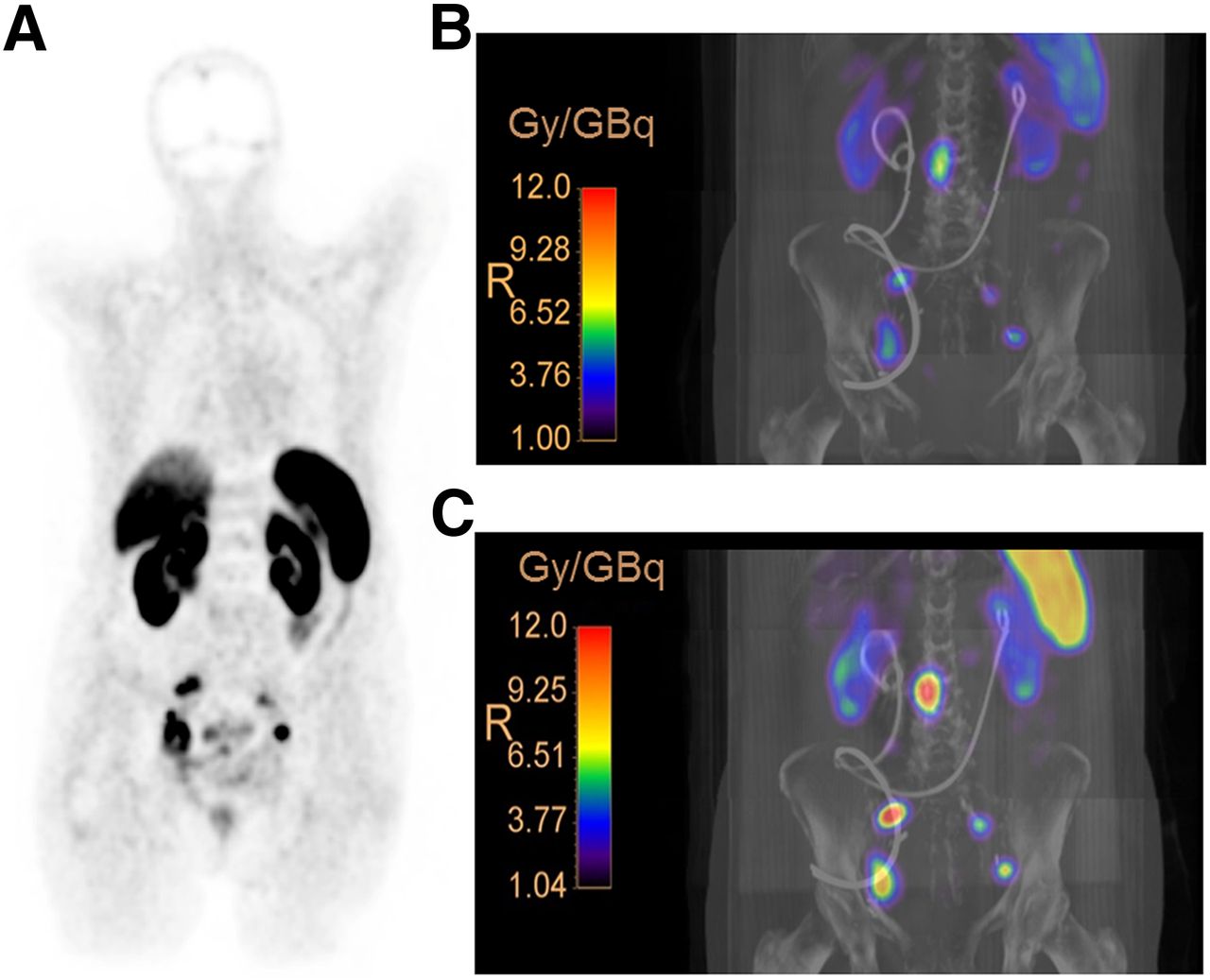
An important evolutionary step is the use of radiolabeled sstr antagonists instead of agonists. Several clinical and preclinical studies (9) suggest significant improvement in the diagnostic sensitivity and therapeutic efficacy of the antagonists. 177Lu-satoreotide tetraxetan (177Lu-OPS201 or 177Lu-DOTA-JR11 [Ipsen], Table 2) shows higher tumor accumulation and absorbed dose (Fig. 1) and higher numbers of DNA double-strand breaks than 177Lu-DOTATATE (10–12). These characteristics are partly due to the higher number of available binding sites and the longer tumor residence time for the antagonist. 177Lu-OPS201 is currently under evaluation in a multicenter phase I/II study (NCT02592707) and in a single-center theranostic study (NCT02609737). Also, 68Ga-satoreotide trizoxetan (68Ga-OPS202 or 68Ga-NODAGA-JR11 [Ipsen], Table 2), showing improved affinity for sstr compared with 68Ga-OPS201, demonstrated substantially higher sensitivity than the agonist 68Ga-DOTATOC in a prospective phase I/II study involving 12 gastroenteropancreatic neuroendocrine tumor patients (13).

Patient with neuroendocrine neoplasia (G3) of bladder with lymph node metastases. (A) 68Ga-DOTATATE PET scan demonstrates progression with pelvic lymph node metastases after surgery. (B and C) Three-dimensional voxel dosimetry after administration of ∼1 GBq of 177Lu-DOTATATE (B) and 177Lu-OPS201 (C) demonstrates increased (∼4 times) tumor dose of 177Lu-OPS201, compared with 177Lu-DOTATATE, implying potential for greater therapeutic efficacy. (Courtesy of Prof. Damian Wild; patient was included in previously reported pilot study (10).)
By introducing the albumin-binding moiety Evans blue, 177Lu-DOTA-EB-TATE was developed as a long-circulating sstr agonist (14). In 8 patients, its prolonged blood circulation resulted in a 7.9-fold increase in tumor dose, compared with 177Lu-DOTATATE, but at the cost of an even greater increase in renal and bone marrow absorbed doses, questioning its potential advantage over 177Lu-DOTATATE.
Use of multivalent or heterovalent vectors to simultaneously target several receptors concomitantly expressed in the same cancer cell is an interesting approach to overcoming tumor heterogeneity, resistance, and change in phenotype during disease progression (15).
New Indications (Non-NEN)
The larger number of binding sites recognized by sstr antagonists may open new horizons for sstr-targeted PRRT. In some nonneuroendocrine neoplasias, the level of binding of antagonists reaches the level of agonists (e.g., 177Lu-DOTATATE) in well-differentiated NEN (16). Indications such as metastatic breast and small cell lung cancers are currently investigated in second line in a basket multicenter phase I/II trial with the theranostic pair 68Ga-OPS202/177Lu-OPS201 (NCT03773133).
Beyond oncologic indications, initial reports suggest a potential role for sstr-targeted PRRT in inflammatory conditions, such as sarcoidosis and atherosclerosis (17,18). In a feasibility study involving only 2 patients with refractory multiorgan involvement of sarcoidosis, 177Lu-DOTATOC showed treatment effects (17), whereas a retrospective analysis of a limited number of oncology patients indicated that 177Lu-DOTATATE reduces atherosclerotic plaque activity (18).
α-Therapy
213Bi-DOTATOC was investigated in 25 metastatic NEN patients progressing after 90Y-/177Lu-DOTATOC therapy (activities ranging from 2.6 to 21 GBq in 1–5 cycles). An interim report on the first 7 patients (8) showed moderate renal and hematologic toxicity, but long-term follow-up data are still pending. 213Bi-DOTATOC treatments resulted in a high proportion of long-lasting antitumor responses, including one complete remission, even with large-volume disease (Fig. 2). However, the current supply limitations for high-activity 225Ac/213Bi generators prevent larger confirmatory prospective studies and have instead motivated the use of 225Ac or 212Pb, which are β− emitters but act as an in vivo generator for the α emitter 212Bi. 212Pb-DOTAMTATE (AlphaMedix; RadioMedix, Inc.) has just entered a phase I study (NCT03466216), and 225Ac-DOTATOC has been administered to 40 patients with progressive NEN (19). The maximum tolerated dose of 225Ac-DOTATOC was established at 40 MBq in a single fraction and 25 MBq in 2 fractions at a 4-mo interval (19). Larger prospective comparative studies are needed to identify the most suitable patients for 225Ac-DOTATOC therapy. Importantly, translocation of radioactive daughter nuclides from the chelator should be considered a potential safety hazard for α emitters with multiple α-emitting daughters.

68Ga-DOTATOC PET/CT images before (left) and after (right) intraarterial therapy with 14 GBq of 213Bi-DOTATOC, demonstrating response of multiple liver lesions. (Reprinted from (8).)
Combination Therapy
Combined use of various radionuclides (e.g., 90Y and 177Lu), sequentially or concomitantly, might optimize radiation delivery across the entire spectrum of lesion size that is often present in one individual. This approach has shown some potential for increasing survival in comparison to the single-radionuclide approach (20).
Integration of PRRT into multimodality-therapy protocols might improve treatment response (21). The use of radiosensitizing chemotherapeutics in combination with 90Y- and 177Lu-DOTATATE has shown additive effects resulting in an unprecedented objective response (22). This may particularly be interesting in bulky, higher-grade, or 18F-FDG–avid NENs, in which the potential risk of long-term hematologic toxicity is balanced by the more aggressive course of the disease. Prospective clinical trials combining PRRT and chemotherapy are under way (NCT02736448, NCT02358356) with agents such as 5-fluorouracil, capecitabine, and temozolomide.
Molecularly targeted therapeutics may be important to attain synergy with PRRT. For instance, concurrent inhibition of the DNA repair mechanisms with a poly-[ADP-ribose]-polymerase 1 inhibitor results in increased DNA double-strand breaks (23).
GASTRIN IN MEDULLARY THYROID CANCER
Virtually all medullary thyroid cancers express the cholecystokinin 2 receptor, for which the endogenous ligand is gastrin. PRRT with radiolabeled gastrin analogs is therefore an attractive treatment option for patients with recurrent or metastatic medullary thyroid cancer. Of large libraries, the analogs 177Lu-PP-F11N (Table 2) and 111In-CP04 (PP-F11, Table 2), as a surrogate for 177Lu- or 90Y-labeled CP04, are currently in early prospective trials (LUMED/NCT02088645 and GRAN-T-MTC/NCT03246659, respectively) to determine safety, maximum tolerated dose, biodistribution, and dosimetry (24,25). Preliminary clinical data identified the stomach as a potential dose-limiting organ, whereas the renal absorbed dose was rather low.
Promising perspectives involve the use of adjuvant protease inhibitors to prevent enzymatic tracer degradation in vivo and improve the tumor uptake and pharmacokinetics of enzymatically vulnerable radiolabeled gastrin analogs (26).
BOMBESIN IN PROSTATE AND BREAST CANCER
GRPR is overexpressed in, among other malignancies, prostate and breast cancers. The 68Ga-labeled GRPR antagonists RM2 and NeoBOMB1 (Table 2) are currently under clinical evaluation in prostate cancer and gastrointestinal stromal tumor. Treatment in humans has not been reported yet but has been evaluated in animal models (27,28). Interestingly, various ongoing studies compare 68Ga-GRPR antagonists with 68Ga-PSMA analogs (29) (NCT03604757, NCT03606837, NCT03698370), which is essential to understand the role of each radiotracer in the management of prostate cancer patients. The fact that GRPR and PSMA seem to be expressed at different stages of the disease, and that the biodistribution of the radiotracers is fundamentally different, may result in 2 complementary rather than competitive theranostic approaches.
Radiolabeled GRPR antagonists may also have a potential in estrogen receptor–positive breast tumors, which represent most breast cancer patients (30).
SUBSTANCE P IN GLIOMA
PRRT of gliomas has been investigated with the substance P analog DOTA-/DOTAGA-SP (Table 2) (31–34), targeting the neurokinin-1 receptor that is overexpressed in World Health Organization grade II–IV gliomas. To overcome the blood–brain barrier and to improve tumor uptake, administration is conducted locoregionally via an implanted catheter connected to a subcutaneous port.
Initial results with 90Y and 177Lu indicated clinical potential (31,32); however, recent studies focused on the α emitters 213Bi and 225Ac, allowing more selective tumor cell irradiation and limiting toxicity to adjacent healthy brain tissue. 213Bi-DOTA-SP has been examined in 61 patients with grade II–IV gliomas (up to a cumulative activity of 14.1 GBq) (33,34). A subgroup analysis in 7 patients with secondary glioblastoma showed a median overall survival of 18.6 mo after conversion to grade IV (33). In another subgroup of 20 patients with recurrent glioblastoma, a median overall survival of 23.6 mo was found, compared with 14.6 mo after standard therapy alone (34). Treatment with the longer-lived 225Ac has been initiated and 20 glioma patients have been enrolled in a dose escalation study (from 10 to 42 MBq) investigating the intratumoral or intercavitary injection of 225Ac-DOTAGA-SP (34). The analysis of therapeutic efficacy and patient recruitment are ongoing.
CXCR4 IN CANCER
CXCR4 is an attractive target for theranostic interventions since it is overexpressed in hematologic malignancies, such as multiple myeloma, leukemia, and non-Hodgkin lymphoma, and in some solid cancers (e.g., lung cancer, adrenocortical cancer, and high-grade NEN). Many high-affinity ligands targeting CXCR4 have been developed, among which the theranostic pair 68Ga-pentixafor/177Lu/90Y-pentixather is the most advanced one (Table 2) (35). Preclinically, efficient eradication of CXCR4-positive human leukemic blasts was shown in a patient-derived xenograft model of acute lymphocytic leukemia (36). In a pilot study, 8 multiple myeloma patients (37) were treated with 177Lu- or 90Y-labeled pentixather (7.6–23.5 GBq or 2.6–6.3 GBq, respectively) and showed a high initial response rate (37).
Given the fact that hematologic cancers are strongly disseminated diseases, 177Lu might not be the optimal radionuclide, and current efforts are directed toward implementing α therapy using 213Bi-/225Ac-pentixather. Thorough evaluation of hematologic safety is indispensable because of the physiologic expression of CXCR4 in hematopoietic stem cells.
FUTURE PERSPECTIVES
Despite the great advances that PRRT represents in the management of NEN, 177Lu-oxodotreotide (also known as 177Lu-DOTATATE and Lutathera [Advanced Accelerator Applications]) is currently the only approved peptide-based radiotherapeutic agent. More effort is needed to expand PRRT beyond NENs, which involves the development of new radiotheranostics and the validation of new targets. Additionally, tumor-specific upregulation of targets by pharmacologic or epigenetic interventions, as well as the use of heterovalent ligands, may be valuable options to enhance tumor uptake and overcome resistance. Furthermore, the use of alternative theranostic pairs of radionuclides, such as radioisotopes of scandium (43/44/47Sc) and terbium (149/152/155/161Tb) (38), might open novel theranostic applications. To further optimize the potential of various radionuclides and to promote synergistic combination therapies, a deeper understanding of all relevant radiobiologic processes is required, including DNA-repair mechanisms and on-target and off-target (bystander and abscopal) effects (39). Finally, more precise image-based dosimetry, to establish dose–effect relationships, and development of biomarkers, such as multitranscript gene blood assays (40), are expected to improve the prediction of outcome after PRRT.
DISCLOSURE
No potential conflict of interest relevant to this article was reported.
Footnotes
Published online Dec. 20, 2018.
- © 2019 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication November 5, 2018.
- Accepted for publication December 14, 2018.





{kind=link}
{kind=link}