


Abstract
In recent years, several new radiotracers and radionuclide therapies have been developed. There is a renaissance in nuclear medicine and molecular imaging today in terms of, for example, the ability to image and treat neuroendocrine and prostate malignancies. To be able to bring a new drug product from bench to bedside and assist patients while also ensuring patient safety, stringent regulations must be met. However, differences in regulatory requirements, often based on jurisdictional politics rather than scientific evidence, can hinder global cooperation, increase expense, and slow progress. In an effort to rise above these differences, nuclear medicine professional organizations, regulators, and international agencies have begun to identify commonalities in the regulations to achieve harmonization. Indeed, a more streamlined approach to radiopharmaceutical drug development across jurisdictions could be achieved through establishing harmonized requirements for preclinical studies and manufacturing standards. This paper provides an educational overview of the regulatory and submission requirements governing investigational radiopharmaceuticals for first-in-human radiopharmaceuticals across the European and North American continents. It is hoped that through ongoing collaboration, regulatory reform and harmonization can become a reality and speed access to the most up-to-date evidence-based patient care for all.
The U.S., European Union (EU), and Canadian regulatory frameworks for human-use applications of radiopharmaceuticals continue to be a major barrier to widespread, affordable patient access to important advances in radiopharmaceutical development. Radiopharmaceuticals are diagnostic imaging agents with unique characteristics, including an extremely small mass dose (typically in the nanogram to microgram range), no pharmacologic effects, a very low incidence of adverse events, and a short shelf-life. This article outlines each country’s regulations and guidelines, focusing on first-in-human use of radiopharmaceuticals before clinical development with full regulatory approval, and suggests possible harmonization enabling more cost-effective and efficient approval.
Problems with access to new radiopharmaceuticals have long been recognized by regulators. In 2004, the Food and Drug Administration (FDA) raised concerns that excessive development costs were preventing new, affordable life-saving drugs from reaching patients (1). That same year, the European Medicines Agency (EMA) published a position paper defining a single dose of a pharmacologically active compound using microdose techniques, with the microdose defined as less than 1/100th the dose calculated to yield a pharmacologic effect, at a dose of no more than 100 μg (2).
The EMA paper encompassed exploratory clinical trials undertaken with a single test drug or several closely related candidates. In 2006, the FDA accepted this definition and published its “Guidance for Industry, Investigators, and Reviewers: Exploratory IND Studies,” adding the definition for a protein product of no more than 30 nanomol (3). The International Council for Harmonization (ICH) guidance M3(R2) on nonclinical safety studies followed, incorporating the same definition, and is used by the FDA, EMA, and Health Canada (4). In August 2018, a new FDA guidance, “Microdose Radiopharmaceutical Diagnostic Drugs: Nonclinical Study Recommendations,” was issued, expanding the guidance to include traditional investigational new drug (IND) phase 2–3 studies (5).
Previously, toxicology studies were performed in laboratories complying with good laboratory practice (GLP); however, recent U.S. changes through pre-IND meetings with the FDA have allowed these studies to be performed in other types of controlled laboratories, such as university comparative anatomy or veterinary medicine departments, which can further reduce nonclinical costs. Although this FDA ruling is not recognized by European or Canadian authorities, a recent position paper from the European Association of Nuclear Medicine (6) and the EMA plans for specific radiopharmaceutical guidance (7) that will hopefully lead to more harmonized and rational approaches to preclinical safety data for new radiopharmaceuticals.
Europe’s new regulation on clinical trials (8) has, for the first time, specifically defined diagnostic radiopharmaceuticals used in clinical trials as being exempt from good manufacturing practice (GMP). Canadian regulations still require GLP and GMP standards to be applied to clinical trials, but this framework can be discussed in consultation meetings.
These changes indicate that regulators recognize the unique nature of radiopharmaceuticals and may tailor nonclinical and GMP requirements. However, further reforms are still necessary, and because of the international nature of drug development and distribution, harmonization between jurisdictions is essential to allow patients to benefit from advances in nuclear medicine imaging and radiotherapeutics. This need is recognized by nuclear medicine scientific societies such as the Society of Nuclear Medicine and Molecular Imaging, the European Association of Nuclear Medicine, and the Canadian Association of Nuclear Medicine, who have initiated efforts to highlight differences and effect positive change. Working with groups such as the International Atomic Energy Association, which has an established goal of facilitating harmonization of regulations and improving access to radiopharmaceuticals, could facilitate this effort.
U.S. Diagnostic Radiopharmaceuticals: Radioactive Drug Research Committee, Exploratory IND (eIND), and IND
In the United States, human administration of research radiopharmaceuticals is subject to FDA regulations. One of the following two mechanisms must be followed: approval of the application by the sponsoring institution’s radioactive drug research committee, or submission of an IND or eIND.
For an application to be filed with the radioactive drug research committee, the research must be considered “basic,” that is, the pharmacology of the drug in humans must be known, and the mass of drug administered must have no pharmacologic or adverse effect, as defined in title 21 of the Code of Federal Regulations, part 361.1 (9). Study data may not be used for clinical decisions.
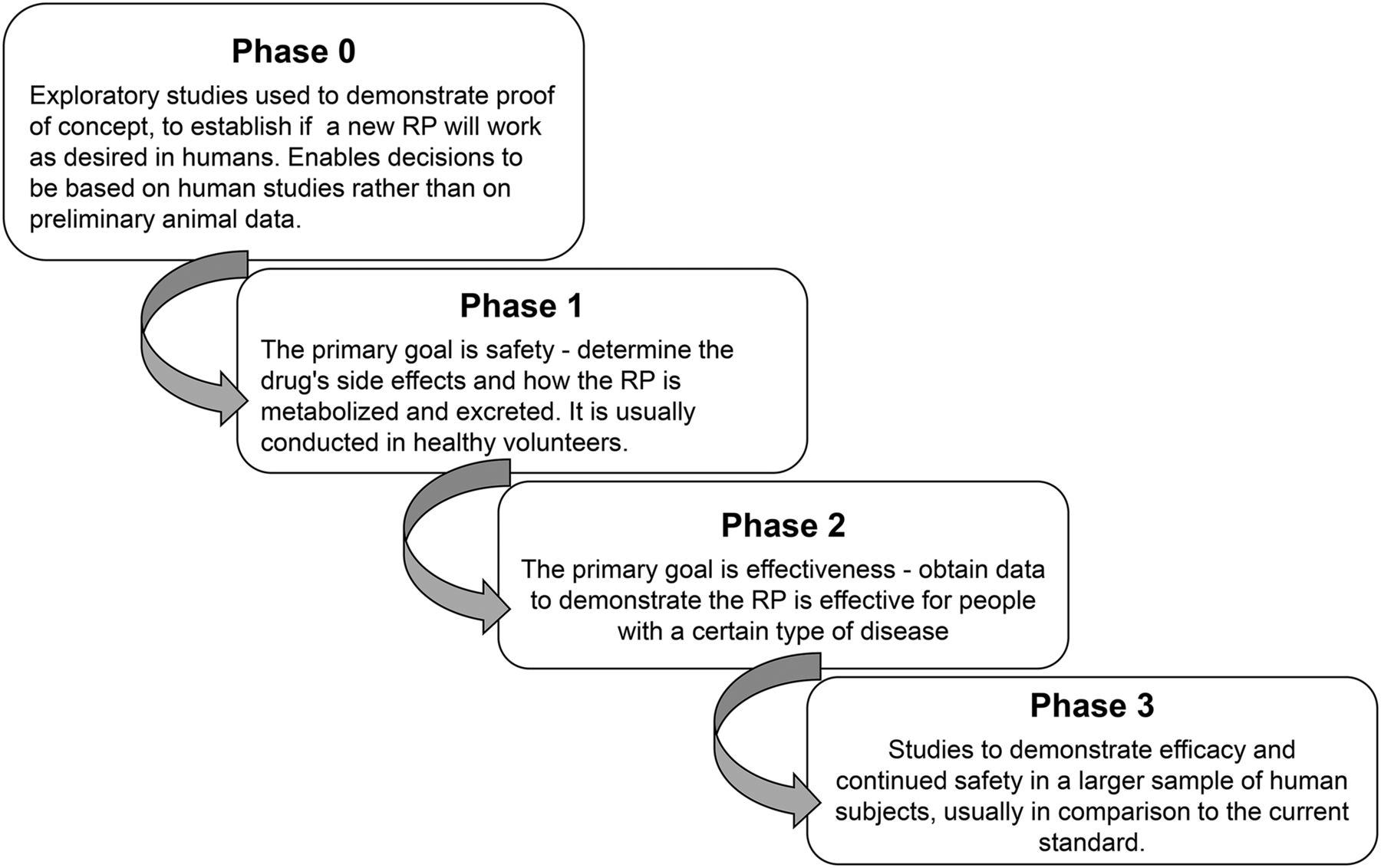
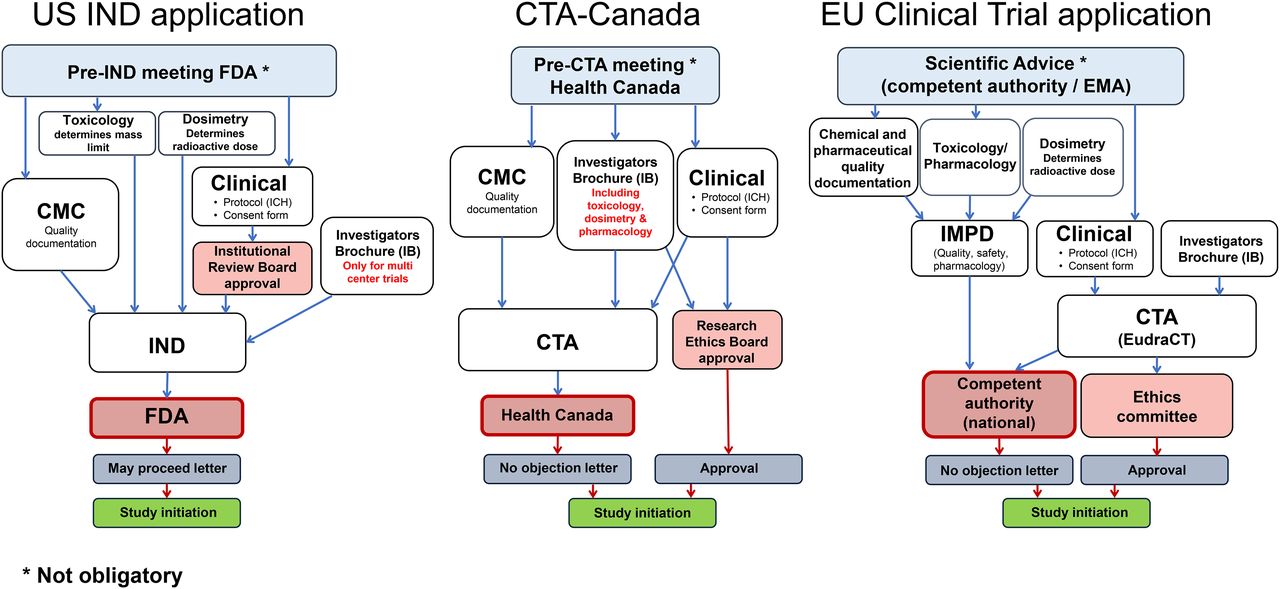
For any first-in-human investigational radiopharmaceutical, an IND or eIND must be filed, as described in title 21 of the Code of Federal Regulations, part 312 (10). These applications include information on pharmacology and toxicology testing from preclinical testing in animals. Traditional IND toxicity studies require 2 mammalian species (both sexes); for microdose toxicity, the FDA accepts a single mammalian species (both sexes), thus reducing costs. A single-species toxicity study costs approximately $70,000–$110,000 when performed in a GLP laboratory. Most toxicology studies are performed in GLP laboratories, but upon discussion, the FDA may allow other types of controlled laboratories to be used, such as university comparative anatomy or veterinary medicine departments. Use of such laboratories also reduces cost. Additionally, the new FDA guidance further reduces or eliminates additional toxicology requirements and clarifies other nonclinical microdose requirements for phase 1–3 studies (Fig. 1) (5).

Phases of clinical trials for radiopharmaceuticals, from first-in-human to marketing authorization. RP = radiopharmaceutical.
The microdose toxicity study should use the intended clinical route of administration. Also, to establish a margin of safety, the sponsor should demonstrate that a dose at least 100 times higher than the proposed human dose does not induce adverse effects in experimental animals, and scaling from animals to humans should be based on body surface area.
U.S. Regulatory Requirements for Production of Research Radiopharmaceuticals
Production of SPECT investigational radiopharmaceuticals requires that title 21 of the Code of Federal Regulations, part 211, “Current Good Manufacturing Practice for Finished Pharmaceuticals” (11), be followed. Phase 1 studies are exempt from part 211 requirements, and the FDA “Guidance for Industry: CGMP for phase 1 Investigational Drugs” provides information on controlled production procedures (12).
IND and eIND PET radiopharmaceutical production requires compliance with title 21 of the Code of Federal Regulations, part 212, “Current Good Manufacturing Practice for Positron Emission Tomography Drugs” (13). The rule allows either the use of U.S. Pharmacopeia Chapter 823 (14) or the use of part 212 for phase 0, 1, or 2 but requires production under part 212 for phase 3, since commercialization of the radiopharmaceutical will usually follow.
U.S. Clinical Trials
To initiate a clinical trial in the United States, an eIND or IND must be submitted to the FDA. Before submission of either document, the FDA encourages applicants to schedule a pre-IND meeting to assist in preparing a development strategy. The request is usually in writing, listing the objectives for the meeting (Tables 1 and 2).
Formal Requirements for Clinical Trials
Documentation Required for Clinical Trials
The eIND and IND have 8 required components. The first is a general investigational clinical plan prepared by the principal investigator. It should include the reason for selecting the compound and the goal of the research study. The second component, an investigator’s brochure, is a comprehensive document summarizing data relevant to studies in human subjects gathered through preclinical and other clinical trials. If an IND involves a single institution, an investigator’s brochure is not needed. The third component is a clinical trial protocol describing the study being performed on humans (Fig. 1).
The fourth component, a document describing production, is called the chemistry, manufacturing, and controls (CMC) document. The CMC information usually includes a standard operating procedure for the radiosynthesis and purification, a list of the raw materials, and a flow diagram of the overall process. The method of radionuclide production must be described, including the target body and the target material. If precursors or nonradioactive standards are produced in-house, a standard operating procedure should be written defining the procedure. The chemicals and glassware should be newly purchased and secured in a controlled area. The final precursor must be fully labeled, including name, lot number, date of production, and expiration time. After synthesis, analytic data such as nuclear MR, mass spectroscopy, and high-pressure liquid chromatography should be obtained for full characterization of the compound. For commercially produced precursors, a certificate of analysis should be obtained from the manufacturer and included in the submission.
The fifth component is a section on toxicology. Quality control specifications need to be developed to define the release of the radiopharmaceutical for human use. The chemical mass allowable in the final product is defined by toxicology studies. Three validation runs are required, with complete quality control for each batch. Stability testing for 3 batches is used to determine the expiration time.
An animal dosimetry section, the sixth component, is required for radiopharmaceuticals but is not defined in the eIND/IND outline requirements. The seventh component, institutional review board approval, must be received from the institution where the investigation will be conducted. The board reviews the clinical protocols, the clinical investigators’ qualifications, and the informed-consent document (15). The final component, the case report form, is a specialized data-collection document that will preserve and maintain the quality and integrity of the data.
As of May 5, 2018, commercial INDs and drug master files must be submitted using the electronic common-technical-document format, whereas paper format is still permitted for noncommercial INDs (16).
The FDA requires 30 d to review and present preclinical, clinical, or CMC-related questions. At the end of this review, if the FDA’s questions are adequately addressed, it will send a “may proceed” letter to the principal investigator or sponsor, who will then submit this letter to the institutional review board (Fig. 2). Table 1 lists the records that must be retained.
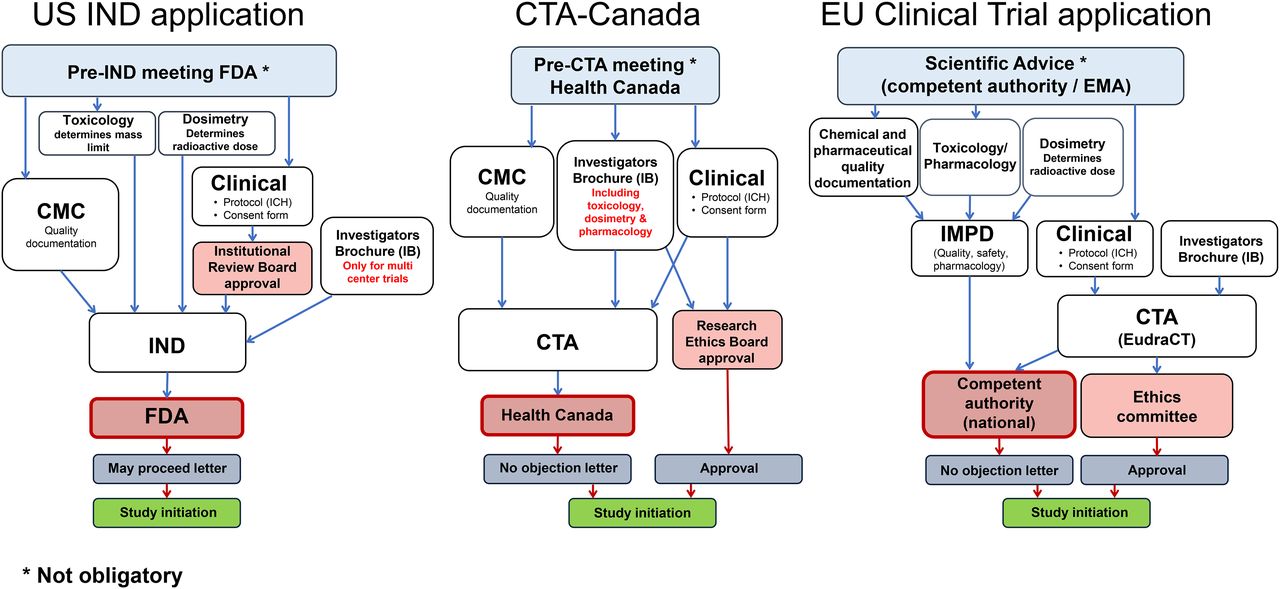
Application process for clinical trial, comparing current situation in United States, Canada, and Europe.
EU Background
The EU is governed by the European Commission and the European Parliament, with the EMA as a decentralized drug agency. Compared with the FDA and Health Canada, the EMA has a more limited mandate on evaluations for European marketing authorization of medicinal products, pharmacovigilance, orphan drugs, and scientific advice.
Not all European countries are members of the EU, and even within the EU, individual member states maintain considerable influence on legislation, leading to a heterogeneous regulatory landscape for radiopharmaceuticals. For example, many EU legislative texts related to pharmaceuticals are so-called European directives that have to be transposed into the national legislation of each member state. This transposition allows for interpretation and can result in differences between nations. Also, certain topics are only rudimentarily regulated by the EU or are exclusively handled and controlled by the national member states. An example is “pharmacy legislation,” which leads to a highly heterogeneous practice, particularly related to radiopharmaceutical preparations. A more detailed overview can be found in previous publications (17,18).
New Radiopharmaceuticals Outside the EU Clinical Trial Track
The introduction and use of new radiopharmaceuticals in Europe currently follows 2 different pathways. One is based on the EU–Clinical Trial legislation, the other on specific national regulations. From a historical perspective, many novel radiopharmaceuticals, in particular for PET but also for radionuclide therapy, have been introduced using specific national pathways. Examples include somatostatin analogs (DOTATOC, DOTATATE) labeled with 68Ga, 90Y, or 177Lu and, more recently, the widespread use of prostate-specific membrane antigen–targeting agents for PET (e.g., labeled with 68Ga or 18F) and therapy (e.g., labeled with 177Lu or 225Ac). This type of introduction highlights the limited legal mandate of the EU for certain topics, including health-care and pharmacy practices, which leads to extreme differences in the application and availability of radiopharmaceuticals across Europe. For example, European directive 2001/83 (19), in its definition, covers only medicinal products that are “prepared industrially or manufactured by a method involving an industrial process,” thereby exempting the “magistral” or “officinal” preparation pathway (i.e., national pharmacy legislation).
With respect to radiopharmaceuticals, there is a wide variation of rules (17). For example, France and Hungary follow a strict policy that allows the use of a radiopharmaceuticals only after they have received marketing authorization or when a clinical trial is initiated. In other countries, such as The Netherlands, Belgium, Austria, and Sweden, national legislation allows the preparation of certain radiopharmaceuticals without marketing authorization based on “pharmacy practice”; that is, involving the hospital pharmacy, which takes responsibility for release. The United Kingdom permits the preparation of radiopharmaceuticals without marketing authorization outside a pharmacy with a so-called Special’s License, whereas in Italy production can be performed in a public hospital when a monograph exists in the European Pharmacopoeia. However, these pathways cannot be used to prepare novel, first-in-human drugs. Poland introduced a special definition of experimental trials, making a distinction from clinical trials as defined in the EU directive. In Germany, the most widely used tool to introduce new radiopharmaceuticals is article 13, section 2b, of the German Drug Law (Arzneimittelgesetz), which allows a physician to produce and administer pharmaceuticals to an individual patient as long as the drug is prepared under the immediate supervision of the physician. At some universities, such applications are reviewed by an ethical board as compassionate use.
Whether these practices must follow GMP is again heterogeneously handled. Some countries do not provide clear regulations, others implement GMP into pharmacy practice, and still others implement GMP within a manufacturing authorization. Sometimes there are variations within a country depending on the local responsible drug inspectorate.
EU Clinical Trial Track
The use of novel radiopharmaceuticals within a clinical trial is more clearly regulated and based on a clear EU mandate. Currently, EU clinical trials legislation is in a transition phase between the old clinical trials directive, 2001/20 (20), and the new clinical trials regulation, 536/2014 (8). The clinical trials have to be conducted according to good clinical practice (GCP) (21), and the preparation of the so-called investigational medicinal product is subject to authorization and must follow dedicated GMP rules (22). A revision of these rules will be applicable with implementation of the new clinical trials regulation (23). Currently, in each individual member state participating in a trial, a clinical trial application (CTA) must be submitted for review and approval both to an ethical committee and to the national competent authority (Fig. 1), with the consequence of national differences in time lines and constraints. The new regulation foresees submission to a central EU portal and database. After application and a validation period of 10 d, one nationally competent authority will review the application within 50 d, valid for all participating member states. This procedure will likely simplify the process and allow coverage of multinational trials within one application. Also included in the new regulation are specific exemption of diagnostic radiopharmaceuticals from manufacturing authorization and GMP, as well as simplifications in the otherwise extensive rules for labeling investigational medicinal products. However, implementation of the central EU portal has been delayed and is currently not expected before 2019. This delay also delays recognition of the rules for radiopharmaceuticals in the new clinical trials regulation (8).
Regardless of whether clinical trials are submitted nationally within the old directive or centrally with the new regulation (Table 1), the main set of documents to be prepared remains the same (Table 2). For initiation of an application, a EudraCT (European Clinical Trials Database) number must be obtained, providing a clear identifier of the trial (www.clinicaltrialsregister.eu). The documentation includes 3 components: the clinical document, the central document, and the investigator’s brochure.
The clinical document, following GCP (24), includes the study protocol, informed consent forms, case report forms, standard operating procedures, logs, contracts, and other documents. In particular for early-phase clinical trials, this document is essential to identify and mitigate risks associated with the investigational medicinal product within the study protocol, such as avoiding simultaneous administration in patients, when the safety profile is not yet established. A dedicated, recently issued guideline from the EMA (25) describes major points in the planning of such trials. Other documents such as informed consent forms often have to follow local and national rules.
The central document, which describes the investigational medicinal product used in the trial, is known as the investigational medicinal product dossier (IMPD). This document, in principle, follows the internationally harmonized common-technical-document format (26). A guideline (27) that details the structure of the first part of the IMPD is related to chemical and pharmaceutical quality documentation and is equivalent to the U.S. CMC information. The European Association of Nuclear Medicine has published guidance to clarify issues specific to radiopharmaceuticals in this part of the IMPD (28). In contrast to the U.S. Pharmacopeia, which does not list research radiopharmaceuticals, the European Pharmacopoeia (29) is an important document to reference for research radiopharmaceuticals used in clinical trials. There are monographs for nonlicensed radiopharmaceuticals, but there are also general monographs, including those for radioactive and cold precursors, which are important in setting quality limits and in establishing analytic methods.
The second part of the IMPD should contain safety data on the investigational medicinal product, including preclinical and, if available, clinical data on pharmacology, such as pharmacokinetics and toxicology. No detailed guidance is provided specifically for radiopharmaceuticals; however, preclinical dosimetry data must be included. For toxicity studies, reference to ICH guideline M3(R2) (4) should be made, describing the currently valid microdosing approach (also see FDA Guidance (3)). This approach is under discussion in Europe because in contrast to the United States, compliance with GLP is required, resulting in very high costs, and the approach does not fully reflect the unique characteristics of radiopharmaceuticals.
Besides the clinical and central documents, an investigator’s brochure is needed. This important document describing the investigational radiopharmaceutical follows a defined structure (24) and provides the principal investigator with all the radiopharmaceutical-related information. The investigator’s brochure is normally a shortened version of the IMPD, prepared by the sponsor. An investigator’s brochure is also required for academic trials, for which the IMPD may be accessible.
It is also important to provide a way to monitor the study to ensure data integrity within the trial, and early-phase trials may require considerably more funds to ensure the required monitoring frequency. Before a CTA is submitted, the competent authority (and in later-stage clinical trials also the EMA) can be approached through a mechanism called “scientific advice,” which typically, similar to in the United States and Canada, is a meeting with regulators discussing predefined questions on the trial. However, in Europe the authorities typically charge fees, even for academic trials.
Canadian Background
Health Canada regulates radiopharmaceuticals, which are defined as schedule C drugs, under the Canadian Food and Drug Act and Regulations (part C, division 3) (30).
Canadian Basic Research Applications: Positron-Emitting Radiopharmaceuticals
Recognition of the importance of PET radiopharmaceuticals as basic research tools has led to a formal pathway providing a reduced regulatory burden for applicable clinical research for which there is minimal risk to research subjects. This regulatory approach (31) bears many similarities to the Radioactive Drug Research Committee system in the United States (9). The imaging agent must have been previously administered to humans, and the study’s purpose must be basic research, not drug development. Subjects may receive no more than a 50-mSv annual radiation dose from the drug (including impurities) and other clinical trial–related procedures. Subjects must be 18 y or older. No more than 30 subjects may be enrolled in the study. Research Ethics Board approval must be obtained. Finally, concomitant drugs used in research must have market authorization.
The application requires a 1- to 2-page study description and attestation that it meets the criteria outlined above plus other requirements (32). There is a 15-d review period by Health Canada. Studies must be conducted according to ICH GCP (24) and GMP. Regulations for expedited adverse drug reactions are applicable. Complete records of results must be maintained according to the requirements specified in the regulations. A similar mechanism is being considered for SPECT radiopharmaceuticals.
Canadian Clinical Trials
With the exception of the “Canadian Basic Research Applications: Positron-Emitting Radiopharmaceuticals” system, any other clinical trial with a PET radiopharmaceutical must be submitted to Health Canada for review in a standard CTA as described in part C, division 5, of the regulations (33) and related guidance (34). Health Canada encourages pre-CTA meetings to discuss submissions and obtain feedback on trial design, supporting preclinical studies, manufacturing validation, and proposed specifications. For a first-in-human study, there must be sufficient preclinical evidence to support the safety and proposed efficacy of the investigational agent (Table 1).
Health Canada has adopted the ICH M3(R2) (4) guideline for preclinical safety testing and has recently released a draft guidance document outlining preclinical requirements for positron-emitting and other radiopharmaceuticals (35). A risk-based approach is generally accepted using studies supporting the dose and dosing frequency in appropriate animal species. Thus, for PET radiopharmaceuticals that qualify for microdosing and will be administered as a single dose, an extended acute toxicology study in rats with a dose 100 times greater than the anticipated human dose could be proposed.
The preclinical package must include an estimate of dosimetry based on preclinical biodistribution data, and it is expected that human biodistribution data will be obtained early in clinical development. Supporting preclinical and clinical data are summarized in the investigator’s brochure with reference to study reports, which are not provided in the CTA but should be available on request.
For CTAs, radiopharmaceuticals are expected to be manufactured according to GMPs (part C, division 2). Additional general guidance for GMPs in Canada (GUI-0001) (36), as well as for radiopharmaceuticals (GUI-0026) (37), PET radiopharmaceuticals (GUI-0071) (38), and clinical trial–specific GMP guidance (GUI-0036) (39), should be reviewed with the GMP regulations. For new agents, validation data typically require 3 consecutive batches at an appropriate scale to demonstrate adequate control of the manufacturing process to reliably produce a safe product meeting predetermined and justified specifications. Analytic methods should be appropriately controlled, and for phase 1 studies, sterility and endotoxin testing must be validated. The proposed shelf life must be supported through stability data, and shipping conditions should be tested if transportation is involved. A description of the source, manufacturing, and specifications for critical raw materials (e.g., isotopes and precursors), with additional information if the precursor is not commercially obtained, is required.
Evidence must be provided that the drug substance and precursor have been characterized and that impurities (potential and actual) have been assessed. The reference standard for analytic assays must be described.
Drug product labeling requirements are defined in part C (divisions 3 and 5) (30,40). If quality information (e.g., precursor or drug product) is proprietary and has been filed by the owner with Health Canada in a master file, the sponsor can cross-reference the master file in the sponsor’s CTA via a letter of access (41).
The clinical trial protocol and informed consent are submitted in the CTA with a protocol synopsis (Protocol Safety and Efficacy Assessment Template–CTA). The protocol and informed consent follow ICH GCP guidance (24) and must have instructions on expedited safety reporting to Health Canada (42). Although case report forms must be used to capture clinical trial data, the template case report form is not required in the submission. For first-in-human studies, the emphasis is on safety data (typically including vital signs, adverse events, electrocardiograms, and lab tests). Healthy volunteers are appropriate for biodistribution data and dosimetry estimates, although relevant patient populations may be used under some circumstances. Secondary objectives on early signs of efficacy are acceptable and may include metrics such as uptake in organs or diseased tissue, and image quality assessment.
The CTA consists of 3 modules, based on the ICH common-technical-document format (26) and further described in Health Canada guidance (Fig. 2) (34). The CTAs for module 1 (clinical and administrative information), module 2 (quality information), and module 3 (supporting quality information) are submitted in “non–electronic common-technical-document electronic-only format,” which comprises the entire submission on a compact disk with a hard-copy cover letter (43). Electronic common-technical-document submissions for CTAs are in the pilot stage.
Health Canada reviews a CTA within 30 d and, once it is cleared, provides a no-objection letter. Approval must also be received from a research ethics board in compliance with C.05.001 (Table 2) (40).
DISCUSSION
Reviewing radiopharmaceutical regulations from 2 continents provides a sense of the complexity surrounding the regulatory regimes that allow first-in-human use. Similarities exist in the requirements of the 3 jurisdictions, including the requirement that GCP be followed and that a clinical trial protocol, informed consent form, and investigator’s brochure be included in the submission. Positive movement includes recognition that radiopharmaceuticals are administered in microdose amounts, the broad similarity between nonclinical and CMC requirements, and the FDA’s endorsement of less stringent requirements for animal toxicity testing and allowing this to occur in non-GLP facilities. However, these are not universal, and the notable differences that exist between jurisdictions are further complicated in Europe by the layered hierarchy.
The renaissance in nuclear medicine of new diagnostic and therapeutic agents is potentially impeded by differing regulatory requirements, which act to ultimately limit patient access to these important advances. Furthermore, because the volume of nuclear medicine radiopharmaceuticals is low compared with therapeutic drugs, such complexity has led to a shrinkage of radiopharmaceutical availability in many countries. This patchwork nature of the regulations runs counter to the globalized environment we are now experiencing.
Extensive documentation is required for investigational submissions. Although use of the common-technical-document format has been undertaken, a harmonized format between regulatory authorities and radiopharmaceutical-specific documentation has not yet been realized. This realization is vital, because reform and standardization will streamline submission preparation and review time and potentially decrease costs. An effort as simple as harmonizing submission document names between countries would be a significant step in more effective communication between jurisdictions.
The recent FDA approval of 68Ga-Netspot (Advanced Accelerator Applications) supported by data from trials in other countries was an important change from historic requirements of country-specific clinical trials; however, use of trial data from other countries requires formalizing and more universal acceptance (43). Moreover, even with this positive step, the 68Ga-Netspot approval in the United States is tied to a single generator manufacturer, leading to a lack of competition and shortages of that generator. Approval of other generators such as those by Isotope Technologies Garching and IDB Holland (iThemba) could ease the shortage, which has limited patient access.
GMP issues and heterogeneity are another area of significant concern. The unique nature of radiopharmaceuticals as radioactive, short-lived, small-batch products administered in low-mass nonphysiologic doses, along with an exemplary historical safety profile, suggests that a more risk-appropriate system could be used. Indeed, Australia has shown that GMP licensing is not an absolute necessity for safe, high-quality radiopharmaceutical products, especially as they relate to clinical trials. Lack of GMP requirements was facilitated by an effective manufacturing culture based on risk assessment of processes for instruments and materials. This culture helps guarantee high-quality products with reduced time and cost needed for radiopharmaceutical development and is particularly applicable for initial clinical trials (e.g., phase 0–1), for which no data are available to demonstrate the need for GMP production of radiopharmaceuticals (44). The EU is following this direction for diagnostic radiopharmaceuticals in the new clinical trial regulation; however, whether national authorities will fully implement a reduction of GMP production requirements is a hot topic of discussion (45).
These regulatory challenges are not unique to radiopharmaceuticals; the entire pharmaceutical sector faces skyrocketing development and deployment costs. Governments and regulators have recognized this problem and have undertaken several initiatives to address it, including formation of the Canada–United States Regulatory Cooperation Council in North America and more widely ranging initiatives such as the ICH, which provides potential avenues for the nuclear medicine community to potentially have an impact. That said, these initiatives were developed to answer questions about much larger pharmaceutical regulations. Radiopharmaceuticals are a small market representing a fraction of the nearly $1 trillion global therapeutic drug market. This lower level of visibility is illustrated by the fact that, to date, the ICH has not directly addressed any issues related specifically to radiopharmaceuticals.
Even with these apparent headwinds, the issues raised in this paper demonstrate the need for this area of drug oversight to be assessed by regulatory bodies including, but not necessarily limited to, the EMA, FDA and Health Canada. The commonalities discussed suggest the potential for a positive impact. The International Atomic Energy Agency (IAEA) has identified these issues as problematic for member nations and initiated efforts to facilitate broader global discussion around harmonization of regulations specific to radiopharmaceuticals. Initial efforts have identified several potential actions the organization can take to improve the situation (44). Identification of these actions is significant, as the IAEA represents an impartial organization comprising developed and developing nations and has expertise on medical isotopes and direct lines of communication with multiple governments and organizations.
The efforts of the IAEA and many of the issues identified provide a potential basis of discussion among the regulatory bodies to find commonality and concurrently address the unique, safe characteristics of radiopharmaceuticals. As part of this effort, the IAEA could provide a conduit to the ICH to emphasize the need for regulators to recognize and use study data and previously obtained approvals from other trusted jurisdictions in their assessment of radiopharmaceuticals. The IAEA could provide an unbiased conduit to link regulators with the nuclear medicine community, encouraging dialogue and increasing mutual understanding of issues in regulatory development and inspection of radiopharmaceuticals.
An educated advocacy on behalf of the nuclear medicine community is vital to push regulators into this domain and attempt some constructive reconciliation of differences. The Society of Nuclear Medicine and Molecular Imaging, the European Association of Nuclear Medicine, and the Canadian Association of Nuclear Medicine can help provide that advocacy.
CONCLUSION
This article explains how differing regulatory practices in Europe and North America adversely affect radiopharmaceutical deployment and development, leading to challenges in patient access to investigational radiopharmaceuticals and to an affordable supply of commercially approved vital diagnostic and therapeutic agents, which can improve and extend lives.
Acknowledgments
We thank Stephanie Nemirov for her assistance in reviewing the manuscript.
Footnotes
Published online Sep. 27, 2018.
Learning Objectives: On successful completion of this activity, participants should be able to (1) understand the requirements for first-in-human radiopharmaceutical applications for the United States, European Union, and Canada; (2) realize the overall need for reduction of the human-use requirements due to lack of toxicity and the need to facilitate patient access; and (3) recognize that harmonization efforts between the U.S. Food and Drug Administration, Health Canada, and the European Medical Authority would streamline efforts in moving new radiopharmaceuticals forward for clinical care.
Financial Disclosure: Dr. Schwarz is an SNMMI past president, a consultant/advisor for Schwarz Consulting, and an employee of Washington University School of Medicine. The authors of this article have indicated no other relevant relationships that could be perceived as a real or apparent conflict of interest.
CME Credit: SNMMI is accredited by the Accreditation Council for Continuing Medical Education (ACCME) to sponsor continuing education for physicians. SNMMI designates each JNM continuing education article for a maximum of 2.0 AMA PRA Category 1 Credits. Physicians should claim only credit commensurate with the extent of their participation in the activity. For CE credit, SAM, and other credit types, participants can access this activity through the SNMMI website (http://www.snmmilearningcenter.org) through February 2022.
- © 2019 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication June 1, 2018.
- Accepted for publication September 7, 2018.





{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.