


Abstract
The use of radioactive sources to deliver cytotoxic ionizing radiation to disease sites dates back to the early 20th century, with the discovery of radium and its physiologic effects. α-emitters are of particular interest in the field of clinical oncology for radiotherapy applications. The first part of this review explored the basic radiochemistry, high cell-killing potency, and availability of α-emitting radionuclides, together with hurdles such as radiolabeling methods and daughter redistribution. The second part of this review will give an overview of the most promising and current uses of α-emitters in preclinical and clinical studies.
The short particle range and high linear energy transfer of α-emitting radionuclides complement the large particle range and low-energy transfer of β-particles. These physical characteristics allow α-particles to deposit the great majority of their energy in the area surrounding the desired targeted tumor cells (<100 μm), enabling them to kill isolated tumor cells. α-emitting radionuclides are therefore of particular interest for the treatment of systemic disease such as leukemia or lymphoma but also minimal disseminated disease comprising small clusters or isolated tumor cells. The past few years have also seen the preclinical or clinical use of α-emitters for the treatment of solid primary and metastatic disease such as glioblastoma or castration-resistant prostate cancer (CRPC) (1,2). Six α-emitters are currently under investigation in preclinical or clinical studies using either the intrinsic targeting properties of the α-emitters or targeted α-therapy (TAT) strategies. Antibodies, peptides, and small molecules have been successfully conjugated to α-particle emitters; however, few of these α-conjugates have been translated to the clinic for evaluation.
The basic physical characteristics of the described α-emitters are covered in part 1 of this review.
PRECLINICAL EVALUATION
The design of preclinical studies plays an important role in the success and translation of α-therapy to the clinical stage (Fig. 1). The following section presents a selection of preclinical studies (published over the last 10 y) with strong translational potential.
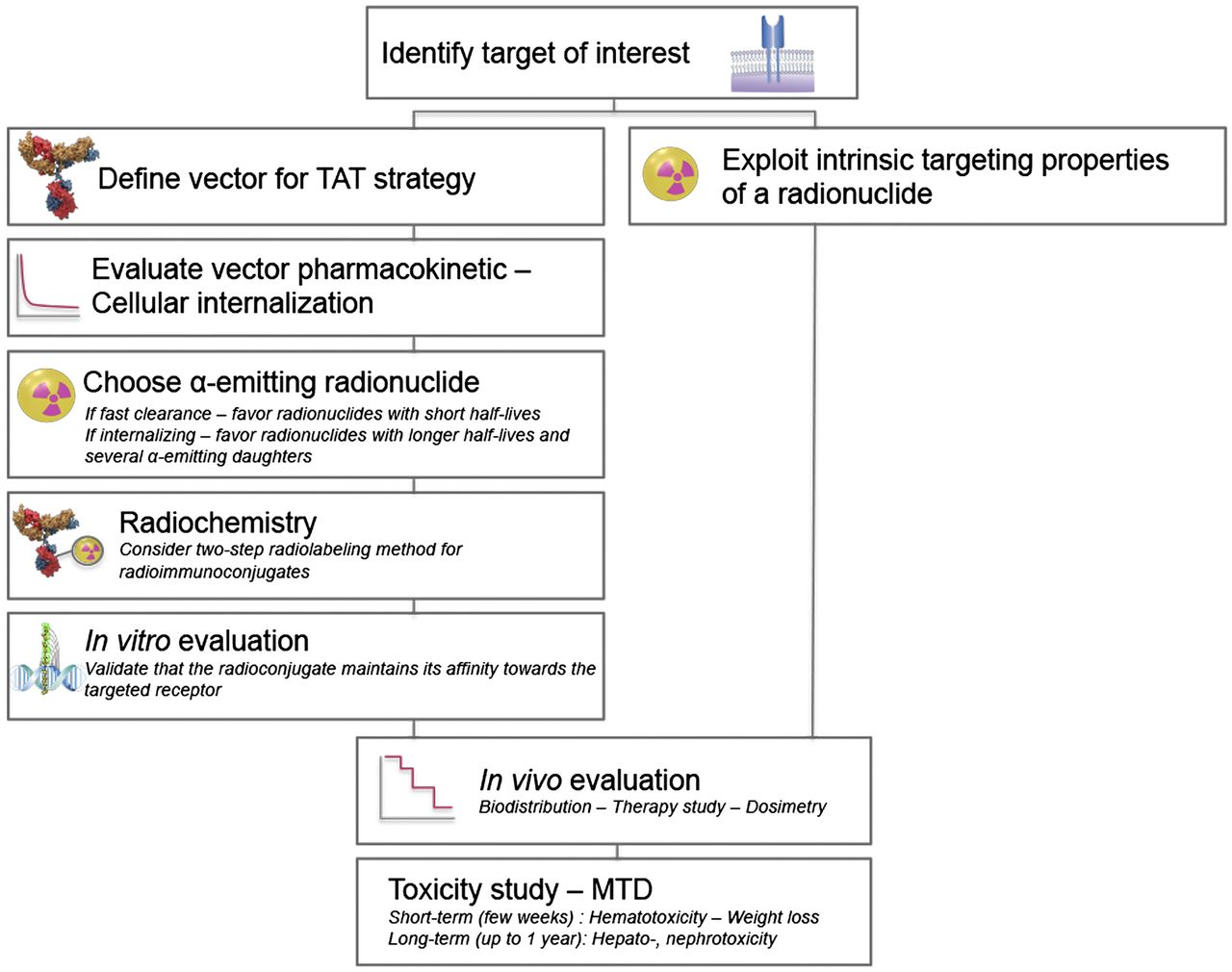
Preclinical study design. TAT = targeted α-therapy; MTD = maximum tolerated dose.
Systemic Cancers: Lymphoma and Leukemia
CD20 is a well-known target for the treatment of B-cell lymphoma. Despite its intermediate half-life, 211At has been conjugated to an anti-CD20 monoclonal antibody and evaluated in 2 lymphoma models: a subcutaneous tumor xenograft and a disseminated model (3). High total doses (1.78 MBq) of 211At-anti-CD20 radioimmunotherapy demonstrated moderate attenuation in tumor growth in a subcutaneous xenograft model. In contrast, a 0.55-MBq total dose resulted in complete disease eradication in 70% of the animals with the disseminated lymphoma model (Fig. 2A) (3). After the 0.55-MBq total dose, universally lethal toxicity was observed within 5 d, with severe weight loss and petechiae. This study highlights that the potential of α-therapy varies with tumor cell accessibility.
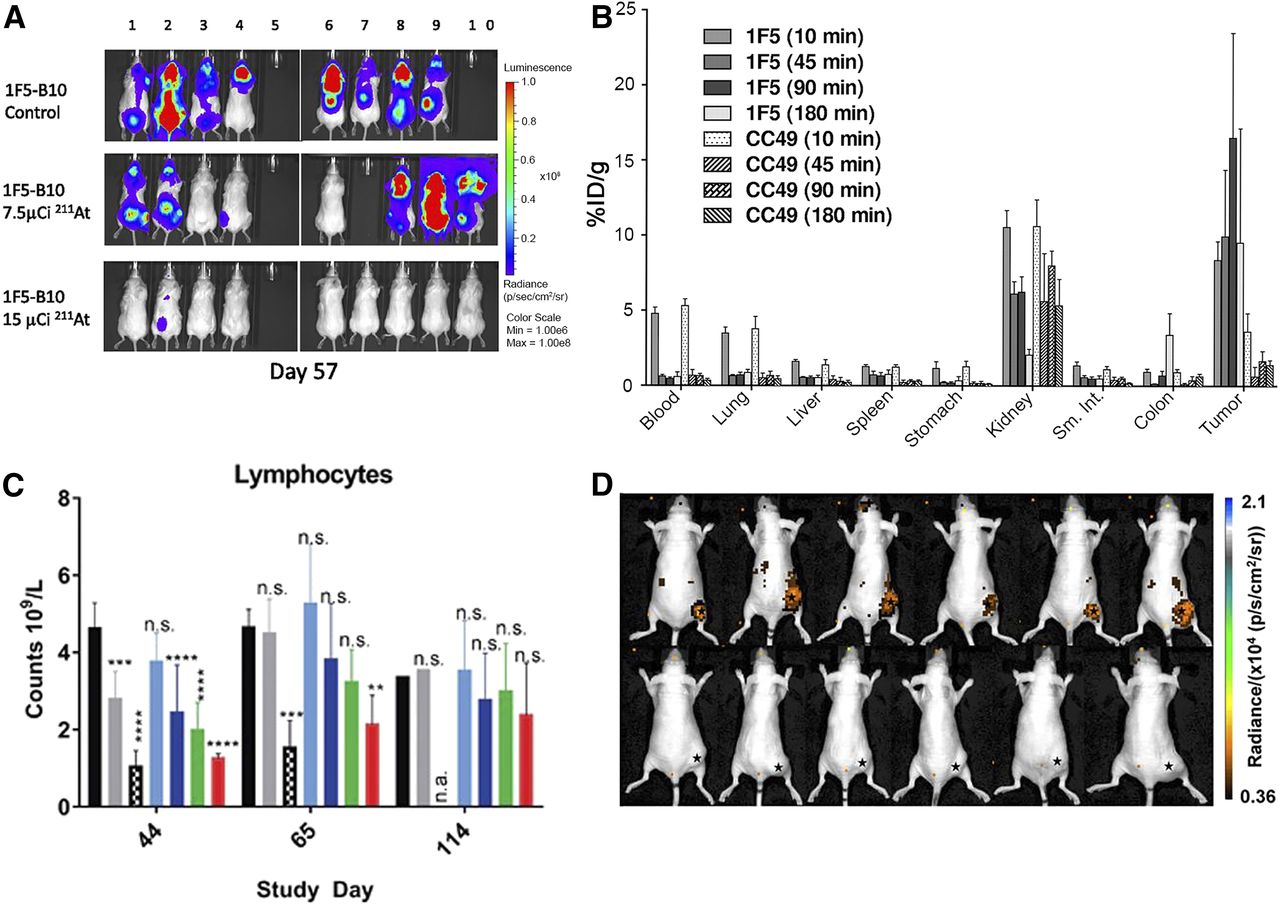
Preclinical studies, potency, imaging, and pitfalls of α-therapy. (A) Bioluminescence imaging of 211At-B10-1F5 (anti-CD20) radioimmunotherapy in Granta-519Luc cell line, a minimal residual disease model of lymphoma. Animals were treated on day 6 with 278 kBq or 555 kBq and received 1 × 107 bone marrow cells intravenously from syngeneic donors 2 d after treatment. (Reprinted with permission of (3).) (B) Biodistribution of 213Bi-DOTA-biotin after pretargeting radioimmunotherapy with 1F5(single-chain variable-region)4-streptavidin (anti-CD20) and CC49(single-chain variable-region)4-streptavidin (negative control). (Reprinted with permission of (7).) (C) Transient lymphocyte decrease after CD70-targeted 227Th-conjugate therapy indicative of myelosuppression (15). (D) Cerenkov imaging 24 h after injection of 225Ac-DOTA-c(RGDyK) in U87MG tumor–bearing mice. Top images show efficient tumor accumulation; bottom images show reduced tumor uptake on blockage and demonstrate specificity of radiotherapy for αvβ3 integrin (17).
The longer-lived α-emitting radioisotope, 227Th, was originally conjugated to antibodies such as trastuzumab and anti-CD20 monoclonal antibody (rituximab), demonstrating significant tumor growth delay and prolonged survival in breast, ovarian, and lymphoma models (4,5). Long-term toxicity (≤1 y) was assessed in mice with human lymphoma Raji xenografts (6). A 1,000 kBq/kg dose for 227Th-rituximab resulted in a significant body weight decrease, with temporary white blood cell and platelet count drops (6). The maximum tolerated dose (MTD) was determined to be between 600 and 1,000 kBq/kg, with a maximum absorbed dose to the bone marrow of 2.1–3.5 Gy (6).
Reducing nonspecific toxicity and hematotoxicity using the pretargeting method was investigated. Pretargeted α-radioimmunotherapy using an anti-CD20 single-chain variable-region streptavidin construct and a 213Bi-DOTA-biotin was reported by Park et al. to treat non-Hodgkin lymphoma (7). A favorable biodistribution profile was obtained using the pretargeting method, with specific tumor uptake of 16.5% ± 7.0% of injected dose per gram at 90 min after injection (Fig. 2B) (7). A therapy study with injections of up to 29.6 MBq (total dose) of 213Bi-DOTA-biotin exhibited a dose-dependent tumor response (7). Three of 10 mice achieved complete remission with the highest dose, and only one showed signs of early toxicity, losing approximately 10% of its body weight. The median survival for the group receiving a total dose of 22.2 MBq of 213Bi-DOTA-biotin was 90 d, compared with 19 d for untreated mice (7).
The anti-CD33 antibody, lintuzumab (HuM195), has a long history of TAT applications for the treatment of acute myeloid leukemia, and the clinical use of this agent will be discussed later in this review. Preclinically, it was first conjugated to 213Bi and 225Ac, and more recently it was conjugated to 227Th (8). In this study, complete tumor regression for up to 20 d was observed in an acute myeloid leukemia xenograft model with a 700 kBq/kg dose of 227Th-radioimmunotherapy (8).
Metastatic Prostate Cancer
The external domain of prostate-specific membrane antigen (PSMA) is a major target of interest for TAT. McDevitt et al. successfully conjugated 213Bi to J591, an anti-PSMA monoclonal antibody, and demonstrated the ability of the developed α-radioimmunotherapy to improve median tumor-free survival together with reduction of prostate-specific antigen (PSA) levels in mice with intramuscular LNCaP xenografts (9). The past decade has also seen the development of numerous small-molecule PSMA inhibitors to diagnose and treat prostate cancer. The 225Ac conjugate will be described later in the clinical section of this review. A 211At-labeled small-PSMA-inhibitor conjugate was recently evaluated in PSMA-positive mouse models, showing significant tumor growth delay and improved survival (10). Long-term toxicity (≤12 mo) revealed nephropathy to be the dose-limiting effect, and the MTD was set to be a 37-kBq total dose in immunocompetent CD1 mice (10).
223Ra is a particularly interesting radionuclide because of its bone-seeking properties, and its antitumor effects were demonstrated in animal models (11). Validation of this radionuclide in both preclinical and clinical studies was extensive, leading to the commercialization and use of this radioisotope in men with metastatic prostate cancer. We will therefore focus our interest on the clinical studies performed with this α-emitter in the clinical section below.
Disseminated Intraperitoneal Disease
212Pb (β-emitter), the decay daughter of 224Ra and parent of 212Bi (α-emitter), is widely exploited as an α-particle nanogenerator. 212Pb was first validated preclinically when conjugated to trastuzumab to treat disseminated human epidermal growth factor receptor 2 (HER-2)–positive peritoneal disease; its clinical application will be discussed in the next section (12). The contribution of internalization in HER-2 TAT in terms of efficacy was compared with a noninternalizing 212Pb-labeled anticarcinoembryonic antigen 35A7 antibody (13). A-431 intraperitoneal xenografted mice that express both HER-2 (low level) and carcinoembryonic antigen (high level) were injected with the same activity of both radioimmunoconjugates. Absorbed dose calculations revealed a higher dose to the tumor for the noninternalizing antibody 212Pb-35A7 (35.5 Gy) than for the internalizing 212Pb-trastuzumab (27.6 Gy) (13). However, the trastuzumab conjugate led to an unexpected longer mean survival (>130 d) (13). This study demonstrates the advantage of internalizing antibodies for therapy of small-volume xenograft tumors but also highlights the need for microdosimetry studies. With 212Pb α-therapy of disseminated intraperitoneal disease well established, the Brechbiel group developed α-therapy targeting HER-1 (also referred to as epidermal growth factor receptor) to provide another possibility or treatment combination for patients with residual tumor tissue after debulking surgery, micrometastatic disease, or disseminated peritoneal disease. Cetuximab was therefore radiolabeled with 212Pb, and total doses of 370–740 MBq were well tolerated by mice, with minimal toxicity (14).
Emerging Targets of Interest: Targeting the Tumor Microenvironment
CD70 belongs to the tumor necrosis factor superfamily and, together with CD27L, plays an important role in T-cell signaling. It is overexpressed on T- and B-cell lymphomas and on several types of solid tumor (i.e., renal cell carcinoma, ovarian and pancreatic adenocarcinoma, breast cancer, and colon cancer). CD70 is notably a promising target for immunotherapy and was also investigated by Hagemann et al. for 227Th-TAT (15). Mice bearing human renal cell carcinoma 789-O subcutaneous xenografts experienced complete growth inhibition with doses of as low as 50 kBq/kg (15). Decreased circulating neutrophils, lymphocytes, and total white blood cells in the treated mice, as compared with the control group, indicated myelosuppression (Fig. 2C). However, this toxicity was transient, and the mice recovered by day 114 (15).
The tumor microenvironment such as the vasculature and the neovascular endothelium are interesting targets for α-therapy. 225Ac-E4G10 antibody conjugate, which targets monomeric vascular endothelial cadherin, recently demonstrated inhibition of tumor growth and improved survival in a high-grade glioblastoma mouse model (1). Mechanistic studies highlighted remodeling of the tumor blood–brain barrier microenvironment by dual depletion of endothelial and perivascular cells (16). Even though more than 20% of injected dose per gram of tissue of 225Ac-E4G10 was observed in the liver at more than 10 d after treatment, no liver toxicity was reported (1).
Integrins play an important role in tumor angiogenesis, and their blockade can inhibit tumor growth or metastasis. Among them, αvβ3 antagonists demonstrate strong potential for the treatment of cancer. A DOTA-(RGDyK) peptide conjugate radiolabeled with 225Ac displays high affinity for αvβ3 integrin (17). Cerenkov imaging performed on U87MG xenografted mice upon injection of 1.9 MBq (total dose) of the radiolabeled peptide showed tumor, liver, and kidney uptake. Ex vivo imaging confirmed the biodistribution (Fig. 2D) (17). Cerenkov radiation is observed with a wide range of medical isotopes, including 225Ac (18). Because α-particles travel with low velocity, 225Ac Cerenkov emissions are postulated to result from the β-decay of 213Bi, 209Tl, and 209Pb. A therapy study using 3 different doses based on the 225Ac-DOTA-(RGDyK) MTD (0.04 MBq; 1, 0.5, and 0.25 MTD) showed high blood urea nitrogen retention in all cohorts, suggesting kidney impairment (17).
α- VERSUS β-STUDIES
The following studies compared the efficacy of α-particle therapy with that of β-particle therapy. 225Ac-labeled antirat HER-2/neu monoclonal antibody (7.16.4) efficacy was compared with 213Bi and 90Y conjugates to treat breast cancer lung metastasis (Fig. 3A) (19). Injection of the MTD resulted in improved median survivals: 50 d for the 90Y-7.16.4 group and 61 d for the 213Bi-7.16.4 group. Of a total of 12 mice, 8 achieved long-term survival (≤1 y) in the 225Ac-7.16.4-treated group (19). The superiority of α-therapy over β-therapy was attributed to delivery of the α-radiation dose locally versus deposition of the β-particle energy mostly outside the metastasis. Because of the pharmacokinetics of the antibody radioconjugates, more than 90% of the 213Bi conjugate decay occurred before the target was reached. The longer physical half-life of 225Ac, together with the emission of 4 net α-particles per decay, therefore resulted in higher absorbed doses, explaining the longer survival of the 225Ac-treated mice. Slow but significant release of 225Ac daughters resulted in long-term renal toxicity (Fig. 3B) (19).

Preclinical α- vs. β-studies. (A) Kaplan–Meier survival curves of neu-N transgenic mice treated with 14.8 kBq of 225Ac-7.16.4, 4.4 MBq of 213Bi-7.16.4, and 4.4 MBq of 90Y-7.16.4, 3 d after intravenous injection of 1 × 105 NT2.5 cells. (Reprinted with permission of (19).) (B, top) Photographs of kidneys from neu-N mouse surviving 1 y after treatment with 14.8 kBq (400 nCi) of 255Ac-7.16.4 compared with healthy neu-N mouse. (B, bottom) Hematoxylin and eosin staining of kidneys from neu-N mice surviving 1 y after treatment with 14.8 kBq (left) and 7.4 + 7.4 kBq (right) of 225Ac-7.16.4. Arrows indicate collapse of cortical tissue due to loss of tubular epithelium in kidney cortex. (Reprinted with permission of (19).) (C) Quantification of γ-H2AX–positive cells after immunofluorescent staining of DNA double-strand breaks in AR42J tumors after treatment with 47 kBq of 225Ac-DOTATOC, 30 MBq of 177Lu-DOTATOC, 1 μg of DOTATOC (unlabeled), or phosphate-buffered saline (20). (D) Tumors showing growth delay after treatment with equitoxic doses of 225Ac-DOTATOC (44 kBq) and 177Lu-DOTATOC (34 MBq) (20).
Peptide receptor radionuclide therapy using the somatostatin analog DOTATOC radiolabeled with 225Ac and 177Lu also compared the potential of α- and β-therapy (20). Equitoxic doses of the α- and β-emitter radiopharmaceuticals were determined using comparative cytotoxicity assessment, and a factor of approximately 700 was applied between the 225Ac- and 177Lu-conjugates. The degree of DNA double-strand breaks was determined using quantification of γ-H2AX levels. The overall percentage of cells with γ-H2AX foci was significantly higher in tumors treated with 48 kBq (total dose) of 225Ac-DOTATOC (35%) than in those treated with 30 MBq (total dose) of 177Lu-DOTATOC (21%) (Fig. 3C) (20). This observation was consistent with a delayed exponential tumor growth of 25 d with 225Ac-DOTATOC versus 21 d with 177Lu-DOTATOC (Fig. 3D) (20).
α-particle emitters have substantial therapeutic potential because they can generate higher levels of DNA double-strand breaks than β-particle emitters. However, α-emitter versus β-emitter studies can be biased by the use of α-emitters with long half-lives and numerous α-emitting progeny that increase the tumor-absorbed dose. Dosimetric considerations, even though challenging with α-emitters, should be a parameter of choice for the determination of the administered activity in such comparative studies. Moreover, parameters such as tumor size, hypoxia, or intratumoral radiopharmaceutical distribution should be considered.
In the clinic, the choice between α- and β-therapy currently depends on the patient tumor burden and previous response to β-therapy. α-therapy is currently offered only at late disease stages, and the efficacy of this therapy is still unknown at earlier disease stages (21). Comparison of the potential of α- versus β-therapy for clinical applications would therefore be premature at this moment. Further evaluation of α-therapy is required at early stage diseases but also in combination with the current standard of care or new therapeutic approaches such as inhibition of DNA repair pathways (e.g., poly(ADP-ribose) polymerase inhibitors) (21). Cocktail approaches of α- and β-therapy should also be evaluated because of the complementary nature of their particle range.
CLINICAL EVALUATION
To our knowledge, 62 clinical trials were registered on clinicaltrials.gov using α-emitters; among them, 52 involved 223Ra-dichloride (Table 1) using the bone-seeking properties of radium. This last section will focus on the clinical achievement and evolution of the most notable clinical studies performed with α-emitting radionuclides.
Examples of Clinical Evaluation of α-Therapy
Advanced Myeloid Leukemia: From 213Bi-Lintuzumab to 225Ac-Lintuzumab
In a phase I dose escalation trial, a humanized anti-CD33 monoclonal antibody, HuM195 (lintuzumab), that targets myeloid leukemia cells was radiolabeled with 213Bi (22). The choice of an α-emitting radionuclide was justified by the significant toxicities, particularly prolonged myelosuppression, observed in previous studies using β-particle emitters. Eighteen patients with primary refractory or relapsed advanced myeloid leukemia were treated with 10.4–37.0 MBq/kg (22). Although all patients developed transient myelosuppression (recovery time of 22 d), no extramedullary toxicity was observed. Of the evaluable patients, 93% showed a reduction in peripheral blood leukemia cells and 78% showed a reduction in bone marrow blasts. However, no patient achieved complete remission (22), likely because of the patients’ large tumor burden (up to 1012 cells). Partial cytoreduction of the tumor burden before the 213Bi treatment was evaluated in a phase I/II trial with sequential treatment with cytarabine (200 mg/m2/d) and 213Bi-HuM195 (18.5–46.3 MBq/kg) (23). A decrease in marrow blasts was reported for all dose levels and a 213Bi dose–response relationship with remission was observed for doses of 37 MBq/kg or higher (23).
225Ac was next considered as an alternative therapeutic radioisotope to 213Bi to take advantage of the greater cytotoxic potential and significantly longer half-life of 225Ac. 225Ac-HuM195 was subsequently evaluated in a phase I study in patients with relapsed or refractory advanced myeloid leukemia (24). Doses of 18.5–148 kBq/kg were administered. Redistribution of daughters in the kidneys was anticipated, but no evidence of radiation-induced nephrotoxicity was seen. Peripheral blasts were eliminated in 63% of the patients but only at doses of 37 kBq/kg or higher. Bone marrow blast reduction was observed in 67% of patients (24). A phase I/II study in advanced myeloid leukemia patients is currently evaluating the MTD and efficacy of the combination of low-dose cytarabine and 225Ac-HuM195. Preliminary results recommend a 74 kBq/kg/fraction dose for the phase II study to limit prolonged myelosuppression (25). Patients receiving 225Ac-HuM195 have also been administered furosemide and spironolactone to prevent potential radiation-induced renal toxicity (25,26). Furosemide has since been discontinued because it was causing dehydration in some patients. However, without furosemide, there has been no renal toxicity observed in patients receiving 225Ac-HuM195.
Neuroendocrine Tumors: 213Bi-DOTATOC for β-Radiation–Refractory Tumors
Kratochwil et al. performed a first-in-human study using a somatostatin analog TAT, 213Bi-DOTATOC, as a therapeutic option for patients with refractory neuroendocrine tumors who were pretreated with β-emitting 90Y/177Lu-DOTATOC (27). Patients with progressive advanced neuroendocrine tumors with liver metastasis were treated with intraarterial infusions, and 1 patient with bone carcinosis was treated with a systemic infusion (Fig. 4A). 213Bi-DOTATOC was administered in cycles with increasing activities (1.0–4.0 GBq, total dose) every 2 mo. All patients showed long-lasting antitumor responses, highlighting the ability of 213Bi-DOTATOC to overcome β-radiation resistance. Renal toxicity was minimized because of a protocol developed with the β-emitting particle radiopeptide therapy, including lysine, arginine, and Gelofusine (Hausmann Laboratories Ltd.) administration (27).

Clinical application of α-radiotherapy. (A, left) 68Ga-DOTATOC PET maximum-intensity projection of neuroendocrine tumor patient showing extensive tumor burden in liver and disseminated bone marrow metastases. (A, right) Significant reduction of liver metastases after administration of 10.5 GBq of 213Bi-DOTATOC into common hepatic artery (27). (B) Kaplan–Meier estimates of overall survival of patients with symptomatic CRPC after treatment with 223Ra-dichloride as compared with placebo group. (Reprinted with permission of (31).) (C) Evolution of 68Ga-PSMA-11 PET/CT scans of patient with metastatic CRPC showing extended peritoneal carcinomatosis and liver metastases. After 2 cycles of 177Lu-PSMA-617, patient showed disease progression and was offered 225Ac-PSMA-617. After 3 cycles, PET/CT indicated complete response and PSA level dropped to immeasurable levels (2). CI = confidence interval.
HER2-Expressing Ovarian Carcinoma: 212Pb-TCMC-Trastuzumab
In patients with HER2-expressing ovarian cancer in which the malignancies were confined mainly to the peritoneal cavity and progressed after multiple therapies, the biodistribution, pharmacokinetics, and safety of 212Pb-TCMC-trastuzumab were assessed in a first-in-human study by Meredith et al. (28). A single intraperitoneal injection of 212Pb-TCMC-trastuzumab (7.4 MBq/m2) after a 4 mg/kg intravenous injection of trastuzumab (28) was evaluated first, followed by a dose escalation study (7.4–21.1 MBq/m2) (29). Furosemide and spirolactone were administered as renal protective agents. Minimal radiopharmaceutical redistribution out of the peritoneal cavity and no significant myelosuppression were observed (28). However, no patient met the criteria for a partial response (29).
Metastatic CRPC: From 223Ra-Dichloride to 225Ac-DOTA-PSMA
With 52 clinical trials registered on clinicaltrials.gov, the use of 223Ra-dichloride (Alpharadin [Bayer] or Xofigo [Bayer]) has impacted the therapeutic landscape of α-radiation therapy. Approximately 80% of the clinical trials involve prostate cancer, and 35% use a combination of drugs and 223Ra-dichloride. The first clinical experiments with 223Ra-dichloride (46–250 kBq/kg) performed on prostate and breast cancer patients reported pain relief and reduction in alkaline phosphatase (30). ALSYMPCA, a phase III randomized double-blind placebo-controlled trial, was performed to evaluate the efficacy and safety of 223Ra-dichloride (50 kBq/kg every 4 wk for a total of 6 cycles) versus placebo plus the standard of care in symptomatic CRPC patients (31). Overall survival was significantly longer with 223Ra-dichloride: 14.9 mo, compared with 11.3 mo with the placebo (Fig. 4B) (31). These primary results led to the Food and Drug Administration approval of 223Ra-dichloride to treat CRPC in May 2013. 223Ra-dichloride has also been shown to prolong the time before an increase in total alkaline phosphatase and PSA occurs (32). The therapy is well tolerated and minimally toxic, and patients have reported a significant improvement in quality of life (32); however, 223Ra-dichloride does not target soft-tissue disease or the circulating component of the disease (late manifestations).
Another promising α-therapeutic option for metastatic CRPC applies a small urea-based PSMA inhibitor (2) already used in the clinic for prostate cancer PET imaging (33) and β-therapy (34). Although promising, this β-therapy is ineffective in about 30% of patients and contraindicated for patients with diffuse red marrow infiltration. 225Ac was considered to overcome β-resistance and reduce hematologic toxicity. Two metastatic-CRPC patients in challenging clinical situations first received 225Ac-PSMA-617 (100 kBq/kg, bimonthly) as salvage therapy after the presence of a PSMA-positive tumor phenotype had been validated by 68Ga-PSMA-11 PET/CT (2). Both patients showed a complete response (Fig. 4C), with a drop in PSA to below the measurable level. Xerostomia was reported in both patients (2). A larger study with 14 metastatic-CRPC patients identified the best compromise between toxicity and antitumor response (35). A treatment activity of 100 kBq/kg was determined to be tolerable yet have significant antitumor activity (35), although a solution to preserve the salivary glands should be investigated. 225Ac-PSMA-617 offers the major advantage of targeting metastases in any tissue and could therefore be used as a complementary option to 223Ra-dichloride therapy. Shortage of 225Ac, however, remains a challenge and limits the evaluation of 225Ac-TAT in large studies.
INTEGRATING α-THERAPY INTO CURRENT AND FUTURE CLINICAL PARADIGMS
The future of α-therapy lies in resolving the hurdles mentioned in part 1 of this review, that is to say, availability, production concerns, and issues associated with daughter redistribution. Solutions to these issues are currently being investigated and should facilitate the broader development of α-emitter radiotherapy with larger, randomized, prospective clinical studies that would ultimately result in more meaningful efficacy and toxicity evaluation.
The future of α-therapy also lies in the proper design of clinical trials and the incorporation of this mode of therapy into the current standard of care or in combination with new therapeutic approaches. An irony of the current status of 223Ra-dichloride is that despite the completion of early-phase studies and ALSYMPCA, the optimal dose and duration of therapy are still not known. Currently, a randomized phase II trial (NCT02023697) is examining men with metastatic CRPC with 2 or more skeletal metastases under 3 separate treatment arms: a standard 223Ra-dichloride regimen (55 kBq/kg, Food and Drug Administration–approved), a high-dose regimen (88 kBq/kg every month for 6 mo), and an extended-duration regimen (55 kBq/kg injections every month for 12 mo). This study will complement a completed smaller single-arm study (NCT01934790) in which patients who had received a 6-mo course of 223Ra-dichloride underwent another 6 mo of the drug at standard doses (55 kBq/kg per month). Preliminary data on 44 patients reveal that 66% of patients completed retreatment with an excellent safety profile (36). The clinical impact of additional treatments still needs to be defined.
The optimization of 223Ra-dichloride treatment is also being explored in combination with tumor-targeting therapy such as androgen receptor axis-directed therapy. A randomized phase III trial (NCT02043678) compared the androgen biosynthesis inhibitor abiraterone acetate and prednisone with abiraterone, prednisone, and 223Ra-dichloride in men with metastatic CRPC who had not received cytotoxic chemotherapy. The Independent Data Monitoring Committee recommended that this trial be unmasked when the committee observed an imbalance in fractures and deaths, favoring treatment with abiraterone/prednisone alone (37). The details of these data are pending, but for now clinicians are increasingly cautious about using a combination of abiraterone, prednisone, and 223Ra-dichloride to treat men with early metastatic CRPC.
The combination of 223Ra-dichloride with chemotherapy is also being explored in men with metastatic CRPC. 223Ra-dichloride has been tested in a phase Ib/IIa study (NCT01106352) in combination with docetaxel. Patients were randomized to either the combination arm or the docetaxel arm on a 2:1 basis. A PSA decline of over 50% occurred in 61% of patients in the combination arm and 54% in the docetaxel arm. Sustained suppression was also apparent on Kaplan–Meier analysis, with a significantly longer time to PSA progression for the combination (6.6 vs. 4.8 mo; P = 0.0198) (38). Several newer trials are exploring 223Ra-dichloride as an adjunct to boost other treatment effects, such as poly(ADP-ribose) polymerase inhibitor (i.e., olaparib (39) or niraparib [NCT03076203)]), or as an immune adjuvant leveraging the abscopal effect to enhance the impact of immunotherapy (223Ra-dichloride in combination with the programmed cell death ligand 1 inhibitor atezolizumab [NCT02814669]). Combinations of tumor and bone targeting offer a promise of amplifying the effects of treatment beyond the host compartment of bone and would allow patients with visceral metastases to receive 223Ra-dichloride, which is not presently permissible in the United States. Dual tumor and bone targeting is also possible with tumor-directed α-emitters (225Ac-PSMA-617), although formal prospective studies for these agents are still needed to define the optimal dose and treatment intervals and to develop toxicity mitigation strategies.
CONCLUSION
The studies described in this review demonstrate that α-emitting radionuclides have the potential to be excellent therapeutic candidates and, along with β-particle therapy, can expand the options for therapy. α-emitting radionuclides are currently considered an alternative at late disease stages when resistance to β-therapy is observed or when the patient presents with extended bone marrow disease; however, applications in earlier disease stages should be evaluated. Together, parts 1 and 2 of this review give a broad overview of α-emitters from basic radiochemistry to clinical use. The future of α-radiotherapy depends on numerous factors; part 1 highlights hurdles and new approaches for wider use of α-emitting radionuclides, and part 2 highlights the importance of clinical trial design in properly determining the optimal dose for α-therapy and incorporating it into standard-of-care protocols.
Footnotes
Published online Mar. 1, 2018.
Learning Objectives: On successful completion of this activity, participants should be able to (1) match α-emitters to potential vectors or targets of interest; (2) highlight the potential of α-emitters in preclinical work; and (3) identify the current clinical uses of α-emitters and the importance of clinical trial design for broader application of α-therapy.
Financial Disclosure: This work is supported by the Radiochemistry and Molecular Imaging Probe Core, which is supported in part by NIH/NCI Cancer Center Support Grant P30 CA008748. Jason Lewis is supported by the Mr. William H. and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research and by the Center for Experimental Therapeutics of Memorial Sloan Kettering Cancer Center. Sophie Poty is supported by the François Wallace Monahan Fellowship from the JLM Benevolent Fund. The authors of this article have indicated no other relevant relationships that could be perceived as a real or apparent conflict of interest.
CME Credit: SNMMI is accredited by the Accreditation Council for Continuing Medical Education (ACCME) to sponsr continuing education for physicians. SNMMI designates each JNM continuing education article for a maximum of 2.0 AMA PRA Category 1 Credits. Physicians should claim only credit commensurate with the extent of their participation in the activity. For CE credit, SAM, and other credit types, participants can access this activity through the SNMMI website (http://www.snmmilearningcenter.org) through July 2021.
- © 2018 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication November 3, 2017.
- Accepted for publication February 3, 2018.





{kind=link}
{kind=link}
{kind=link}
{kind=link}