


Abstract
The clinical impact and accessibility of 68Ga tracers for the prostate-specific membrane antigen (PSMA) and other targets would be greatly enhanced by the availability of a simple, 1-step kit-based labeling process. Radiopharmacy staff are accustomed to such procedures in the daily preparation of 99mTc radiopharmaceuticals. Currently, chelating agents used in 68Ga radiopharmaceuticals do not meet this ideal. The aim of this study was to develop and evaluate preclinically a 68Ga radiotracer for imaging PSMA expression that could be radiolabeled simply by addition of 68Ga generator eluate to a cold kit. Methods: A conjugate of a tris(hydroxypyridinone) (THP) chelator with the established urea-based PSMA inhibitor was synthesized and radiolabeled with 68Ga by adding generator eluate directly to a vial containing the cold precursors THP-PSMA and sodium bicarbonate, with no further manipulation. It was analyzed after 5 min by instant thin-layer chromatography and high-performance liquid chromatography. The product was subjected to in vitro studies to determine PSMA affinity using PSMA-expressing DU145-PSMA cells, with their nonexpressing analog DU145 as a control. In vivo PET imaging and ex vivo biodistribution studies were performed in mice bearing xenografts of the same cell lines, comparing 68Ga-THP-PSMA with 68Ga-HBED-CC-PSMA. Results: Radiolabeling was complete (>95%) within 5 min at room temperature, showing a single radioactive species by high-performance liquid chromatography that was stable in human serum for more than 6 h and showed specific binding to PSMA-expressing cells (concentration giving 50% inhibition of 361 ± 60 nM). In vivo PET imaging showed specific uptake in PSMA-expressing tumors, reaching 5.6 ± 1.2 percentage injected dose per cubic centimeter at 40–60 min and rapid clearance from blood to kidney and bladder. The tumor uptake, biodistribution, and pharmacokinetics were not significantly different from those of 68Ga-HBED-CC-PSMA except for reduced uptake in the spleen. Conclusion: 68Ga-THP-PSMA has equivalent imaging properties but greatly simplified radiolabeling compared with other 68Ga-PSMA conjugates. THP offers the prospect of rapid, simple, 1-step, room-temperature syringe-and-vial radiolabeling of 68Ga radiopharmaceuticals.
Infrastructure in radiopharmacies has been developed around 99mTc-labeling protocols, in which a generator is eluted multiple times per day to produce diverse tracers by reconstituting commercially available cold kits compatible with good manufacturing practice (1). This is typically a high-throughput environment where speed, simplicity, volume, and reproducibility of radiolabeling are paramount. The growth of 18F and 11C PET tracers, with which a kit model is not compatible because of the need for an on-site cyclotron and more complex synthetic chemistry, has spawned more diverse, complex, and costly infrastructure to support PET. Nevertheless, the generator and kit approach retains its appeal and in principle, 68Ga generators compatible with good manufacturing practice now available are amenable to kit production if a simple, mild chelation step can be achieved (2,3). This would make 68Ga tracers widely available without the complex and costly infrastructure associated with 18F and 11C tracer production.
This concept was suggested more than 2 decades ago (4), but despite several recent reports of kit-based 68Ga tracer production (5,6), an ideal 1-step procedure for 68Ga radiolabeling, matching the simplicity typical of long-established 99mTc-labeling procedures requiring only the addition of generator eluate to the kit vial, has not yet been achieved. Toward this end, the chelator must meet several criteria: its labeling should reach completion (>95%) quickly (<5 min) at room temperature and be unaffected by common trace metals, without additional steps to concentrate, buffer, or purify. Its complex should resist in vivo transchelation (e.g., by transferrin), and conjugation and radiolabeling should not induce mixtures of diastereomers, enantiomers, or geometric isomers, nor adverse pharmacokinetics, for example, delayed clearance or nonspecific binding. The current generation of 68Ga chelators do not meet these criteria. For example, the widely adopted macrocycle DOTA (7), while complexing Ga3+ with extraordinarily high kinetic stability, has very slow complexing kinetics, necessitating heat (e.g., 90°C, followed by a suitable cooling period), a large amount of the biomolecule, and low pH. Low yields (<95%) necessitate a purification step. These factors add process complexity, limit specific activity, and may damage the biomolecule. Conversely, the fast chelation rate of HBED-CC makes radiolabeling of 68Ga-HBED-CC-PSMA (PSMA is prostate-specific membrane antigen) possible at room temperature, but produces an undesirable mixture of cis/trans geometric isomers distinguishable by high-performance liquid chromatography (HPLC) (5,8,9). Thus, a heating step is still required, to reduce the number of isomers and increase the yield of 1 to approximately 90% (9). Clinical radiosynthesis of tracers based on these chelators is currently performed on cartridge-based synthesis modules, taking 35 min and typically affording 80% ± 5% decay-corrected radiochemical yield (9).
Recently, several groups have introduced new 68Ga3+ chelators that address these issues but none eliminate all of them. NOTA, TRAP, and DEDPA are promising but, like DOTA, require acidic conditions and are vulnerable to competition from contaminating trace metals. The DATA series of chelators show rapid, room-temperature labeling at pH 5; the DATAPPh variant can be labeled in 15 min at pH 7 but requires preprocessed eluate (10). A class of chelator that promises to meet the requirements for kit-based labeling is the tris(hydroxypyridinone) (THP) system; it can complex 68Ga rapidly at room temperature and neutral pH, with high yield and purity. Its performance has previously been evaluated against a range of common chelators (11), including HBED, demonstrating superior radiolabeling properties under milder conditions. THP has also been functionalized for conjugation to peptides and proteins while retaining the required mild radiolabeling and in vivo targeting properties (11–15).
Here we evaluate a THP bioconjugate targeting PSMA (overexpressed in prostate cancer), incorporating a small urea-linked dipeptide pharmacophore (Fig. 1) (7,8,16). A 68Ga-labeled conjugate of this targeting moiety with HBED-CC has shown outstanding clinical promise in several trials in patients with prostate cancer (17,18) but is subject to the production difficulties outlined above. The aims of this work were to determine the potential of 68Ga-THP-PSMA to achieve 1-step kit-based labeling of a radiopharmaceutical intended for PSMA imaging and to evaluate preclinically the resulting tracer.

MATERIALS AND METHODS
All reagents and consumables were purchased from Sigma Aldrich or Fischer Scientific, with the exception of Fmoc-Lys-Dde-COOH (Bachem AG), PMPA (Enzo Life Sciences), DOTA-PSMA (PSMA-617), and HBED-CC-PSMA DKFZ-11 (ABX). Severe combined immunodeficient (SCID)/beige mice were purchased from Charles River. These cells were cultured in RMPI 1640 medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and penicillin/streptomycin. To prepare for experiments, cells were grown at 37°C in an incubator with humidified air equilibrated with 5% CO2.
1H- and 13C-NMR spectra were acquired on a Bruker Avance III 700 spectrometer operating at 700 MHz (1H frequency), equipped with a quadruple-resonance QCI cryoprobe. High-resolution liquid chromatography electrospray ionization mass spectrometry (LC-ESI-MS) was performed in positive-ion mode on an Agilent 6520 Accurate-Mass Q-TOF LC/MS connected to an Agilent 1200 HPLC. All HPLC utilized an Agilent 1200 LC with in-line radio (sodium iodide γ-detector) and ultraviolet detection (220 nm analytical, 280 nm preparative). Data were analyzed using Laura software (version 4.0.2.75; LabLogic Systems Ltd.).
HPLC Methods
The chemical purity of THP-PSMA and radiolabeling of 68/67Ga-THP-PSMA were assessed using an Agilent Eclipse XDB C18 5-μm 4.6 × 150 mm column with an isocratic mobile phase (87.5% H2O, 12.5% acetonitrile [ACN], 0.075% trifluoroacetic acid [TFA], 0.05% triethylamine), with a flow rate of 1 mL/min. 68/67Ga radiolabeling of HBED-CC-PSMA and DOTA-PSMA was assessed with the same HPLC equipment but an alternative mobile phase: A = H2O with 0.05% TFA, B = ACN with 0.05% TFA, gradient: 0–5 min = 90% A, 5–15 minutes = ramp to 50% B (4%/min), 15–20 min = 90% A, with a flow rate of 1 mL/min. Serum stability was assessed using size-exclusion chromatography with a Phenomenex BioSep 5-μm size-exclusion chromatography s2000 column (300 × 7.8 mm) with phosphate-buffered saline (PBS) as the mobile phase and a flow rate of 1 mL/min.
Instant Thin-Layer Chromatography (iTLC) Methods
Radiolabeling for all 67/68Ga-PSMA radiotracers was assessed by iTLC utilizing Varian ITLC SGI0001 strips (10-cm length) with a mobile phase of 1 M ammonium acetate in water/methanol (1:1). iTLC plates were scanned with a Lab-Logic mini-Scan TLC reader and analyzed with Laura software. Radioactivity counting was performed with a γ-counter (LKB Wallac; PerkinElmer) for cell studies and in vivo biodistribution.
Conversion of 67Ga-Citrate Solution to 67Ga-Chloride in 0.1 M HCl
Conversion of 67Ga-citrate to 67Ga-chloride is required because the presence of citrate reduces the radiolabeling efficiency of 67Ga with a number of chelators. 67Ga-citrate (6.49 mM citrate; Mallinckrodt) was passed over a Silica Light Sep-Pak cartridge (120-mg sorbent, 55- to 105-μm particle size) at 1 mL/min to capture the radiometal on the cartridge. This process was repeated 3 times whereupon approximately 90% of the activity remained on the cartridge. After being washed with 5 mL of H2O, the 67Ga was eluted with 400 μL of 0.1 M HCl and collected in 50-μL fractions. Fractions with the highest activity concentration (75 MBq/50 μL) were used for labeling (activity concentration, 1.5 GBq/mL).
Synthesis of THP-PSMA
Synthesis of H-Lys-(Dde)-2CT-Resin (Compound 3)
2-chlorotrityl resin (350-mg equivalent to a loading of 0.35 mmol) was inserted in a fritted polypropylene syringe, and a solution of Fmoc-Lys-Dde-COOH (600 mg, 1.1 mmol) (1, Supplemental Fig. 1 [supplemental materials are available at http://jnm.snmjournals.org]) and excess N,N-diisopropylethylamine (DIPEA) in dichloromethane (DCM) was added. After being stirred overnight to form compound 2, the resin was washed with DCM and subsequently treated with methanol for 10 min. Thereafter, the resin was washed with dimethylformamide (DMF) and DCM and finally dried under vacuum. Resuspension of the beads in a solution of piperidine/DMF (20%:80%) for 20 min followed by separation of the resin from the supernatant by suction filtration and washing with DMF and DCM produced resin-bound compound 3.
Synthesis of Resin-Bound THP-PSMA (Compound 9)
Bis(tert-butyl) ester L-glutamate hydrochloride (444 mg, 1.5 mmol) was dissolved in anhydrous DCM (100 mL) containing DIPEA (750 μL, 4.3 mmol). This solution was added dropwise to a 3-neck round bottom flask containing a solution of triphosgene (150 mg, 0.5 mmol) in anhydrous DCM (5 mL) at 0°C under a nitrogen atmosphere. The reaction solution was then allowed to warm to room temperature and stirred for 1 h. The previously prepared H-Lys-(Dde)-2CT-resin (3, Supplemental Fig. 1) (250 mg, equivalent to a loading of 0.25 mmol) was added and stirred overnight to form compound 6 (Supplemental Fig. 1). The resin was separated from the supernatant by suction filtration in a fritted polypropylene syringe, washed, and dried. The resin was suspended in a mixture of 2% (v/v) hydrazine hydrate in 2 mL of DMF and stirred for 15 min, and then washed with DMF. This step was repeated 4 times to produce compound 7 (Supplemental Fig. 1). Compound 7 (100-mg equivalent to a loading of 0.1 mmol) was added to a solution of glutaric anhydride (114 mg, 1 mmol) and DIPEA (358 μL, 2 mmol) dissolved in 2 mL of DMF, and the mixture was stirred for 3 h to produce compound 8 (Supplemental Fig. 1). Compound 8 (50 mg, equivalent to a loading of 0.05 mmol) was suspended in a solution of 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (20 mg, 0.05 mmol) and DIPEA (18 μL, 0.1 mmol) in anhydrous DMF (0.5 mL), and in situ activation of the carboxylate was allowed to proceed for 10 min. A solution of THP-NH2 (70 mg, 0.09 mmol), synthesized by a previously published method (12–14), in DMF/DMSO (2.5 mL/2 mL) containing DIPEA (18 μL, 0.1 mmol) was then added to this suspension and stirred for 36 h, to furnish compound 9 (Supplemental Fig. 1).
Cleavage of THP-PSMA (Compound 10) from Resin
Cleavage and removal of the side-chain protecting groups were performed by treating the resin-bound compound 9 with TFA for 3 h at room temperature in the presence of phenol (5% w/v), water (5% v/v), and triisopropylsilane (2% v/v) as scavengers. The filtrate was collected and the resin washed with TFA and DCM. The solution was concentrated to less than 0.5 mL at 40°C under a gentle stream of nitrogen. Ice-cold diethyl ether was added to precipitate THP-PSMA (10, Supplemental Fig. 1). The suspension was centrifuged, supernatant decanted off, precipitate washed with diethyl ether, and finally dissolved in aqueous ACN and lyophilized. Crude THP-PSMA was purified by semipreparative reversed-phase HPLC to give a TFA salt (C54H77N11O19.(CF3COO)3) (molecular weight, 1,526.33). Semipreparative reversed-phase HPLC was conducted using an Agilent Eclipse XDB C18 5-μm 21.2 × 150 mm column with the concentration of mobile phase B increasing at a rate of 1%/min (A = H2O with 0.2% TFA, B = ACN with 0.2% TFA, starting from 100% A at time 0; flow rate, 5 mL/min). The yield of THP-PSMA(CF3COO)3 was 4 mg, 2.6 μmol, 5.2% yield from resin loading. The analytical HPLC Rt was 13 m 23 s, and purity was greater than 98%. The “Results” section provides analytical data.
Preparation of Lyophilized Kits
Kits for 1-step radiolabeling were prepared by lyophilizing an aqueous solution (5.25 mL) containing sodium bicarbonate (44 mg), sodium phosphate monobasic (8.6 mg), sodium phosphate dibasic heptahydrate (8.9 mg), and THP-PSMA (40 μg, 26 nmol) in a plastic vial affording a white powder.
68Ga-THP-PSMA Radiolabeling
Radiolabeling and radioanalysis (HPLC and iTLC) of 67/68Ga-DOTA-PSMA (PSMA-617) (8), 67/68/natGa-HBED-CC-PSMA (DKFZ-PSMA-11) (9,10), and 67/natGa-THP-PSMA were performed as follows.
Initial 68Ga radiolabeling optimization was performed with an Eckert and Ziegler (E&Z Radiopharma GmbH) 68Ge/68Ga generator, producing 120–400 MBq of 68Ga. Eluates (0.1 M HCl, 5 mL [Sigma Aldrich], HPCE grade) were fractionated (10 × 0.5 mL) but not preconditioned to concentrate 68Ga or remove trace metal contaminants. Typically, 68Ga3+ (5–75 MBq, 250 μL of the hottest fraction) was added to a mixture containing THP-PSMA in a range of concentrations (5–0.01 μg in 3 μL) in sodium bicarbonate solution (1 M, 26 μL), producing a solution with a pH of 6.5–7.5. Radiochemical yield was evaluated after 5 min at room temperature using HPLC and iTLC (HPLC: 68Ga-THP-PSMA Rt = 10.9 min, unbound 68Ga Rt = 2.32 min; iTLC: 68Ga-THP-PSMA Rf = 0.8–1, unbound 68Ga Rf = 0). For in vivo studies, THP-PSMA (2 μg) was labeled with 68Ga as described above; a greater than 95% radiochemical purity was consistently achieved.
To evaluate labeling using the entire eluate without fractionation, E&Z 68Ge/68Ga generator eluate (5 mL 0.1 M HCl, 122–202 MBq) or Galli EO (IRE ELiT) 68Ge/68Ga generator eluate (1.1 mL eluate diluted to 5 mL with 0.1 M HCl, 600–660 MBq) was added directly into a vented freeze-dried kit vial. After being mixed, carbon dioxide evolution visibly ceased after 15 s, providing a transparent, colorless solution at pH 6–7 with a final concentration of 5.25 μM THP-PSMA. iTLC was performed 5 min and HPLC 10 min after reconstitution. Decay-corrected radiochemical yield determined by each method was greater than 95% (n = 3 per generator).
Preparation of 67Ga and 68Ga PSMA Tracers for Biological Evaluation
For 67Ga-THP-PSMA for in vitro studies, 10 μL of a 2 mg/mL solution (20 μg, 13.1 nmol THP-PSMA [molecular weight, 1,526.33]) was added to 50 μL of 67Ga in 0.1 M HCl (converted from 67Ga citrate) and the pH adjusted to 6.5–7.5 with 4.5 μL of 1 M sodium bicarbonate. Labeling was assessed after 5 min at room temperature by iTLC and HPLC. Before being added to cells, the tracer was diluted to 50 nM in PBS.
For DOTA-PSMA for in vitro studies, 50 μL of 67Ga or 68Ga in 0.1 M HCl, 5.25 μL of 1 M HCl, and 20 μL of 2.1 M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) were combined, producing a solution at pH 3.5. For 67Ga labeling, 20 μg (13.3 nmol) of DOTA-PSMA (ABX; molecular weight, 1,498.18) were added; for 68Ga labeling, 2 μg (1.3 nmol) of DOTA PSMA were added. The solution was heated to 95°C for 30 min, and then labeling was assessed by iTLC and HPLC. Before being added to cells, the tracer was diluted to 50 nM in PBS.
For HBED-CC-PSMA for in vitro studies, 50 μL of 67Ga or 68Ga in 0.1 M HCl, 5 μL of 1 M HCl, and 50 μL of 2.1 M HEPES were combined, producing a solution at pH 4.5. For 67Ga labeling, 29 μg (20.6 nmol) of HBED-CC-PSMA (ABX; molecular weight, 1,403.09) were added; for 68Ga labeling, 2 μg (1.4 nmol) of HBED-CC-PSMA were added. The solution was heated to 95°C for 10 min, and then labeling was assessed by iTLC and HPLC. Before being added to cells, the tracer was diluted to 50 nM in PBS.
For HBED-CC-PSMA for in vivo studies, 68Ga eluate (200 μL, 35–50 MBq) was added to a pre-prepared solution of HBED-CC-PSMA (15 μL, 105 μM, 2.2 μg of HBED-CC-PSMA) and sodium bicarbonate (19 μL, 1 M) and heated at 90°C for 10 min (pH 3–4). A radiochemical yield of greater than 95% was consistently achieved.
Preparation of natGa Chelated PSMA Tracers
For natGa-THP-PSMA, 124 μL of 0.93 mM THP-PSMA were added to 93 μL of 6.4 mg/mL natGa(NO3)3.X(H2O) dissolved in 0.1 M HCl (a 20-fold excess of gallium over THP-PSMA). The pH was adjusted to 6.5–7.5 with 14 μL of 1 M sodium bicarbonate, and 231 μL of PBS were then added. The solution was sonicated for 15 min, producing a final solution of 0.25 mM natGa-THP-PSMA. This solution was diluted in PBS to produce the range of concentrations required for the concentration giving 50% inhibition (IC50). This complex was characterized with LC-ESI-MS: m/z [C54H74N11O19Ga + H]+ observed monoisotopic peak = 1250.45092, calculated = 1250.44909. [C54H74N11O19Ga + 2H]2+ observed monoisotopic peak = 625.72846, calculated = 625.72818.
For natGa-HBED-CC-PSMA,124 μL of 0.93 mM HBED-CC-PSMA were added to 93 μL of 6.4 mg/mL natGa(NO3)3.X(H2O) dissolved in 0.1 M HCl (a 20-fold excess of gallium over HBED-CC-PSMA). The pH was adjusted to 4 with 13 μL of 1 M sodium bicarbonate, and the solution was heated to 95°C for 10 min. The pH was adjusted to 6.5–7.5 with 1 M sodium bicarbonate followed by sonication for 15 min, producing a final solution of 0.5 mM natGa-HBED-CC-PSMA. This solution was diluted in PBS to produce the range of concentrations required for the IC50.
Log POCT/PBS
67Ga-labeled radiotracer (50 μL, 10 μM; specific activity, 1.5 MBq/nmol) was added to a preequilibrated mixture of 500 μL of octanol (OCT) and 450 μL of PBS. The mixture was mixed for 30 min and the phases separated by centrifugation (10,000 rpm, 10 min). Aliquots from each phase were γ-counted.
Serum Stability
68Ga-THP-PSMA was labeled at 5 MBq/nmol as described above. Twenty microliters were added to 180 μL of human serum, giving a final THP-PSMA concentration of 2.5 μM. Samples were incubated at 37°C and monitored over 6 h by size-exclusion HPLC. 68Ga-THP-PSMA without serum and unchelated 68Ga3+ incubated with serum were analyzed similarly.
Cell Uptake and Binding Affinity Assays
To determine the cellular uptake of each tracer at 37°C and 4°C over time, and whether a plateau (equilibrium) state is reached, PSMA-expressing cells DU145-PSMA and non–PSMA-expressing cells DU145 (19) were seeded in a 24-well plate (0.25 × 106 cells/well), 1 d before the assay. Two minutes before incubation, the medium was replaced with 245 μL of fresh medium at 37°C or 4°C, then 5 μL of one of the 67Ga PSMA tracers (specific activity, 0.75–2.2 MBq/nmol) were added, giving a final concentration of 1 nM. Plates were incubated at 37°C or 4°C. At each time point, supernatant was removed and cells were washed with 3 × 0.25 mL of PBS to determine the unbound fraction, followed by an acid wash (0.5 M glycine, pH 2.5, at 4°C, 5 min) to determine cell surface–bound activity. Cells were lysed with 1 M NaOH to determine activity internalized by the cells. Fractions were γ-counted.
To determine the IC50, competitive binding studies were performed with DU145-PSMA cells with 1 nM 68Ga-DOTA-PSMA as the probe, blocking with natGa-THP-PSMA or natGa-HBED-CC-PSMA over a range of concentrations. Cells (0.25 × 106 per well) were seeded in a 24-well plate, 1 d before the assay. Increasing concentrations of natGa-THP-PSMA or natGa-HBED-CC-PSMA followed by 1 nM 68Ga-DOTA-PSMA (specific activity, 5–7.5MBq/nmol, diluted in PBS) were added to the cells (total volume, 250 μL). After 30-min incubation at 37°C, the cells were washed with PBS (3 × 0.25 mL), lysed with NaOH (1 M, 0.25 mL), and the wells washed with PBS (0.25 mL). The activity present in supernatant and lysate was measured by γ-counting. Data were analyzed using GraphPad Prism (GraphPad Software) and a 1-site–fit log IC50 algorithm.
Because of the poor solubility of natGa-THP-PSMA above 0.25 mM, an alternative measure of affinity was developed, allowing the relative affinity of 2 gallium PSMA tracers to be determined simultaneously without the natGa complex, mitigating solubility difficulties and minimizing variance across samples arising from different cell numbers or radiotracer batches. Two different PSMA tracers, one labeled with 67Ga and the other with 68Ga, were simultaneously incubated with DU145-PSMA cells in a single well at 4°C for 2 h (noninternalizing conditions selected to best represent the equilibrium state from the cell uptake studies). The tracers compared were 67/68Ga-DOTA-PSMA, 67/68Ga-HBED-CC-PSMA, and 67/68Ga-THP-PSMA. Each 67Ga tracer was compared with each 68Ga tracer, and affinity ratios were obtained by measuring total and nonspecific binding for each tracer (γ-counting) and calculating the ratio of their specific binding.
Mouse Model of Prostate Cancer
Animal studies complied with the guideline on responsibility in the use of animals in bioscience research of the U.K. Research Councils’ and Medical Research Charities’, under U.K. Home Office project and personal licenses. Subcutaneous prostate cancer xenografts were produced in SCID/beige mice (male, 5–12 wk) by injecting 4 × 106 DU145-PSMA or DU145 cells in the right flank. Imaging was performed once the tumor had reached 5–10 mm in diameter (1–4 wk after inoculation).
PET Scanning
PET imaging was performed on 4 groups of mice (n = 3 each) under isoflurane anesthesia with a BioScan nanoPET-CT PLUS (Mediso). Mice were imaged with CT before radiotracer administration, and dynamic PET data were collected for the first hour after injection. Mice were then euthanized and organs harvested, weighed, and γ-counted. Mice bearing DU145-PSMA tumors were administered either 68Ga-THP-PSMA (5–15 MBq, 50–140 μL, 0.4–0.9 μg, group 1) or 68Ga-HBED-CC-PSMA (5–15 MBq, 50–140 μL, 0.6–1.3 μg, group 2) by tail vein injection. A third group (group 3) was coadministered 68Ga-THP-PSMA and the PSMA-inhibitor 2-(phosphonomethyl)pentane-1,5-dioic acid (PMPA) (50 μg, Enzo Life Sciences) (20,21). Mice bearing DU145 tumors were administered 68Ga-THP-PSMA (group 4).
Imaging Protocol
PET/CT Acquisition
The PET/CT scans were obtained on a BioScan nanoPET-CT PLUS (Mediso) scanner using their proprietary acquisition software (Nucline, version 2.00). CT was performed with an x-ray tube voltage of 45 kVp, 600 ms of exposure time, and 360° projections. This scan took 10 min to obtain. Dynamic PET scans were acquired within a 94.7-mm field of view from 0 to 60 min after tail vein injection of the 68Ga-PSMA tracer. Acquisition took place in 1–5 coincidence mode with a coincidence window of 5 ns and a 400- to 600-keV energy window. The dynamic PET data were reconstructed using Nucline software (version 2.00). Data were allocated to both 20- and 2-min bins using 0.4 mm3 voxels for PET and 0.21 mm3 for CT. Image processing and analysis were performed using Vivoquant software (version 1.23). Before analysis, both CT and PET images were realigned and processed to a voxel size of 0.21 mm3 and the PET output calibrated to display MBq per voxel. Regions of interest (ROIs) for each data file were produced using 3 different techniques: fixed-volume ROIs, Otsu thresholding, and freehand segmentation from the CT image. Fixed-volume ROIs were used for muscle (17.2 mm3 sphere within the thigh muscle) and blood pool (2.2 mm3 sphere in left ventricle). These ROIs were positioned manually and drawn in triplicate within each organ. Otsu thresholding was used to determine the total activity deriving from the kidneys or the bladder. This method was not suitable for tumor uptake because of the close proximity of kidney signal. Total tumor volume was drawn manually from the CT image, defined 3 times per animal, and the average value from these 3 ROIs determined. Percentage injected dose (%ID) per cubic centimeter (%ID/cm3) values for each ROI were calculated using the activity and volume of the ROI and the ID as the total activity within the image, excluding activity within the tail. Time–activity curves were produced from the 2-min bins, and total uptake was determined from the 20-min binned data at the 40- to 60-min time point. Static images were produced from dynamic data between 40 and 60 min and scaled between 0 and 25 %ID/cm3.
Biodistribution Studies
Additional 68Ga (THP-PSMA) biodistribution and blocking studies were performed in mice bearing DU145 or DU145-PSMA tumors without imaging or continuous anesthesia. 68Ga (THP-PSMA) (3.5–12.7 MBq, 0.7–0.9 μg THP-PSMA) was administered via tail vein injection under isoflurane anesthesia. After injection anesthesia ceased and 1 h after injection the animals were euthanized by cervical dislocation and organs harvested, weighed, and counted (n = 3 for each group).
Statistical Analysis
Data were analyzed in GraphPad Prism 5 (version 5.04) and expressed as mean ± SD. Student t tests were used to determine statistical significance, with a P value of less than 0.05 considered significant.
RESULTS
Synthesis of THP-PSMA
Supplemental Figure 1 shows the route used to synthesize THP-PSMA. The resin-bound PSMA inhibitor (compound 7) was prepared by in situ formation of a bis(tert-butyl) glutamate isocyanate (compound 5) followed by coupling with resin-bound protected lysine (compound 3). After deprotection and coupling with glutaric anhydride, the intermediate bearing a pendant carboxylate (compound 8) was activated and formed an amide bond with the free amine of a THP derivative (22). Cleavage from the solid support and glutamate deprotection using trifluoro acetic acid formed THP-PSMA (Fig. 1). Purification by semipreparative HPLC afforded THP-PSMA as a trifluoro acetic acid salt, yield 4 mg, 2.6 μmol, 5.2% yield from resin loading; (C54H77N11O19.(CF3COO)3) molecular weight, 1526.33, HPLC: Rt = 13.4 min, greater than 98% purity. LC-ESI-MS, 1H, and 13C NMR data are shown in Supplemental Table 1.
Radiolabeling
The THP chelator enabled radiolabeling with unmodified generator eluate, in a single step. 68Ga3+ (5–75 MBq) in aqueous HCl (0.1 M, 250 μL) was added to preprepared THP-PSMA (5–0.01 μg in 3 μL of H2O) in sodium bicarbonate solution (1 M, 27 μL). After 5 min, pH was 6.5–7.5. Radiochemical purity, determined by HPLC and iTLC as a function of THP-PSMA concentration is shown in Figure 2. Specific activities between 15 and 45 MBq/nmol were consistently achieved with the labeling conditions used for in vivo work (2 μg, 1.3 nmol THP-PSMA, 250 μL 20–60 MBq of 68Ga eluate, >95% radiochemical purity).

(A) Dependence of radiochemical purity of 68Ga-THP-PSMA on mass of THP-PSMA in 280 μL, as measured by HPLC (red) and iTLC (black) after 5 min; n = 3, mean ± SD. (B) HPLC (λ = 220 nm) of THP-PSMA (blue, RT = 13.38 min) and natGa-THP-PSMA (black, RT = 10.92 min, excess Ga(NO3)3 is present with RT = 1.47 min), and radio-HPLC of 68Ga-THP-PSMA (red, RT = 11.05 min) labeled using 1-step kit and analyzed 10 min after reconstitution. (C) iTLC of 68Ga-THP-PSMA labeled using 1-step kit, analyzed 5 min after reconstitution (Rf unchelated 68Ga = 0, Rf 68Ga-THP-PSMA = 0.8–1).
1-Step Kit for Radiolabeling
When kit vials containing lyophilized THP-PSMA (40 μg, 26 nmol), sodium bicarbonate (44 mg), and sodium phosphate buffer (17.5 mg) were used, radiosynthesis of 68Ga-THP-PSMA was achieved in 1 step by direct addition of 5 mL of generator eluate (0.1 M HCl, 122–202 MBq from the E&Z generator or 600–660 MBq from the IRE generator). After 5 min, the pH was 6–7 and iTLC confirmed radiochemical purity above 95% with specific activities of up to 22 MBq/nmol using the IRE generator (Fig. 2; Supplemental Fig. 2). iTLC analysis up to 3 h after reconstitution showed no instability or autoradiolysis (Supplemental Fig. 2).
Lipophilicity and Serum Stability
The log POCT/PBS values of the 3 68Ga-PSMA complexes at pH 7.4 were similar: −5.35 ± 0.1 for 67Ga-THP-PSMA (n = 6), −5.40 ± 0.2 for 67Ga-HBED-CC-PSMA (n = 6), and −5.40 ± 0.1 for 67Ga-DOTA-PSMA (n = 5), indicating that all tracers are hydrophilic and lipophilicity is unlikely to underlie differences in in vivo performance. In serum, 68Ga-THP-PSMA (Supplemental Fig. 3) showed minimal transchelation (<2%) to proteins after 6-h incubation.
In Vitro Cell Uptake and Binding Affinity Assays
Uptake of 67Ga-DOTA-PSMA, 67Ga-HBED-CC-PSMA, and 67Ga-THP-PSMA in DU145-PSMA and DU145 cells at 37°C and 4°C is shown in Figures 3A and Supplemental Figure 4. All 3 tracers showed time-dependent accumulation in PSMA-expressing cells, but low uptake in non–PSMA-expressing cells, confirming that uptake is PSMA-mediated. At 37°C, uptake continued to increase with time, preventing measurement of equilibrium binding parameters such as the dissociation constant (Kd), but at 4°C, a plateau was reached for all tracers after approximately 2 h. Relative affinity was therefore measured after 2 h at 4°C. Results showed that the Kd of 67/68Ga-THP-PSMA was 10.4 ± 3.4 times higher than that of 67/68Ga-HBED-CC-PSMA and 8.6 ± 3.4 times that of 67/68Ga-DOTA-PSMA, in agreement with directly measured IC50 values of Ga-THP-PSMA (361 ± 60 nM) and Ga-HBED-CC-PSMA (34.3 ± 4.1 nM) (Fig. 3B; Supplemental Fig. 5).
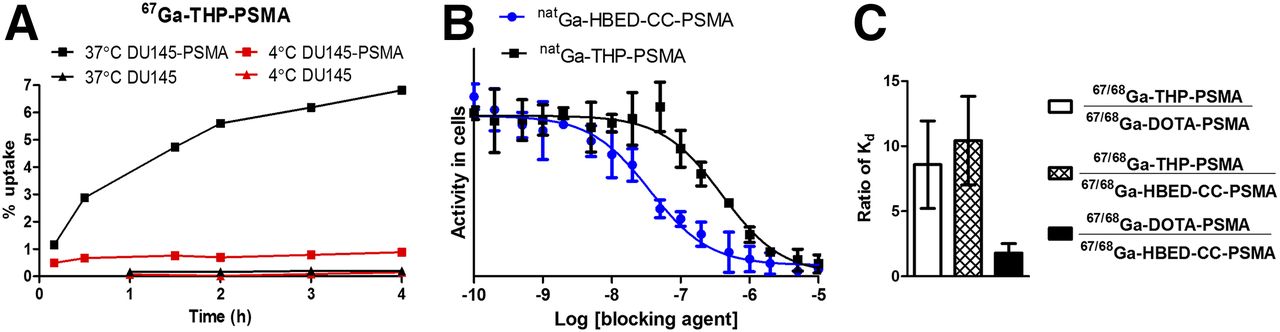
(A) 67Ga-THP-PSMA uptake over time at 4°C and 37°C, 1 × 106 cells/mL with DU145-PSMA or DU145 cells. (B) Representative IC50 experiment for natGa-THP-PSMA (black) or natGa-HBED-CC-PSMA (blue) with 1 nM 68Ga-DOTA-PSMA as probe (n = 3 for each concentration). IC50 values in main text are mean of at least 3 experiments. (C) Ratio of Kd of 2 tracers incubated at 1 nM with DU145-PSMA cells at 4°C for 2 h. Ratios were calculated from specific binding obtained by incubating 67Ga version of tracer in same well as 68Ga version of its comparator, and vice versa. Total wells (n = 34) for comparison of 67/68Ga-THP-PSMA with 67/68Ga-HBED-CC-PSMA or 67/68Ga-DOTA-PSMA and n = 18 for comparison of 67/68Ga-HBED-CC-PSMA with 67/68Ga-DOTA-PSMA; mean ± SD.
PET Imaging and Biodistribution
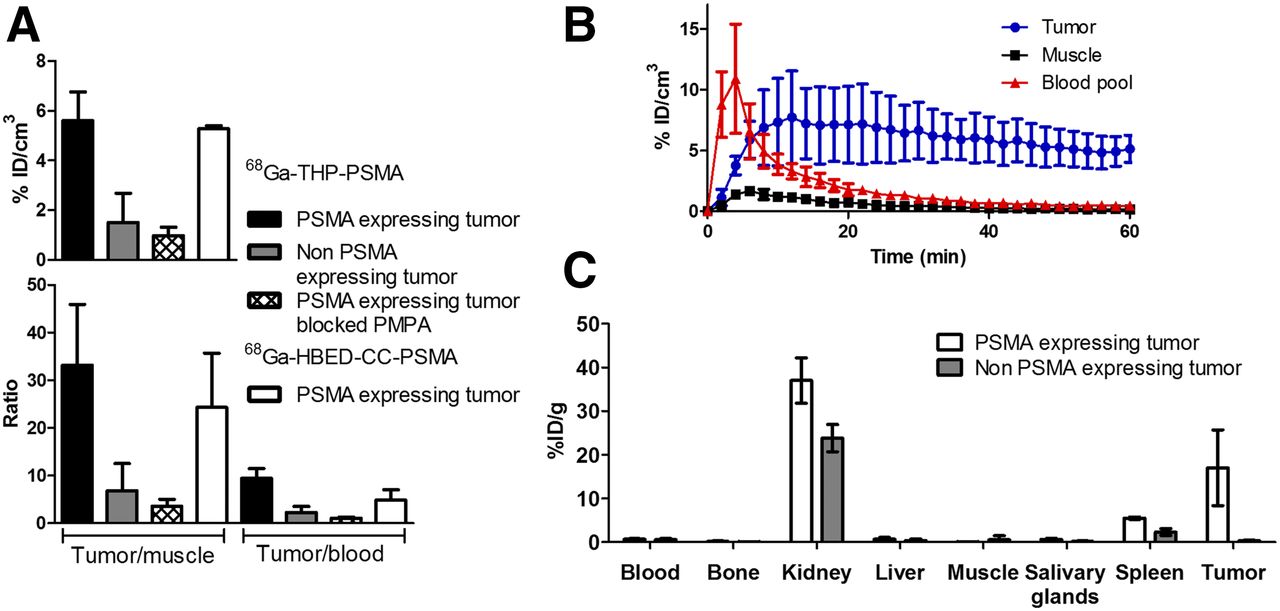
PET/CT scanning of 68Ga-THP-PSMA in SCID/beige mice bearing PSMA-positive xenografts (DU145-PSMA, group 1) showed that excretion of 68Ga-THP-PSMA was rapid and exclusively renal, with most activity associated with bladder and kidneys by 60 min (Fig. 4; Supplemental Fig. 6). Blood clearance, represented by blood pool in the left ventricle, was rapid, decreasing to 1 %ID/cm3 within 30 min (Fig. 5B). With images scaled between 0 and 25 %ID/cm3, tumors were clearly delineated (Fig. 4). Analysis of the 40- to 60-min postinjection images gave a tumor uptake value of 5.6 ± 1.2 %ID/cm3 (Fig. 5B). Specificity was confirmed both by mice bearing a PSMA-negative but otherwise similar tumor (DU145, group 4) with uptake of 1.5 ± 1.2 %ID/cm3 (P < 0.05 compared with group 1), and by blocking experiments in mice bearing a DU145-PSMA tumor where PMPA was coadministered with 68Ga-THP-PSMA (group 3), giving a tumor uptake of 1.0 ± 0.4 %ID/cm3 (P < 0.05 compared with group 1).

Representative PET/CT images of mice bearing xenografts at 40–60 min after injection with PET images scaled from 0%–25 %ID/cm3. (A) 68Ga-THP-PSMA in DU145-PSMA tumor (group 1). (B) 68Ga-THP-PSMA in DU145 tumor (group 4). (C) 68Ga-THP-PSMA in DU145-PSMA tumor blocked with PMPA (group 3). (D) 68Ga-HBED-CC-PSMA in DU145-PSMA tumor (group 2).

(A) PET/CT-derived %ID/cm3 in tumor and tumor-to-blood and tumor-to-muscle ratio 40–60 min after injection. 68Ga-THP-PSMA in DU145-PSMA tumor (black), 68Ga-THP-PSMA in DU145-PSMA tumor blocked with PMPA (hatched), 68Ga-THP-PSMA in DU145 tumor (gray), or 68Ga-HBED-CC-PSMA in DU145-PSMA tumor (white) (n = 3, mean ± SD). (B) PET-derived time–activity curves of mice bearing DU145-PSMA tumors, imaged with 68Ga-THP-PSMA for 1 h after injection: blood pool (left ventricle) (red), tumor (blue), and leg muscle (black); (n = 3, mean ± SD). (C) Ex vivo biodistribution of 68Ga-THP-PSMA in mice with DU145-PSMA (white) or DU145 tumor (gray), culled 1 h after injection (n = 3, mean ± SD). Unlike PET-imaged animals, these were not anesthetized between injection and euthanasia.
For comparison with an established PET tracer, mice bearing DU145-PSMA tumors were imaged with 68Ga-HBED-CC-PSMA (group 2) (8,9,17). Time–activity curves for 68Ga-HBED-CC-PSMA (group 2) and 68Ga-THP-PSMA (group 1) showed similar blood clearance, tumor uptake, and renal excretion (Fig. 5). Image analysis of 40- to 60-min PET images revealed that tumor uptake of 68Ga-THP-PSMA (5.3 ± 0.1 %ID/cm3) was not significantly different from that of 68Ga-HBED-CC-PSMA (5.6 ± 1.2 %ID/cm3).
Ex vivo biodistribution data, summarized in Figure 5C and Supplemental Table 2, were consistent with PET image analysis, confirming renal excretion and excellent specificity of 68Ga-THP-PSMA for PSMA-expressing tumors. 68Ga-THP-PSMA showed ex vivo biodistribution similar to 68Ga-HBED-CC-PSMA, apart from markedly lower spleen uptake (3.7 ± 1.3 and 17.6 ± 6.1 %ID dose per gram, respectively). Importantly, anesthesia appeared to severely affect tracer uptake in the kidney and excretion to bladder, with large variation in kidney uptake across all groups, so comparison of kidney and bladder activity should be interpreted with caution.
DISCUSSION
The aim of this study was to develop a 68Ga radiotracer that targets PSMA with simple and high-yielding radiolabeling procedures suitable for development into a single-step kit-formulated radiopharmaceutical compatible with good manufacturing practice, requiring only addition of unprocessed, unfractionated generator eluate to a single vial. The latter requirement was met by incorporating THP. A THP-PSMA conjugate has been synthesized and characterized, and could be readily radiolabeled with 68Ga (and 67Ga). Radiolabeling yields of greater than 95% and specific activity of 15–45 MBq/nmol with unprocessed generator-produced 68Ga were achieved in 1 step at pH 7 and ambient temperature within 5 min without further purification. On the basis of this, we developed a lyophilized kit, which can be labeled/reconstituted simply by adding 5 mL of raw generator eluate. 1-step kits can be used only with generators with less than 0.001% 68Ge breakthrough; of these, the E&Z has the largest elution volume (5 mL, 0.1 M HCl) and the kit was designed accordingly. Within the range used, specific activity was limited only by the activity available from the generator; the highest observed was 22 MBq/nmol with an elution of 660 MBq in 5 mL. Higher specific activities have been obtained in a clinical setting and will be reported alongside first-in-human studies. These radiolabeling properties demonstrate that kit-based radiolabeling of 68Ga tracers, analogous to the simple manipulations to which radiopharmacy staff producing 99mTc radiopharmaceuticals are accustomed, and requiring equipment no more complex than a shielded syringe and vial, is entirely feasible with an appropriate chelator. As well as rapid and simple radiolabeling, the coordination properties of THP endow its conjugates with other properties well suited to radiopharmaceutical application. It is highly selective for tripositive metal ions with ionic radius similar to that of Fe3+ and Ga3+ (23), potentially reducing the need to remove contaminating metal ions in raw generator eluate. Unlike HBED-CC, the tripodal design of THP restricts the number of geometric isomers that form on Ga3+ coordination: on reversed-phase HPLC, 68Ga-THP-PSMA elutes as a single radioactive species (Fig. 2B) shown by LC-MS to be a 1:1 complex with gallium (Supplemental Table 1). Any isomerism (such as the Δ/Λ isomerism possible with tripodal complexes) is subject to equilibration that is rapid compared with the timescale of HPLC and in vivo processes, and hence biologically irrelevant. The lipophilicity is low and comparable to that of established 68Ga-PSMA ligands.
Although THP-PSMA was rationally designed on the basis of previous literature (8,21,24,25), we have not yet attempted to optimize the specific target affinity of THP-PSMA bioconjugates. In vitro experiments demonstrate that 68Ga-THP-PSMA binds specifically to PSMA, although with weaker affinity than 68Ga-HBED-CC-PSMA and 68Ga-DOTA-PSMA. The simplicity of radiolabeling, excellent serum stability, and in vitro PSMA binding justified further evaluation of the new tracer in vivo, which showed that incorporation of THP into bioconjugates confers no THP-specific adverse pharmacokinetics. The rapid renal excretion, low nonspecific uptake of 68Ga-THP-PSMA in nontarget tissues, and high, specific radioactivity concentration in PSMA-expressing tumors are clinically desirable features. In the preclinical model used here, tumor uptake and pharmacokinetic properties of 68Ga-THP-PSMA and 68Ga-HBED-CC-PSMA are indistinguishable except that spleen uptake of 68Ga-THP-PSMA is lower by almost a factor of 5. Although PSMA-targeting radiopharmaceuticals generally appear to share high uptake in spleen in mice, murine spleen is not believed to express PSMA (26). It seems likely therefore that some other target capable of binding the PSMA tracers is present in the spleen and that 68Ga-THP-PSMA has enhanced ability to distinguish PSMA from this alternative target.
CONCLUSION
Use of THP as the 68Ga3+ chelator facilitates rapid chelation under mild conditions and produces a PSMA-targeted bioconjugate that can be labeled in 1 step by reconstitution of a kit with unprocessed generator eluate. Labeling requires only a generator, a cold-kit vial, a syringe, quality control facilities, and shielding. Kit-based labeling can be performed in a few minutes, using the full volume of unprocessed generator eluate (5 mL), without postsynthesis purification, achieving a greater than 95% radiochemical purity. In vivo, 68Ga THP-PSMA accumulates in PSMA-expressing tumors, with good tumor-to-background ratio delineation of PSMA-positive tumor lesions similar to 68Ga-HBED-CC-PSMA. Kit-based radiolabeling of 68Ga radiopharmaceuticals is feasible with THP and would facilitate wider and more economical use of 68Ga in hospitals, hence benefitting more patients.
DISCLOSURE
Philip J. Blower, Robert C. Hider, and Greg E. Mullen are named inventors on related patents. Greg E. Mullen and Levente K. Meszaros are current employees of Theragnostics Ltd. Jennifer D. Young is funded by the King’s College London and Imperial College London EPSRC Centre for Doctoral Training in Medical Imaging (EP/L015226/1) and Theragnostics Limited. We acknowledge support from KCL and UCL Comprehensive Cancer Imaging Centre funded by CRUK and EPSRC in association with the MRC and DoH (England), and the NIRH Biomedical Research Centre award to Guy’s and St Thomas’ NHS Foundation Trust in partnership with King’s College London and King’s College Hospital NHS Foundation Trust. PET scanning equipment was funded by an equipment grant from the Wellcome Trust. No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Apr. 13, 2017.
- © 2017 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication February 21, 2017.
- Accepted for publication April 3, 2017.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- 68Ga-Bisphosphonates for the Imaging of Extraosseous Calcification by Positron Emission Tomography
- A Phase II, Open-Label Study to Assess Safety and Management Change Using 68Ga-THP PSMA PET/CT in Patients with High-Risk Primary Prostate Cancer or Biochemical Recurrence After Radical Treatment: The PRONOUNCED Study
- Cold Kit for Prostate-Specific Membrane Antigen (PSMA) PET Imaging: Phase 1 Study of 68Ga-Tris(Hydroxypyridinone)-PSMA PET/CT in Patients with Prostate Cancer