


Abstract
Castration-resistant prostate cancer (CRPC) is the lethal form of prostate cancer, and more than 26,000 men will die from this disease in 2016. The pathophysiology of CRPC is clearly multifactorial, but most often, androgen receptor (AR) upregulation is associated with its earliest beginnings and the AR increase is part of the multimolecular complex including downstream effector proteins linked to AR (AR-axis) responsible for rapid proliferation and malignant features of the malignant cell. In both animal models and patients, glycolysis (Warburg effect) is also an early manifestation of CRPC transformation. At Memorial Sloan Kettering Cancer Center, we have focused our energies on imaging studies of the AR-axis in CRPC, using 18F-FDG, 18F-16β-fluoro-5α-dihydrotestosterone (18F-FDHT), and a variety of radiolabeled antibodies targeting downstream effectors, such as prostate-specific membrane antigen (PSMA). Small-molecular-weight PSMA-targeting agents are not part of this review. In this review, we will focus on molecular imaging of the AR-axis in metastatic CRPC (mCRPC) and discuss our personal experience with these tracers. Our goal is to put these radiopharmaceuticals in the context of mCRPC biology and diagnosis (e.g., 18F-FDHT).
- molecular imaging
- oncology: GU
- radioimmunoimaging
- androgen receptor–axis imaging
- CRPC
- castration-resistant prostate cancer
Prostate cancer, when detected before spread from the prostate gland, may be completely eradicated by surgery or local radiotherapy. Approximately one third of patients will fail primary treatment, and a rising prostate-specific antigen (PSA) level will herald the onset of recurrent or metastatic tumor. At this stage, PSA levels will usually decline with hormonal castration to levels of androgen hormone below 50 ng/dL. Typically, in castrate patients, PSA begins to rise again within approximately 16–18 mo (median) despite castration, heralding the onset of CRPC.
MOLECULAR IMAGING OF AR WITH 18F-FDHT
Discovery and Clinical Translation of 18F-FDHT
John Katzenellenbogen at the University of Illinois (Urbana-Champaign) and Michael Welch at Washington University (St. Louis) developed steroid-based radioligands with sufficient affinity to hormone receptors (i.e., estrogen receptor/AR) to quantify receptor status in breast and prostate cancer patients using whole-body PET. The highly versatile and easily obtained isotope 18F with its 110-min half-life was chosen for labeling (1–3). In vitro and in vivo biologic data demonstrated that 18F-FDHT was the best choice for AR imaging in prostate cancer in light of its good balance of in vivo stability, ease of production, and similar binding affinity for AR, while retaining selectivity from other nuclear receptors (e.g., estrogen receptor, glucocorticoid receptor, and progesterone receptor) (Supplemental Fig. 1 [supplemental materials are available at http://jnm.snmjournals.org]).
The study that set the stage for clinical translation was imaging in baboons showing good, saturable focal uptake in AR-positive tissue, with an excellent tumor-to-background ratio (4). About 8 y later, the first in-human PET imaging studies in castrate patients were reported by Memorial Sloan Kettering Cancer Center and Washington University (5–7). The production methodology developed by Washington University continues to be used to this day, though recent improvements have been made in the radiosynthetic chemistry (8) and to the production process by automation (9,10).
Pharmacokinetics of 18F-FDHT
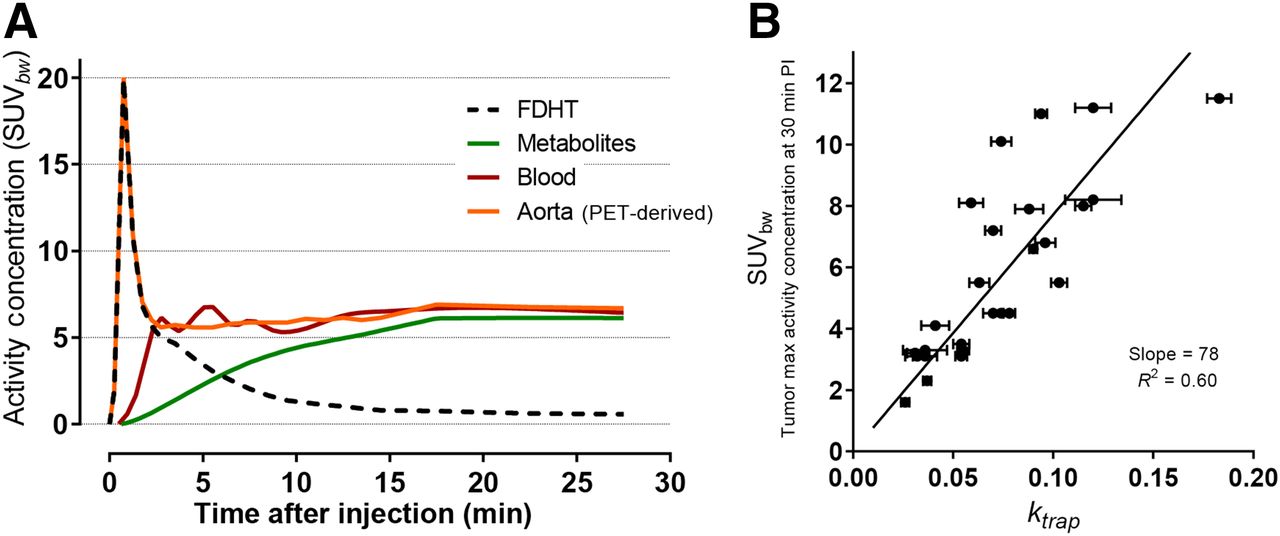
Administered as an intravenous infusion in the brachial vein, a typical dose of 185–370 MBq (5–10 mCi) of 18F-FDHT (1–4 nmol; 0.3–1.2 μg mass) rapidly binds to serum proteins and albumin- and sex hormone–binding globulin-bound state in blood, as one would expect for a hydrophobic steroid (Fig. 1) (6). In men, most intact 18F-FDHT has a plasma half-life of only 5–7 min and clears from the bloodstream in 15 min (Fig. 2A) (6). Via primarily hepatic metabolism, radiometabolites appear over the same timeframe, and the system plateaus at 20 min after injection. The metabolites are excreted in the bile, and a portion is refluxed into the blood. Fortunately, the metabolites do not compete with 18F-FDHT binding to AR.
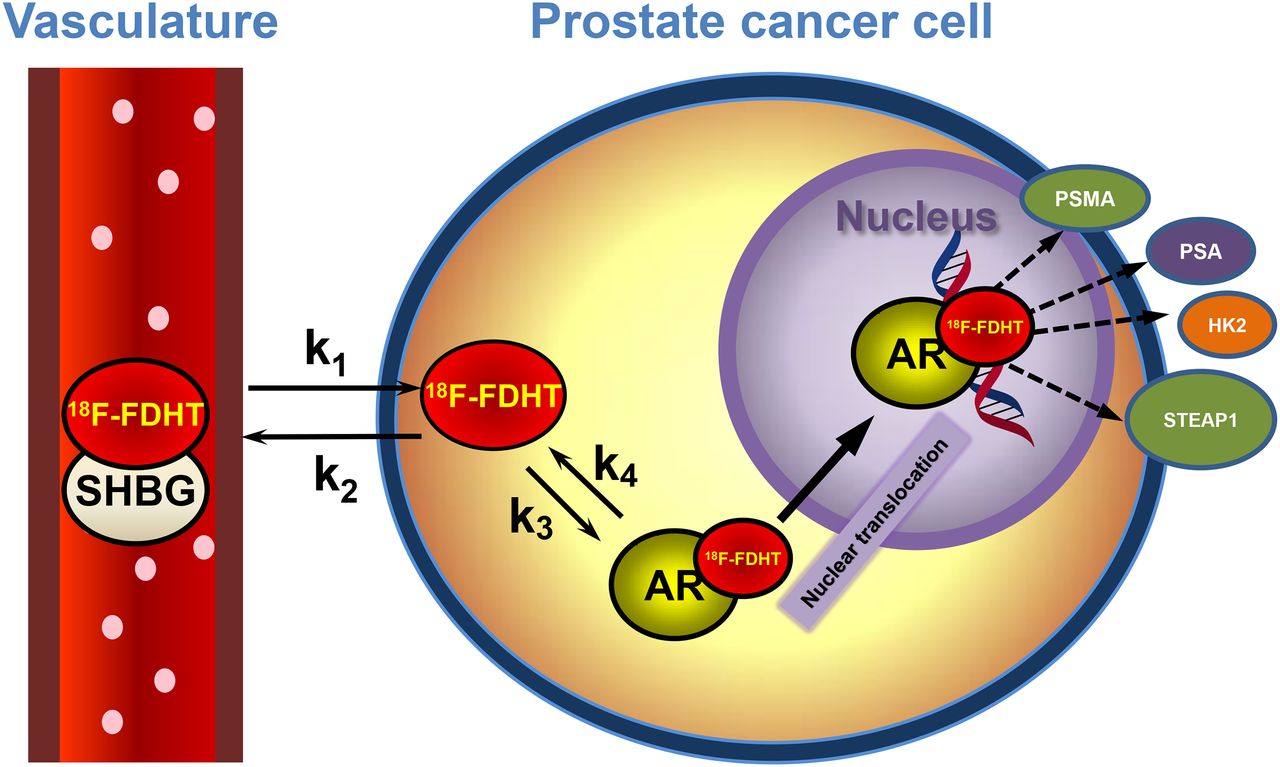
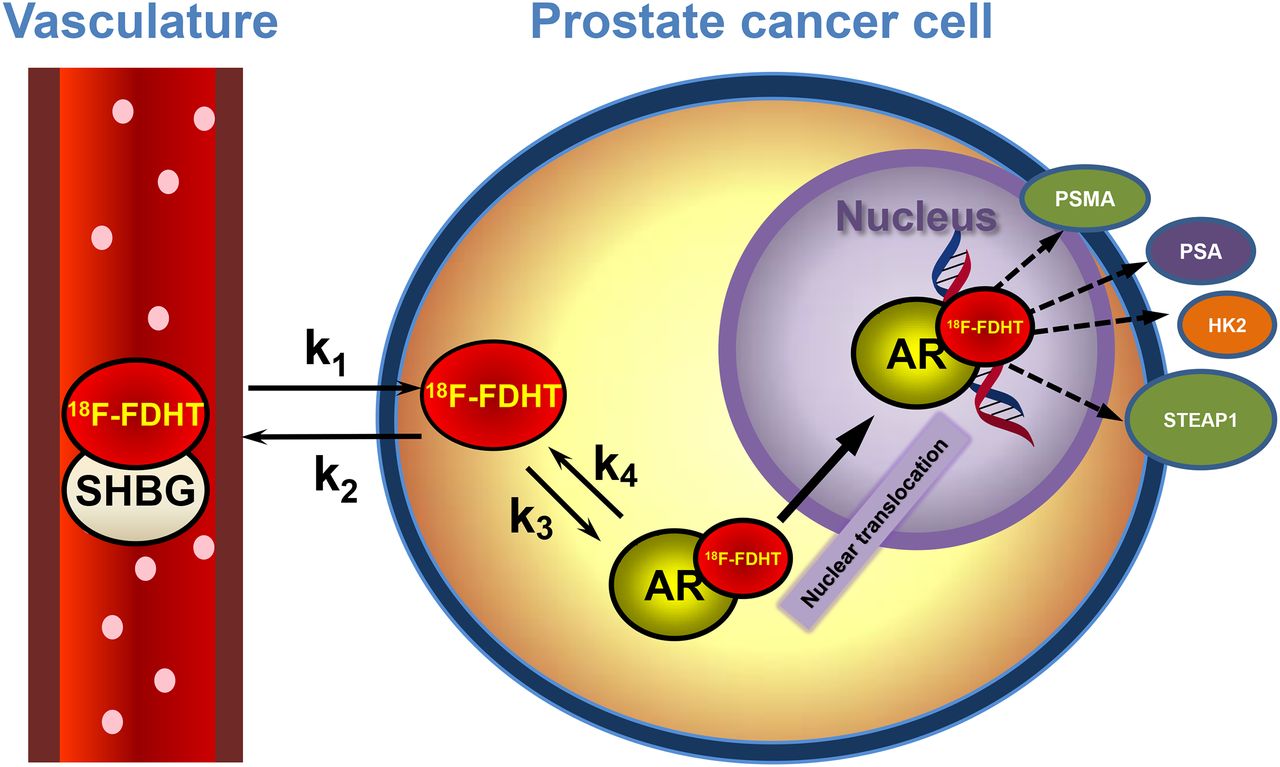
AR-axis PET imaging. 18F-FDHT is high-affinity DHT analog binding AR in cytoplasm. 18F-FDHT–AR complex localizes to nucleus, where it binds hormone-responsive elements on DNA, activating androgen-linked proteins such as PSA (secreted), PSMA (membrane), HK2, and STEAP. These may be imaged with 89Zr-labeled antibodies.

(A) Time–activity curves of 18F-FDHT in mCRPC patient in body weight–corrected SUV units from PET region of interest–derived data (aorta) or blood sample data (11). (B) Scatterplot showing weak relationship between unidirectional rate constant, ktrap, and tumor SUVbw (at 30 min; normalized by body weight) for 18F-FDHT PET in patients with prostate cancer. This linear regression analysis was constrained through origin. (Adapted with permission of (11).)
Beattie et al. applied linear compartmental pharmacokinetic modeling to 18F-FDHT uptake into tumor to a simple 2-compartment model in which the destination compartment was considered a permanent, unidirectional parameter, ktrap, attributed to intramolecular transport and binding to AR; a population-based input function was calculated by averaging aorta region-of-interest data from dynamic PET scans of a 25-patient cohort (11) (Fig. 2B). This kinetic modeling approach shows that uptake by PET imaging correlates with AR expression levels. In a recent study supported by the Movember GAP2 project, these findings were confirmed and refined by Kramer et al. at VU Amsterdam, who reported an improvement over using body weight–normalized SUV in dynamic 18F-FDHT PET images of 31 lesions in a 4-patient cohort. They observed a high correlation (R2 = 0.97) between SUVAUC,PP (SUV normalized to 18F-FDHT plasma area under the curve) and an irreversible trapping constant in a 2-tissue-compartment model, using an input function derived from both PET and venous blood sample data to correct for metabolites (12). Such studies may make it more practical to obtain biologically relevant quantitation of receptor number.
Clinical Molecular Imaging of AR in Men
18F-FDHT binds with high affinity to AR-expressing tumor tissues in men. Patient selection should include serum androgen measurements, because AR imaging is practical only in castrate patients (currently) with testosterone levels below 50 ng/dL. The active radiotracer 18F-FDHT is an agonist of AR and an analog of DHT, the most common androgen in prostate and prostate cancer at the cellular level in men. The 18F-FDHT uptake, as quantified by SUVmax or related SUV parameter, plateaus within about 30 min, with no further uptake thereafter. A single patient dose at the conservative 5-cGy threshold for diagnostic studies corresponded to 331 MBq (8.9 mCi) (7).
An example of a PET imaging study using 18F-FDHT is shown in Figure 3. Normally, subjects are studied within the same time window (a few days), with 18F-FDG as well, because we have found that there is considerable heterogeneity of biochemical features of individual CRPC lesions, when glycolysis and AR expression are compared. In addition, the companion CT provides anatomic orientation to lesion site.
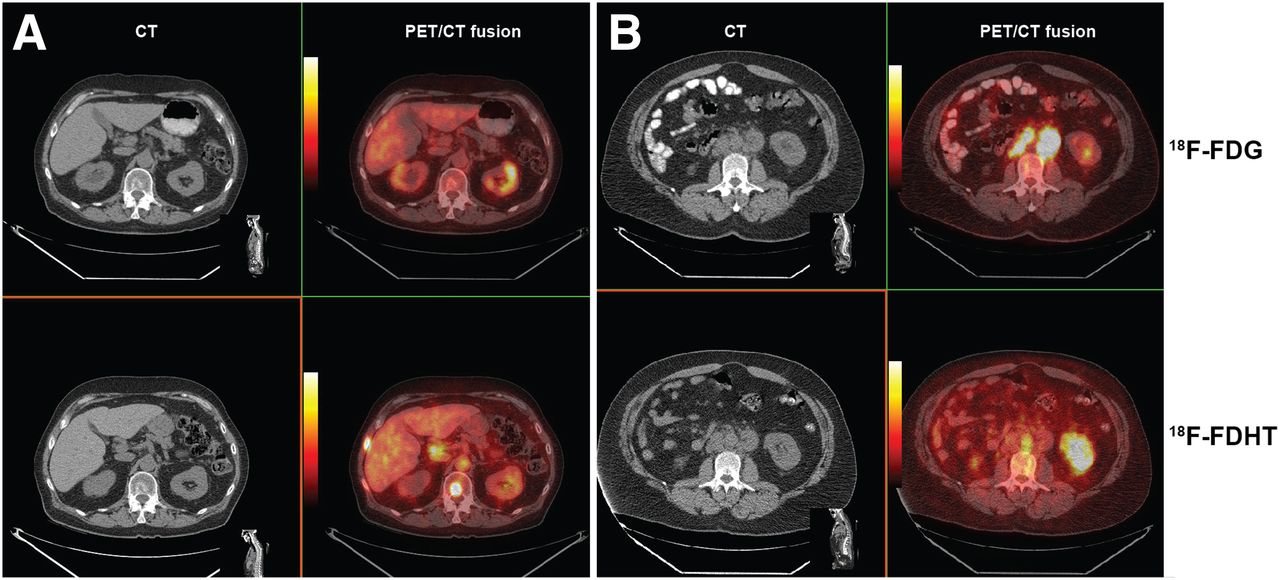
(A) CT and PET/CT fusion images showing relationship of CT findings and molecular imaging changes. CT scan shows osteoblastic rim around metastasis in the T-12 vertebrae body. 18F-FDG image (top row) shows no localization in tumor-bearing site, whereas 18F-FDHT image obtained 24 h later shows high-intensity uptake (bottom row). (B) CT and PET/CT fusion images in another patient with bulky metastasis in periaortic lymph nodes. 18F-FDG imaging (top) shows avid uptake in bulky tumor-bearing lymph nodes, whereas 18F-FDHT imaging performed 24 h later shows no nodal uptake (bottom). Note aortic activity of bound metabolite.
Hitting the Target: In Vivo Displacement of 18F-FDHT by AR Inhibitor Drugs
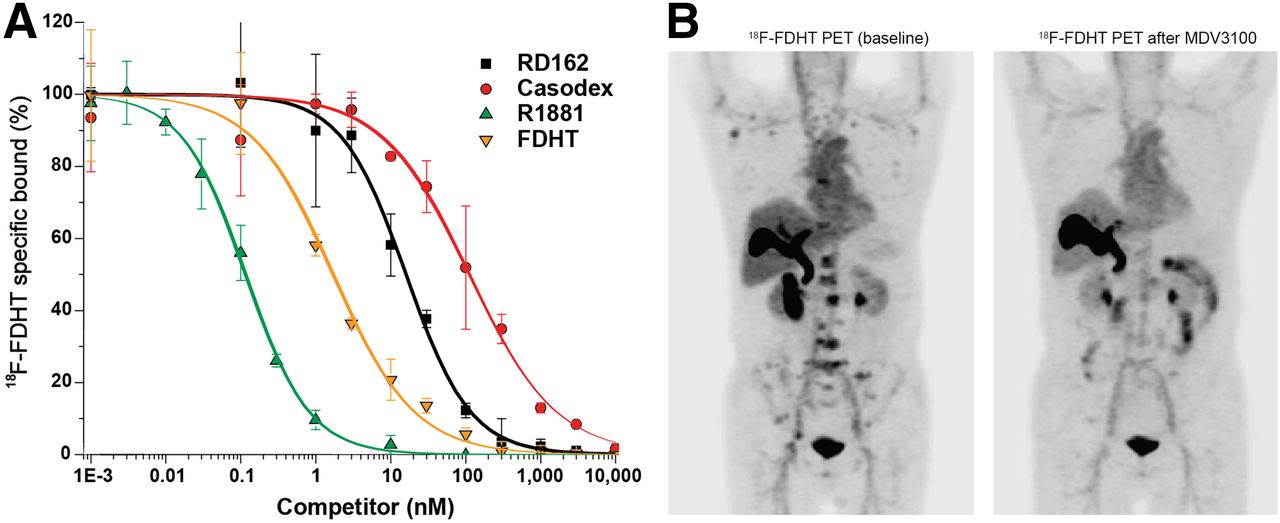
In the last few years, high-affinity AR inhibitors have been developed and approved by the U.S. Food and Drug Administration for use in CRPC on the basis of prolonged survival. One of these, enzalutamide, has been studied in men, using 18F-FDHT imaging to show whether or not the AR blockade of 18F-FDHT uptake is occurring, as would be expected for effective therapy. Figure 4 shows in vitro and in vivo studies in support of the concept that 18F-FDHT may be used as a probe to demonstrate the high-affinity binding requisite for pharmacologic AR blockade in vivo (13).

(A) In vitro displacement studies showing expected differences in range of displacement of 18F-FDHT, in proportion to binding affinity of several drugs that bind to AR in this assay. 18F-FDHT and DHT (natural androgen) have a binding affinity similar to AR. Also shown are competitor drugs with higher affinity (R1881) and lower affinity (bicalutamide and RD162). (B) 18F-FDHT PET imaging in patient with mCRPC. Note 18F-FDHT uptake in patient’s bones before treatment (left) and after therapeutic doses of daily enzalutamide (right).
Determining Biologically Effective Dose for AR Blockade
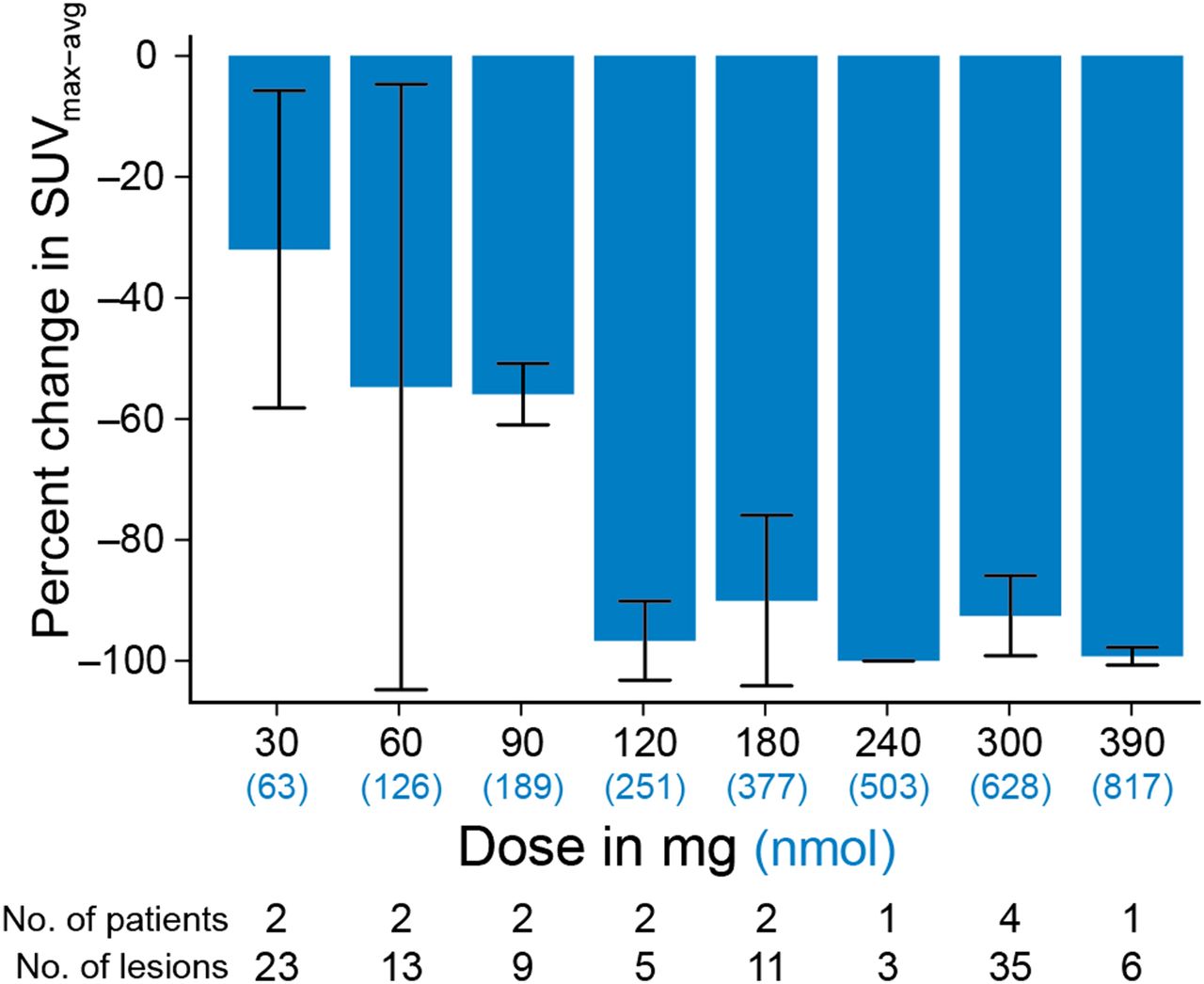
In a first-in-human study of optimizing dosing schedules for a novel second-generation AR blockade drug, apalutamide, 18F-FDHT was also used to explore progressive AR blockade in a phase I trial of dose escalation (Fig. 5) (14). Sixteen patients received a baseline and follow-up 18F-FDHT scan during the course of dose escalation, with doses ranging from 30 to 390 mg. A background-corrected SUVmax was determined as previously described (15), and all lesions in individual patients were averaged. A mean of the SUVmax parameter at each drug level was then correlated with dose level, with saturation levels beginning at 120 mg. Because seizures had been observed at highest doses of other AR inhibitors, a final phase II dose of 240 mg was chosen to ensure saturation and minimize neurotoxicity.
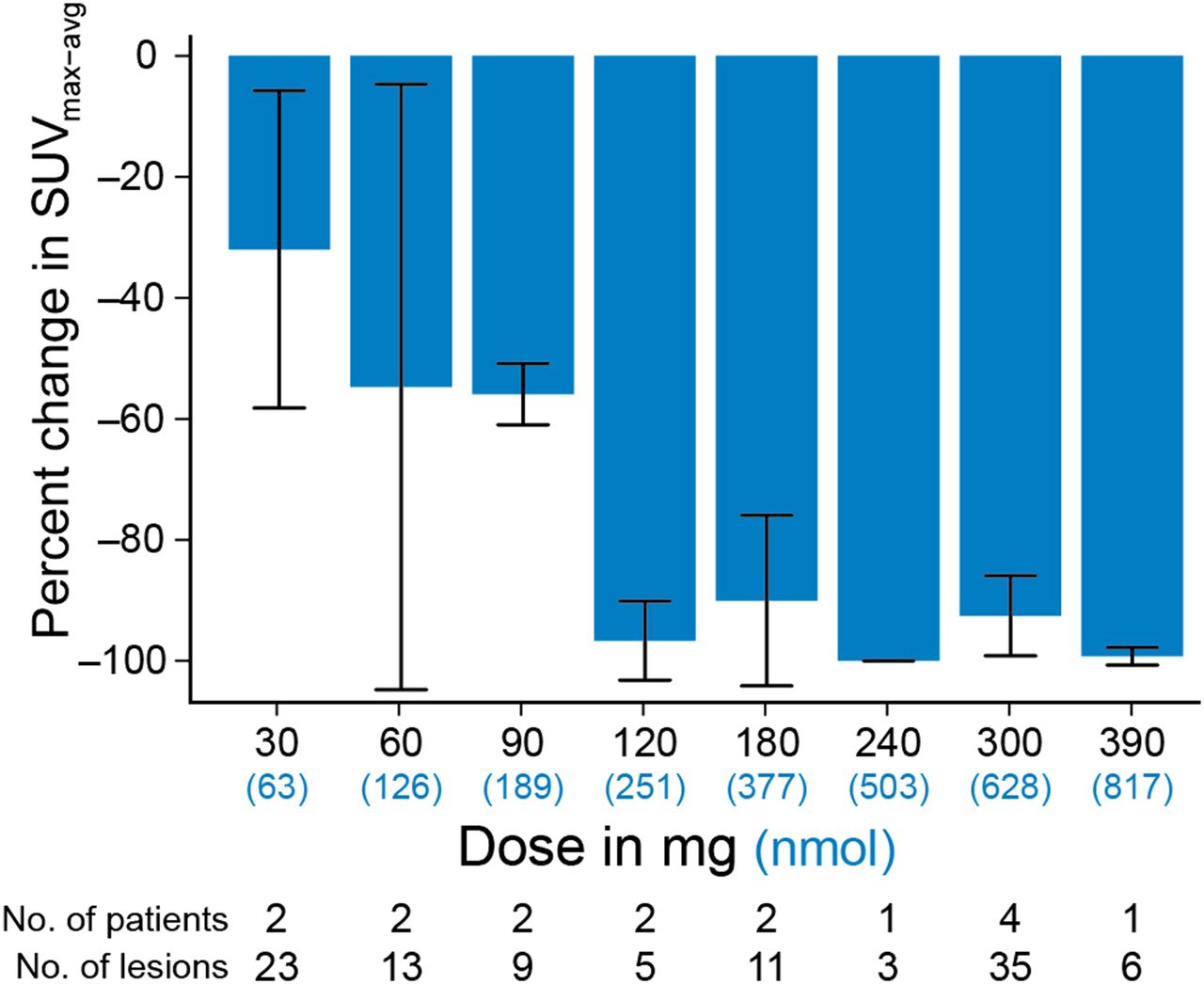
Percentage change from baseline in SUVmean average in cohorts of patients with mCRPC who were being treated with apalutamide, an AR inhibitor. Progressive decline in follow-up SUV is consistent with progressive saturation of AR at tumor site. (Modified from Figure 1 and reprinted with permission of (14).)
PSMA IMAGING WITH RADIOLABELED ANTIBODIES
AR-Directed Agents and PSMA Targeting with Antibodies
PSMA is a well-established marker for prostate cancer that is highly expressed in prostate cancer cells. Although highly specific for prostate, it is also expressed in the cells of the small intestine, proximal renal tubules, and salivary glands (16,17). However, the level of expression in the prostate cells is 100- to 1,000-fold higher than in nonprostate tissue (18,19). PSMA is a type II integral cell surface membrane protein that is not secreted, thereby making it an ideal target for monoclonal antibody (mAb) imaging or therapy. PSMA has been found to have folate hydrolase and glutamate carboxypeptidase activity (20–22). PSMA upregulation correlates with increased aggressiveness and recurrence (23,24) and higher mortality (25), suggesting a functional role of PSMA in prostate cancer progression.
PSMA-targeted imaging with 111In-labeled mAb was approved by the Food and Drug Administration as 7E11/CYT-356 Capromab (ProstaScint) to detect disease in patients (26–29). Initial clinical enthusiasm was tempered by the fact that 7E11 targets the internal portion of the PSMA molecule, which is less accessible, and targeting was suboptimal in intact cells versus necrotic tissue, and viable tumor was missed (30,31).
Given the limitation of Capromab, targeting of the extracellular domain of PSMA was explored (31–33) and found to be more effective. J591 binds to the external domain of PSMA with high-affinity binding to prostate cancer cells in tissue culture and animal models (33,34).
Imaging with Radiolabeled J591
For human studies, J591 has been conjugated with a metal chelating agent (e.g., diethylene triamine pentaacetic acid or DOTA) to enable labeling with radiometals such as 111In or 177Lu (35). More recently, J591 has been radiolabeled with a PET radioisotope, 89Zr, using the siderophore desferrioxamine as a chelator (36,37).
Initial phase I studies of huJ591 trace-labeled with 111In using a DOTA chelate showed that repetitive dosing was well tolerated, with total doses of up to 500 mg/m2 without the development of a human antihumanized (deimmunized) antibody response (38,39). No dose-limiting toxicity occurred, and the maximum tolerated dose was not reached. Excellent tumor targeting occurred at all dose levels of mAb. No mAb targeting to sites other than those involved by prostate cancer was observed, although, as seen in other trials using radiometals, the liver is the primary site of metabolism. Percentage injected dose in the liver diminished with increasing dose of antibody, and higher doses were associated with longer plasma clearance times (40).
In a dose escalation study, 111In-J591 antibody was given in combination with cold J591 administered in escalating doses from 25 to 100 mg. Dose-dependent plasma clearance of the antibody occurred more slowly as antibody mass increased up to 100 mg (41). The mean biologic half-life was 0.96, 1.9, 2.75, and 3.47 d for the 10-, 25-, 50-, and 100-mg doses, respectively. Hepatic saturation was achieved at a 10- to 25-mg dose of antibody, and the optimal tradeoff between increased J591 circulation and liver uptake was obtained with 25 mg of total antibody dose (41).
In a detailed analysis of lesion targeting with 111In-J591, both bone and soft-tissue lesions were targeted well, with localization in about 94% of skeletal lesions detected by conventional imaging (42). Visualization of lesions was better in delayed images and at later infusions of higher antibody masses. Imaging was similarly high with 177Lu-J591 (43).
PET allows for superior image resolution and ability to quantify uptake as compared with single-photon imaging, enabling better dosimetry estimations for normal organs and tumors in vivo. 89Zr has been increasingly used for immuno-PET imaging because of its favorable decay characteristics, including a radioactive half-life of 78.4 h, which is more suitable for imaging antibody uptake. Chelation methodology using desferrioxamine has been described for labeling antibodies (37,44), and preclinical studies with 89Zr-J591 have demonstrated excellent targeting of prostate cancer in vivo (44).
In a first-in-human phase I study with 89Zr-J591 in prostate cancer patients, the clearance of 89Zr-J591 from serum was biexponential, with biologic half-lives of 7.0 ± 4.5 h (range, 1.1–14 h) and 62 ± 13 h (range, 51–89 h). Whole-body clearance was monoexponential, with a mean half-life of 219 ± 48 h (range, 153–317 h). Dosimetric estimates to critical organs showed highest dose to the liver (208 ± 40.5 cGy/GBq [7.7 ± 1.5 cGy/mCi]), with lesser doses to renal cortex (95 ± 10.9 cGy/GBq [3.5 ± 0.4 cGy/mCi]) and bone marrow (32 ± 5.3 cGy/GBq [1.2 ± 0.2 cGy/mCi]). The optimal time for PET imaging after injection was 7 ± 1 d (45).
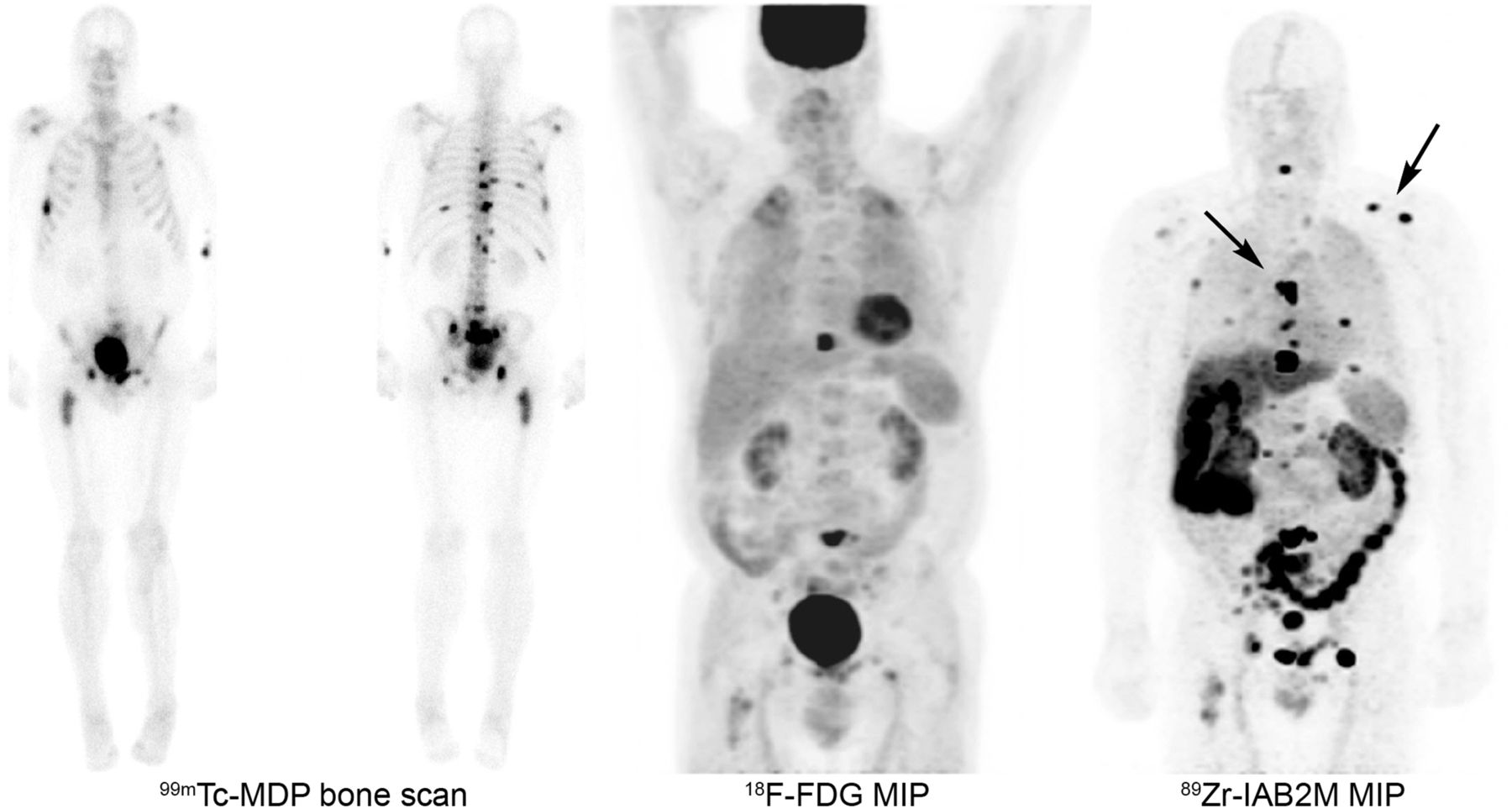
A follow-up analysis of lesion targeting in 50 patients with CRPC imaged using 89Zr-J591 PET/CT showed good localization of both bone and soft-tissue lesions (46). Higher targeting, including both uptake and lesion detection, was seen for bone lesions than for soft-tissue lesions. The median SUV was significantly higher for bone lesions than for soft-tissue lesions (8.9 vs. 4.8, respectively; P < 0.00003). In a comprehensive comparative analysis with conventional imaging, 89Zr-J591 detected more osseous sites relative to conventional methylene diphosphonate bone scanning and CT scanning (Fig. 6). However, soft-tissue disease detection was inferior to CT scanning. Pathology correlation showed a high overall accuracy of 89Zr-J591 (95.2%) for osseous lesions and slightly lower (60%) for soft-tissue lesions.

A 74-y-old man with prostate carcinoma with PSA 8.8. Bone scan showed multiple bone lesions that were stable. 18F-FDG PET scan showed mild uptake in some but not all bone lesions. 89Zr-J591 PET imaging (upper, arrows) showed more extensive uptake and involvement of skeletal system, with uptake in a number of lesions not clearly seen on bone scan and that were also non–18F-FDG-avid (lower). MIP = maximum-intensity projection.
Bayesian analysis was used to predict the number of positive lesions among the unbiopsied sites for each modality separately for bone and soft tissue (46). The results showed the highest predicted number of positive findings for J591 in osseous lesions, as compared with other conventional modalities (Supplemental Table 1).
ANTI-PSMA MINIBODY IMAGING
It has been shown that antibody fragments such as minibodies and diabodies clear faster and may allow for early imaging of tumor sites (47,48). Fragments below 60 kDa are filtered through the glomerular system, leading to significant kidney excretion (49–52), which is not ideal for prostate cancer imaging agents; on the other hand, minibodies are slightly larger and do not have high renal clearance. IAB2M is an 80-kDa minibody genetically engineered from the parent humanized anti-PSMA mAb J591, consisting of a bivalent homodimer, with each monomer comprising a single-chain variable fragment (scFv) linked to a human IgG1 CH3 domain, that targets the extracellular domain of PSMA (Supplemental Fig. 2). IAB2M lacks Fc receptor interaction domains on the minibody that make it pharmacologically inert to Fc-mediated effector functions (50–56).
In a phase I first-in-human study evaluating PET imaging with 89Zr- IAB2M (57), there was rapid clearance of the minibody from the blood. Lesions were seen as early as 24 h after injection, with most lesions seen by 48 h. Targeting to both bone and soft-tissue lesions (Fig. 7) and high correlation with pathology was observed.

A 68-y-old man with prostate carcinoma with increasing level of PSA 24. Bone scan showed multiple stable bone lesions. 18F-FDG PET scan showed mild uptake in a few lesions. 89Zr-IAB2M PET imaging showed more lesions in skeletal system, with visualization of lesions not seen on bone scan or 18F-FDG (arrows).
CONCLUSION
Prostate cancer is biologically heterogeneous, with clinical behavior ranging from more benign to lethal. Prostate cancer imaging with radionuclides has come a long way in the past decade, with the introduction of a variety of new agents that provide detection of early and late metastases for many of these clinical states, particularly early recurrence with PSA. Several reviews provide summaries of the role of molecular imaging agents for prostate cancer (e.g., Kircher et al. (58)). Our group has focused principally on mCRPC, the lethal form of the disease, with tracers most relevant to AR-axis stimulation and growth, such as 18F-FDHT, 18F-FDG, and radioantibodies that target a variety of downstream effector proteins that can be used to monitor the response of tumor to treatment as well as the status of AR-axis activity in late stages of the disease, through PET imaging of antibodies. In this review, we have emphasized our work with AR imaging and PSMA-targeted antibodies.
From a clinical point of view, 18F-FDHT has proven to be highly accurate for detecting the presence of AR (data not shown), which in turn is the most common driver of the CRPC state. The utility of 18F-FDG comes from the ability to monitor treatment response in CRPC. 18F-FDHT has proven uniquely useful in documenting the interaction of second-generation AR-axis inhibitors with AR binding in human tumors during clinical trials, and in this way has facilitated acceptance by regulatory agencies of these drugs for therapy in CRPC.
The explosion of interest in small molecules targeting PSMA, particularly in Europe (59), is one of the reasons we emphasized our studies with J591, a high-affinity anti-PSMA antibody. PSMA antibody imaging and therapy with radioantibodies is feasible and provides a specific technique to evaluate viable disease. Given certain practical limitations of longer circulation times of the antibody, methods to reduce circulation times and use of smaller molecules would be more suitable and should be further explored. PSMA-targeted therapeutics may offer another option for therapy especially in those with refractory disease (supplemental materials).
One final point: new targeting agents have recently been approved by the Food and Drug Administration, such as 18F-fluciclovine (60), 11C-choline (61), and 68Ga-PSMA (59). All of these agents provide high-quality images and are documented to be useful for image-guided biopsy to detect occult tumors, especially in PSA recurrence after primary treatment of prostate cancer. Often, we are called on to choose the best molecular imaging agent, which is a difficult question; enthusiasm based on superb images is not proof. Instead, we propose considering biopsy-based approaches with careful statistics to estimate error rates, such as the Bayesian method illustrated in Supplemental Table 1. In a clinical research setting, in which we can perform a battery of tests on the same patients, comparison of 95% confidence for accuracy of individual tests allows us to rank-order the effectiveness of a new test—in this case, 89Zr-J591—compared with standard tests, such as CT, 18F-FDG, and 99mTc bone scanning. Each of these exciting new small molecules will have to be studied in a similar fashion, perhaps head-to-head, to choose which of these newly introduced imaging tracers will prove to be most effective.
DISCLOSURE
No potential conflict of interest relevant to this article was reported.
- © 2016 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication July 14, 2016.
- Accepted for publication August 15, 2016.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}