


Abstract
Targeted diagnosis and therapy enable precise tumor detection and treatment. Successful examples for precise tumor targeting are diagnostic and therapeutic radioligands. However, patients with tumors expressing low levels of the relevant molecular targets are deemed ineligible for such targeted approaches. Methods: We performed a screen for drugs that upregulate the somatostatin receptor subtype 2 (sstr2). Then, we characterized the effects of these drugs on transcriptional, translational, and functional levels in vitro and in vivo. Results: We identified 9 drugs that act as epigenetic modifiers, including the inhibitor of DNA methyltransferase decitabine as well as the inhibitors of histone deacetylase tacedinaline and romidepsin. In vitro, these drugs upregulated sstr2 on transcriptional, translational, and functional levels in a time- and dose-dependent manner. Thereby, their combinations revealed synergistic effects. In vivo, drug-based sstr2 upregulation improved the tumor-to-background and tumor-to-kidney ratios, which are the key determinants of successful sstr2-targeted imaging and radiopeptide therapy. Conclusion: We present an approach that uses epigenetic modifiers to improve sstr2 targeting in vitro and in vivo. Translation of this method into the clinic may potentially convert patients ineligible for targeted imaging and therapy to eligible candidates.
Somatostatin analogs are valuable tools for targeted imaging and therapy of somatostatin receptor subtype 2 (sstr2)–expressing malignancies such as neuroendocrine tumors and meningiomas (1–4). sstr2-targeted imaging identifies tumors undetectable with conventional imaging modalities (5,6), and sstr2-targeted therapy achieves responses in tumors that failed all previous treatments (7).
However, several patients are not eligible for these targeted approaches. Patients with neuroendocrine tumors or meningiomas that express marginal sstr2 levels (8) or that lost sstr2 expression during tumor progression (9), or patients with other tumor entities that normally express low sstr2 levels, such as prostate cancer (10), currently cannot benefit from sstr2-targeted imaging and therapy.
Herein, we describe a method to enable targeted therapies for these patients. We screened for drugs that upregulate sstr2 (Fig. 1), characterized their efficacy in vitro (Fig. 2), assessed synergistic effects (Fig. 3), and validated the upregulating effects in vivo (Fig. 4).

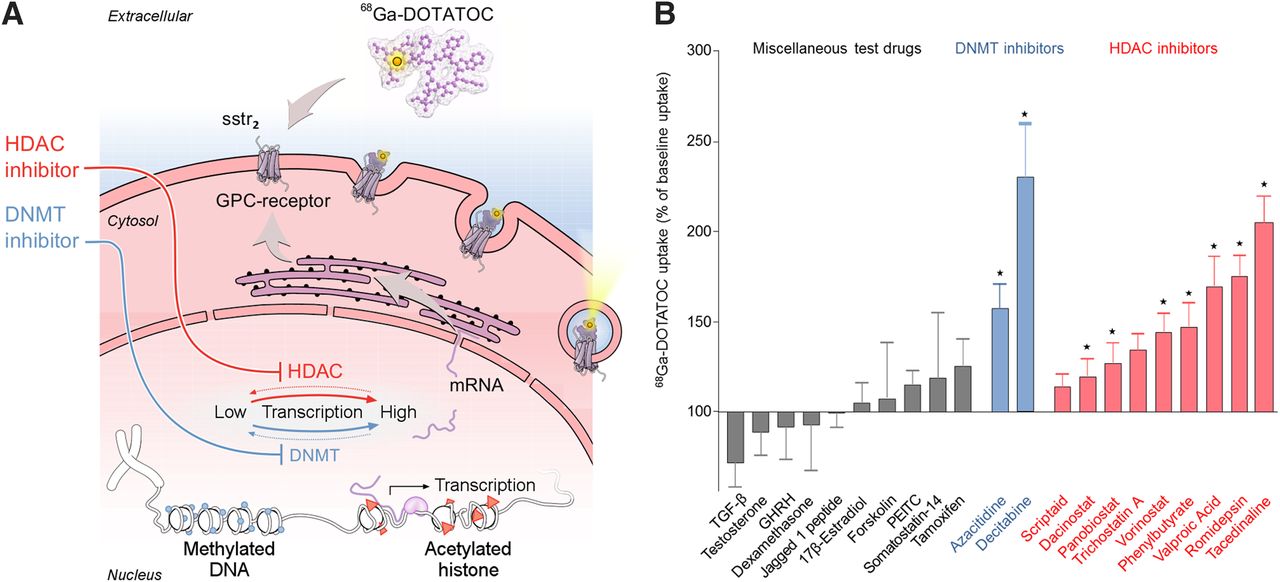
Screen revealed drugs that upregulate targets for molecular diagnosis and therapy. (A) Principle of drug screen: cells were tested for their baseline uptake of 68Ga-DOTATOC. Subsequently, they were treated with test drugs (Supplemental Table 1), including inhibitors of DNMT and HDAC. DNMT inhibitors and HDAC inhibitors reduce DNA methylation and stimulate histone acetylation, respectively, which induces upregulation of gene expression. Upregulation of target sstr2 led to increased internalization of its ligand 68Ga-DOTATOC. Readout of screen was sstr2-mediated internalization of 68Ga-DOTATOC, monitored via its γ-emission. (B) Drug-induced changes in 68Ga-DOTATOC uptake, normalized to baseline uptake (=100%) in untreated cells. Statistical significance was tested using Student t test and is indicated (*P < 0.05). GHRH = growth hormone–releasing hormone; PEITC = phenethyl isothiocyanate; TGF-β = transforming growth factor β.
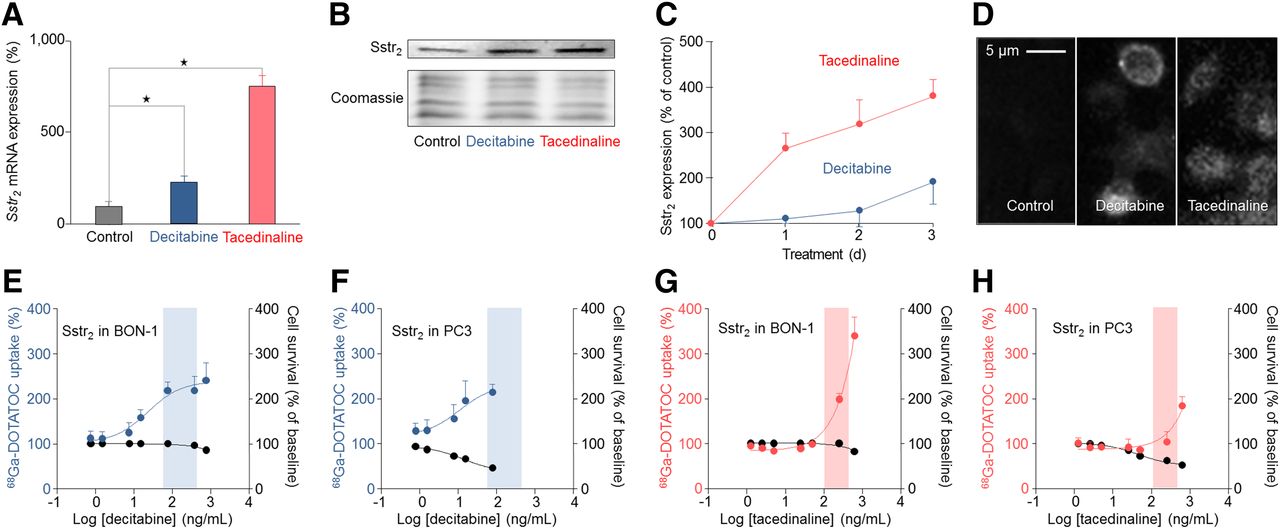
Drugs identified by screen improve molecular targeting. qRT-PCR and Western blot, ELISA, and immunocytochemistry with sstr2-specific antibodies were performed in BON-1 and PC3 cells to assess effects of decitabine and tacedinaline on sstr2 expression versus untreated controls. Decitabine and tacedinaline were tested at their therapeutic serum concentrations. Cell survival and uptake assays at different drug concentrations were performed to assess dose-dependent toxicity and changes in sstr2 expression. All results are expressed as changes in percentage and normalized to results in untreated cells. (A) qRT-PCR showing sstr2 messenger RNA expression in untreated BON-1 cells and BON-1 cells treated with decitabine (75 ng/mL) or tacedinaline (500 ng/mL). (B) Western blot showing sstr2 expression in untreated BON-1 cells and BON-1 cells treated with decitabine (75 ng/mL) or tacedinaline (500 ng/mL). Coomassie staining displays remaining protein after blotting. (C) ELISA showing sstr2 expression over a time period of 1, 2, and 3 d in BON-1 cells treated with decitabine (75 ng/mL) or tacedinaline (500 ng/mL). (D) Immunocytochemistry showing sstr2 expression in untreated BON-1 cells and BON-1 cells treated with decitabine (75 ng/mL) or tacedinaline (500 ng/mL) for 3 d. Dose-dependent effects of decitabine on uptake of sstr2 ligand 68Ga-DOTATOC in BON-1 cells (E) and PC3 cells (F). Dose-dependent effects of tacedinaline on uptake of sstr2 ligand 68Ga-DOTATOC in BON-1 cells (G) and PC3 cells (H). Therapeutic serum concentration of decitabine (65–460 ng/mL) and tacedinaline (135–570 ng/mL) is indicated by highlighted areas. Dose-dependent cell survival representing toxicity of decitabine and tacedinaline is shown as black curves.
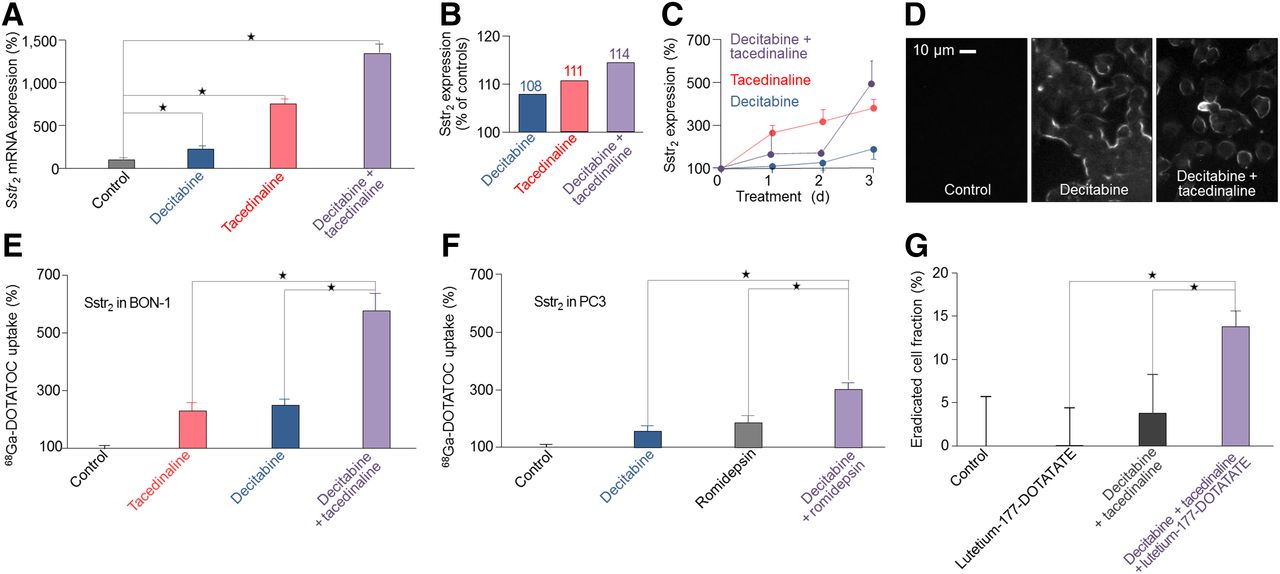
Combination treatment is superior to single-drug treatment. qRT-PCR and Western blot, ELISA, and immunocytochemistry with sstr2-specific antibodies were performed to assess effects of combination treatment on sstr2 messenger RNA and sstr2 expression compared with effects of single-drug treatment. Uptake assays were performed to assess changes in sstr2 expression for single-drug treatment and combination treatment. All results are expressed as changes in percentage and normalized to results in untreated cells. (A) qRT-PCR showing sstr2 messenger RNA expression in untreated BON-1 cells and BON-1 cells treated with decitabine (75 ng/mL), tacedinaline (500 ng/mL), or decitabine (75 ng/mL) plus tacedinaline (500 ng/mL). (B) Quantification of Western blot showing sstr2 expression in untreated BON-1 cells and BON-1 cells treated with decitabine (75 ng/mL), tacedinaline (500 ng/mL), or decitabine (75 ng/mL) plus tacedinaline (500 ng/mL) for 3 d. (C) ELISA showing sstr2 expression over time period of 1, 2, and 3 d in BON-1 treated with decitabine (75 ng/mL), tacedinaline (500 ng/mL), or decitabine (75 ng/mL) plus tacedinaline (500 ng/mL). (D) Immunocytochemistry showing sstr2 in untreated BON-1 cells and BON-1 cells treated with decitabine (75 ng/mL) alone and with decitabine (75 ng/mL) plus tacedinaline (500 ng/mL) over 3 d. sstr2-targeting experiments show 68Ga-DOTATOC uptake in BON-1 cells treated with decitabine, tacedinaline, and their combination (E) and in PC3 cells treated with decitabine, romidepsin, and their combination (F). (G) Treatment studies assessing eradication fraction in untreated BON-1 cells, BON-1 cells treated with 5 MBq/mL of 177Lu-DOTATATE, BON-1 cells treated with decitabine plus tacedinaline, and BON-1 cells treated with 177Lu-DOTATATE after sstr2 upregulation with decitabine plus tacedinaline. Statistical significance was tested using Student t test and is indicated (*P < 0.05).

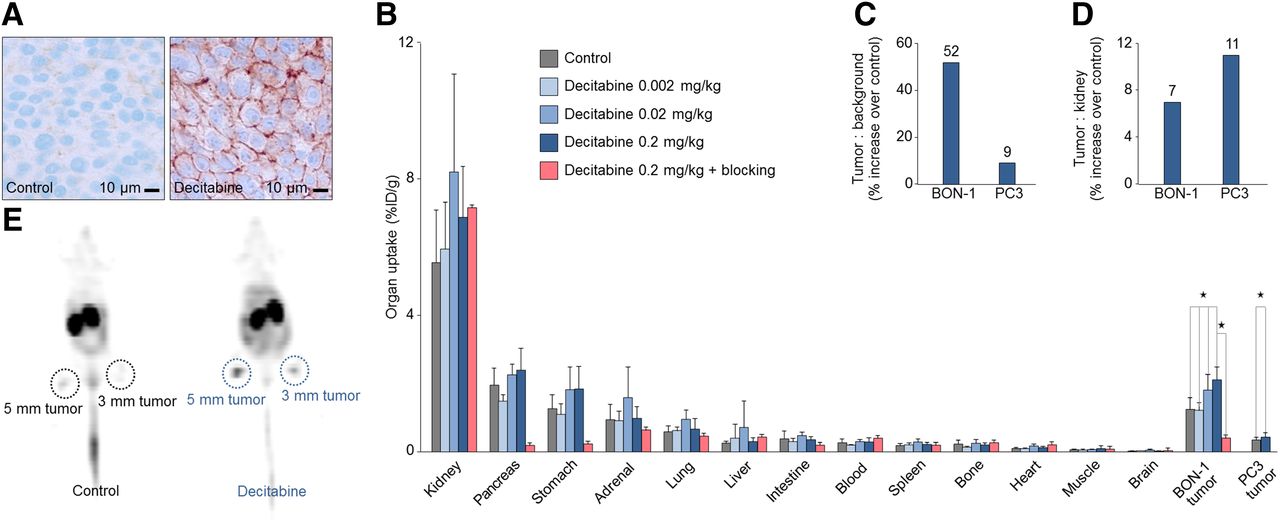
Target upregulation improves molecular imaging in vivo. In vivo experiments were performed in BON-1 and PC3 tumor–bearing nude mice. Mice received subcutaneous injections of decitabine over 9 d. (A) Immunohistochemistry with UMB-1 antisstr2 monoclonal antibody in BON-1 tumors with and without decitabine treatment (n = 4). (B) Biodistribution studies of 68Ga-DOTATOC uptake in all relevant organs, BON-1 tumors (n = 21), and PC3 tumors (n = 30). Dose-escalation studies were performed with 0.002, 0.02, and 0.2 mg/kg decitabine (n = 20). To evaluate specific sstr2 binding blocking experiments were performed by coapplication of excess of nonradiolabeled DOTATOC to compete with 68Ga-DOTATOC binding (n = 5). (C) Tumor-to-background ratios of 68Ga-DOTATOC uptake with and without decitabine treatment in BON-1 tumors, representing key factor for imaging contrast in sstr2-targeted imaging (n = 21). (D) Tumor-to-kidney ratios of 68Ga-DOTATOC uptake with and without decitabine treatment in BON-1 tumors, representing measure of therapeutic index in sstr2-targeted radiopeptide therapy (n = 30). (E) 68Ga-DOTATOC PET with and without decitabine treatment. Statistical significance was tested using Student t test and ANOVA and is indicated (*P < 0.05).
For these studies, we tracked and quantified the biodistribution and pharmacokinetics of radiolabeled somatostatin analogs and were thereby able to monitor changes in the expression of the relevant target, sstr2.
MATERIALS AND METHODS
All reagents and antibodies that we used are listed in the supplemental materials, together with a more detailed description of all experimental procedures (supplemental materials are available at http://jnm.snmjournals.org).
Cell Models
We used the human pancreatic neuroendocrine tumor cells BON-1, which express sstr2 at low to medium levels (11); the human prostate cancer cells PC3, which also express sstr2 at low to medium levels (11); the human pancreatic islet cell carcinoma cells QGP1 (12), which express sstr2 at low levels; and the rat pancreatic acinar cells AR42J, which express sstr2 at high levels (13). We cultured all cells at 37°C and 5% CO2 in Dulbecco modified Eagle medium or RPMI containing GlutaMAX (Thermo Fischer Scientific), 10% (v/v) fetal bovine serum, penicillin (100 U/mL), and streptomycin (100 μg/mL).
Radiolabeling
We used the short half-life positron-emitter 68Ga for biodistribution studies and PET imaging. We eluted 68Ga from a commercially available 68Ge/68Ga generator and purified it as previously described (14). We then incubated the 68Ga solution with DOTATOC at 0.1 mg/mL in Dulbecco phosphate-buffered saline at a ratio of 1:1 (v/v) for 10 min at 95°C. We determined the radiolabeling yield by radio–thin-layer chromatography and used only batches with radiochemical purities of greater than 97%.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
We performed real-time qRT-PCR to determine messenger RNA levels using the primer Hs00265624_s1 for human sstr2, as previously described (15).
Western Blotting
We cultured cells with or without the test drug, and prepared cell lysates. Then, we analyzed the amount of sstr2 protein in the lysates via Western blot, using the sstr2-specific antibody A01591 (Genscript #A01591) in combination with the secondary antibody goat antirabbit IRDye 680 and goat antimouse IRDye 800 coupled to infrared dyes. We quantified all Western blots and coomassie-stained gels using a LI-COR Odyssey scanner system.
Enzyme-Linked Immunosorbent Assay (ELISA)
We cultured cells with or without the test drugs and washed, fixed, and permeabilized them. Then, we assessed sstr2 expression using the primary sstr2-specific antibody UMB-1 and a secondary antibody goat antirabbit IgG horseradish peroxidase conjugate, by adding the horseradish peroxidase substrate and measuring the absorbance at 415 nm. We normalized the specific absorbance to the total amount of protein and set the normalized specific absorbance value of the untreated cells to 100%.
Immunofluorescence Microscopy
We cultured cells with or without the test drugs and washed, fixed, and permeabilized them. Then, we performed immunofluorescence microscopy as previously described (16), using the primary sstr2-specific antibody UMB-1 in combination with the secondary antibody Alexa Fluor 488 goat antirabbit IgG (H-L). Subsequently, we imaged the cells using a Nikon Eclipse TS 100 immunofluorescence microscope and a Nikon DS-Fi1 camera.
Immunohistochemistry
We processed BON-1 xenografts as conventional, 2-μm (sstr2)-thick paraffin wax sections using the sstr2-specific primary antibody UMB-1. We performed immunohistochemical staining using the compact polymer dextran-peroxidase complex method (EnVision), which yields a brown staining signal (17). In addition, we used hemalum for counterstaining of the samples.
Uptake Assay
We performed all uptake experiments as previously described (18). In brief, we cultured cells with or without the test drugs and incubated them with 68Ga-DOTATOC. Subsequently we lysed the cells and determined the amount of intracellular 68Ga using a γ-counter. We normalized the cell-associated radioactivity with the relative amount of protein reflecting the cell number and used untreated cells to measure the baseline uptake of radioactivity.
Drug Screening
To identify drugs that upregulate sstr2, we designed an in vitro screening based on cellular 68Ga-DOTATOC uptake as readout of sstr2 function (Fig. 1A). To increase the chance of clinically useful results, we included drugs that are already in clinical use and that have a potential to upregulate sstr2, for example, as they already demonstrated epigenetic modulation, especially of sstr2 gene expression. We screened all drugs at their therapeutic serum concentrations to facilitate potential clinical translation (Fig. 1B). We used BON-1 cells for screening and PC3 cells for validation.
In Vitro Treatment
We cultured cells with or without the test drugs and then incubated them with 177Lu-DOTATOC. After 2 h, we washed and trypsinized the cells, reseeded them at 50,000 cells per milliliter, and cultivated them for an additional 12 h. Then, we lysed the cells and determined the amount of protein reflecting the number of cells.
Animal Studies
We treated Nude-Foxn1nu mice carrying BON-1 or PC3 xenografts with decitabine or phosphate-buffered saline over a period of 9 d. Then, we injected 10 MBq of 68Ga-DOTATOC via the tail vain and performed PET/CT imaging or biodistribution studies 1 h later. For the latter, we collected all organs, measured the uptake of 68Ga-DOTATOC via a γ-counter, and computed all organ activity concentrations as percentage decay-corrected injected activity per gram of tissue. All animal experiments were conducted after approval by the local authorities and in accordance with the national regulations for animal treatment.
Statistical Analysis
We expressed all data as mean ± SD and compared results for 2 or more groups via the Student t test or ANOVA, respectively, using SPSS 21. We established all dose–effect curves using the Boltzmann method in Prism 5. We considered P values of less than 0.05 to indicate statistical significance.
RESULTS
The screen identified the inhibitor of DNA methyltransferase (DNMT) decitabine and the inhibitors of histone deacetylase (HDAC) tacedinaline and romidepsin as most efficacious for upregulating sstr2 in BON-1 cells (Fig. 1B; Supplemental Fig. 1).
Drugs Identified by Screen Improve Molecular Targeting
We tested the effects of these drugs on the transcription, translation, and function of sstr2 (Fig. 2). In line with the screening results, qRT-PCR analysis demonstrated that decitabine and tacedinaline increased sstr2 gene expression (Fig. 2A), Western blot confirmed upregulation of sstr2 protein expression (Fig. 2B), ELISA showed an increase in sstr2 expression over time (Fig. 2C), and immunocytochemistry confirmed membrane localization of upregulated sstr2 (Fig. 2D). We then evaluated the effects of decitabine on sstr2 function and cell toxicity in various cell models. Decitabine increased 68Ga-DOTATOC uptake within its therapeutic serum concentration in BON-1 and PC3 (Figs. 2E and 2F), whereas it had no significant effect on sstr2 function in QGP1 and AR42J cells (Supplemental Fig. 2).
We then assessed whether tacedinaline has effects similar to those of decitabine on sstr2 function and cell toxicity. In BON-1, tacedinaline induced a dose-dependent increase of 68Ga-DOTATOC uptake, albeit to lower levels than those achieved with decitabine, and without reaching a plateau within the therapeutic serum concentration (Fig. 2G).
In PC3, tacedinaline at therapeutic serum concentrations had no significant effect on 68Ga-DOTATOC uptake (Fig. 2H). Overall, tacedinaline showed toxicity similar to decitabine in BON-1 (Figs. 2E and 2G) and lower toxicity in PC3 (Figs. 2F and 2H). Decitabine and tacedinaline upregulated sstr2 with similar efficacy, whereas tacedinaline treatment in PC3 did not upregulate sstr2.
Combination Treatment Is Superior to Single-Drug Treatment
Because the drugs identified by the screen have different mechanisms of action, we evaluated potential synergistic effects to increase the efficacy of target upregulation (Fig. 3). qRT-PCR analysis revealed upregulation of sstr2 gene expression by decitabine plus tacedinaline (Fig. 3A), with stronger effects than those of either drug alone (Fig. 2A). Western blot confirmed upregulation of sstr2 protein (Fig. 3B); ELISA showed increased sstr2 expression over time (Fig. 3C), with stronger effects than those of either drug alone (Fig. 2C); and immunocytochemistry confirmed membrane localization of upregulated sstr2 (Fig. 3D).
Subsequently, we tested for the most efficacious combinations of drugs identified by the screen on the functional level and found that decitabine, tacedinaline, and romidepsin upregulation of sstr2 depended on the cell model. In BON-1, decitabine plus tacedinaline was most efficacious in upregulating sstr2, superior to either single drug (Fig. 3E). Conversely, in PC3, tacedinaline as a single drug or in combination treatments had no effects on sstr2 function. In these cells, the most efficacious combination was decitabine plus romidepsin (Fig. 3F). Next, we studied whether sstr2 upregulation with decitabine plus tacedinaline can improve sstr2-targeted therapy with 177Lu-DOTATATE. In BON-1 cells, 177Lu-DOTATATE had no detectable effects without pretreatment; however, we found significant effects after pretreatment with decitabine plus tacedinaline (Fig. 3G).
Target Upregulation Improves Molecular Imaging In Vivo
We then evaluated the observed effects in vivo. We used decitabine, which was the most efficacious drug in the in vitro experiments (Fig. 4). Immunohistochemistry of BON-1 tumors in athymic nude mice undergoing systemic decitabine treatment confirmed upregulation of sstr2 protein (Fig. 4A). On the functional level, decitabine significantly and dose-dependently increased 68Ga-DOTATOC uptake in both human cancer models, BON-1 and PC3 (Fig. 4B).
We confirmed sstr2-mediated effects in vivo by performing blocking experiments, which significantly reduced 68Ga-DOTATOC uptake in tumors and organs with high sstr2 expression, for example, the pancreas and stomach (Fig. 4B) (19). In contrast, the blocking did not decrease the kidney uptake. Importantly, in both tumor models, decitabine improved the overall biodistribution (Supplemental Fig. 5B), especially by increasing the tumor-to-background (Fig. 4C) and tumor-to-kidney ratio (Fig. 4D), thereby indicating its potential for improving tumor detectability and treatability.
Finally, we used receptor-targeted imaging with 68Ga-DOTATOC to visualize decitabine-based sstr2 upregulation in vivo. Decitabine increased tumor uptake and tumor-to-background ratio and thereby converted almost undetectable BON-1 tumors into detectable tumors (PET, Fig. 4E; PET/CT, Supplemental Fig. 5A).
DISCUSSION
Our screen identified the DNMT inhibitor decitabine and the HDAC inhibitors romidepsin and tacedinaline as relevant modulators of sstr2 expression. Tacedinaline recently underwent clinical phase 3 testing, whereas decitabine and romidepsin are already in clinical routine for treating myelodysplastic syndrome and T-cell lymphoma (20–22). All 3 drugs are known to modulate cellular transcriptional activity. Specifically, DNMT inhibitors enhance gene transcription by inducing DNA hypomethylation, whereas HDAC inhibitors enhance transcriptional activity by increasing histone acetylation (23,24).
Epigenetic modulation has a high clinical potential (25), yet little is known about the epigenetic control of sstr2. Previous studies suggested effects on a transcriptional level via DNMT and HDAC inhibition (26,27) and on a translational level via HDAC inhibition (28). In the present study, we confirmed these findings and expanded the principle to the functional level in vitro and in vivo. Thereby, we used BON-1 and PC3 cells, which represent a clinical scenario of low sstr2 expression and low 68Ga-DOTATOC uptake. In addition, we used AR42J cells, which had high sstr2 expression, and QGP1 cells, which had an undetectable sstr2 expression. BON-1, AR42J, and QGP1 are commonly used neuroendocrine tumor models, whereas PC3 is a commonly used prostate cancer model with expression of sstr2 (11).
The present study indicates that the potential for improving targeted approaches with DNMT and HDAC inhibitors might vary considerably among different tumors. DNMT inhibition upregulated sstr2 in BON-1 and in PC3, whereas HDAC inhibition upregulated sstr2 in BON-1 but not in PC3. However, DNMT and HDAC inhibition had no significant effects in AR42J and QGP1. Consequently, epigenetic upregulation of sstr2 might have the highest potential in tumors with a measurable but not already maximally amplified target expression.
Furthermore, epigenetic upregulation of drug targets might benefit from a combination of DNMT and HDAC inhibition. The combined use of DNMT and HDAC inhibitors already demonstrated antitumor effects superior to those of single drug treatment (20), and similarly, we found synergistic effects for their application as epigenetic modifiers. These synergistic effects largely depended on the tumor model and target molecule, which is in line with the clinical observation that cancer treatment with DNMT and HDAC inhibitors shows varying efficiency among tumor types (29). The present findings indicate that target upregulation might require tailoring to tumor type, tumor characteristics, and target molecule to optimize the efficacy of our approach.
The feasibility of our approach could be most efficiently tested with a low-dose regimen of a single drug, for example, decitabine, in patients with progressive metastasized neuroendocrine tumors that demonstrate low sstr2 expression and low 68Ga-DOTATATE uptake. Such a translational study should use 68Ga-DOTATATE PET before and after decitabine treatment to verify functional upregulation of sstr2 and thereby stratify patients to 177Lu-DOTATATE treatment after DNMT inhibition.
A clinical study would be feasible and safe because of the established use of decitabine and 68Ga-DOTATATE. Furthermore, clinical translation would be promising due to the improvements of tumor-to-background and tumor-to-kidney ratios, which are key elements for successful targeted imaging and treatment and which significantly exceed the improvements that the most frequently used sstr2 radioligand DOTATATE provided over its predecessors (18). Successful clinical translation of epigenetic target upregulation might provide patients with neuroendocrine tumors with low sstr2 expression, who have failed all currently available treatments, with new therapeutic options.
Finally, successful translation has a potential impact beyond sstr2 targeting, because reversible DNA hypermethylation has been described for promoters of clinically relevant target structures other than sstr2 (27), including the sodium-iodine symporter in differentiated thyroid cancer (30), CD-20 in lymphoma (31), and human epidermal growth factor receptor 2 in breast cancer (32). Thus, drug-based epigenetic modulation is a promising strategy to facilitate molecularly targeted diagnostics and therapies for a wide range of oncologic patients.
CONCLUSION
We present an approach that uses epigenetic modifiers to improve sstr2 targeting in vitro and in vivo. The presented method is easily transferrable into the clinic and might provide patients who have failed various treatment regimens with new therapeutic options.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. Our work was supported by the OPO-Foundation, Braun-Foundation, Gebauer-Foundation, and Stiftung zur Krebsbekämpfung. Moreover, Martin A. Walter (31003A_156731) and Aurel Perren (310030_144236) were supported by the Swiss National Science Foundation. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Ramtin Khalafi for technical assistance, Andrew Q. Tran for medical illustrating assistance, and Duncan Marriott and Caroline Spencer (Rx Communications, Mold, U.K.) for medical writing assistance, which was funded by the University Hospital Bern. Finally, we thank Nadia Sanchez, Rebecca Dumont, and Johannes Czernin for the valuable discussions and helpful comments on the manuscript.
Footnotes
↵* Contributed equally to this work.
Published online Jun. 30, 2016.
- © 2016 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication August 4, 2015.
- Accepted for publication April 22, 2016.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Study protocol of LANTana: a phase Ib study to investigate epigenetic modification of somatostatin receptor-2 with ASTX727 to improve therapeutic outcome with [177Lu]Lu-DOTA-TATE in patients with metastatic neuroendocrine tumours, UK
- 225Ac-MACROPATATE: A Novel {alpha}-Particle Peptide Receptor Radionuclide Therapy for Neuroendocrine Tumors
- Heterogeneity of SSTR2 Expression Assessed by 68Ga-DOTATOC PET/CT Using Coefficient of Variation in Patients with Neuroendocrine Tumors
- Upregulation of Somatostatin Receptor Type 2 in a Receptor-Deficient In Vivo Pancreatic Neuroendocrine Tumor Model Improves Tumor Response to Targeted 177Lu-DOTATATE
- 177Lu-DOTA-EB-TATE, a Radiolabeled Analogue of Somatostatin Receptor Type 2, for the Imaging and Treatment of Thyroid Cancer
- Combination Strategies to Improve Targeted Radionuclide Therapy
- Combination of 5-Fluorouracil with Epigenetic Modifiers Induces Radiosensitization, Somatostatin Receptor 2 Expression, and Radioligand Binding in Neuroendocrine Tumor Cells In Vitro
- Establishment of the First Well-differentiated Human Pancreatic Neuroendocrine Tumor Model
- Genetic and epigenetic drivers of neuroendocrine tumours (NET)