


Abstract
Bradykinin B1 receptor (B1R) is a G-protein–coupled receptor that is overexpressed in a variety of cancers. B1R is not expressed in healthy tissues, making it an attractive cancer imaging marker. Previously, we reported selective uptake of 68Ga-P03034 (68Ga-DOTA-dPEG2-Lys-Arg-Pro-Hyp-Gly-Cha-Ser-Pro-Leu) in B1R-positive (B1R+) HEK293T::hB1R tumor xenografts in mice. In this study, we compare 68Ga-P03034 with 68Ga-labeled P04158 (68Ga-DOTA-dPEG2-Lys-Lys-Arg-Pro-Hyp-Gly-Igl-Ser-D-Igl-Oic) and Z02090 (68Ga-DOTA-dPEG2-Lys-Lys-Arg-Pro-Hyp-Gly-Cpg-Ser-D-Tic-Cpg) derived from 2 potent B1R antagonists, B9858 and B9958, respectively, for imaging B1R expression with PET. Methods: Peptide sequences were assembled on solid-phase. Cold standards were prepared by incubating DOTA-conjugated peptides with GaCl3. Binding affinity was measured via competition binding assays using hB1R-expressing Chinese hamster ovary-K1 cell membranes. 68Ga labeling was performed in N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) buffer with microwave heating and purified by high-performance liquid chromatography. Imaging/biodistribution studies were performed in mice bearing wild-type HEK293T (B1R−) and B1R+ HEK293T::hB1R tumors. Results: P03034, P04158, and Z02090 bound B1R with high affinity, with Ki values at 16.0 ± 2.9, 1.5 ± 1.9, and 1.1 ± 0.8 nM, respectively. 68Ga-labeled P03034, P04159, and Z02090 were obtained in greater than 50% decay-corrected radiochemical yields with more than 99% radiochemical purity. Biodistribution studies showed that all three 68Ga-labeled tracers cleared rapidly from the blood and normal tissues, with excretion mainly via the renal pathway. At 1 h after injection, only the kidneys, bladders, and B1R+ HEK293T::hB1R tumors were clearly visualized in PET images. Uptake values of 68Ga-labeled P03034, P04158, and Z02090 in B1R+ tumors were 2.17 ± 0.49, 19.6 ± 4.50, and 14.4 ± 1.63 percentage injected dose per gram, respectively. Uptake ratios of B1R+ to B1R− tumor, blood, and muscle were 6.23 ± 1.69, 5.72 ± 2.20, and 25.5 ± 13.1 for 68Ga-P03034; 34.5 ± 10.5, 19.2 ± 8.21, and 66.1 ± 17.0 for 68Ga-P04158; and 29.3 ± 9.68, 29.9 ± 5.58, and 124 ± 28.1 for 68Ga-Z02090, respectively. Conclusion: All three 68Ga-labeled B1R-targeting peptides generated specific and high-contrasted images of B1R+ tumors xenografted in mice. With significantly higher tumor uptake and target-to-nontarget ratios, 68Ga-labeled P04158 and Z02090 are superior to P03034 for imaging B1R expression with PET.
Bradykinin (BK) B1 and B2 receptors (B1R and B2R, respectively) are G-protein–coupled receptors and have long been known to function in pain and inflammatory pathways (1,2). The peptides BK and kallidin (Lys-BK) are produced by enzymatic cleavage of kininogens and act as endogenous agonists for the constitutively expressed and widely distributed B2R (3). Removal of the C-terminal Arg from BK and kallidin by carboxypeptidases generates [des-Arg9]BK and [des-Arg10]kallidin, respectively, which are the natural agonists for the inducible B1R (3). BK and related peptides have been shown to induce angiogenesis (4), cell proliferation (5–9), migration, and metastasis (10–12) through activation of B1R and B2R. Although B1R is not constitutively expressed in most tissues, its expression has been shown to be upregulated in various cancers.
By examining human breast tumors, Molina et al. (13) found that B1R was highly expressed in ductal carcinoma in situ (4/4) and invasive ductal carcinoma (11/13). Using immunohistochemical staining on 16 prostate cancer biopsies, Taub et al. (14) reported that B1R was detected in all prostatic intraepithelial neoplasia and malignant lesions but not in benign prostate tissues. By looking at different subtypes, Chee et al. (15) reported that B1R was expressed in most of the lung cancers (adenocarcinoma, 6/6; squamous cell carcinoma, 5/6; large cell carcinoma, 5/6; small cell carcinoma, 6/6; carcinoid tumor, 4/6). Recently Toledo et al. (16) reported that B1R was highly expressed in gallbladder cancer (intestinal metaplasia, 24/24; carcinoma in situ, 10/10; intestinal adenoma, 6/6; pyloric adenoma, 10/15) but not in normal gallbladder epithelial cells. The expression of B1R has also been found in chondrosarcomas (17), colorectal adenomas (18), melanomas (19), clear-cell renal carcinomas (20), esophageal squamous cell carcinomas (21), and astrocytic tumors (22). Because B1R is not expressed in most tissues under normal conditions, B1R serves as an excellent imaging marker for cancer diagnosis.
Previously we reported the synthesis of 68Ga-P03083 (23), a B1R-targeting tracer derived from the potent antagonist [Leu9,des-Arg10]kallidin (24). Despite its high binding affinity to B1R (Ki = 2.6 nM, Table 1), 68Ga-P03083 was ineffective at visualizing tumor xenografts derived from B1R-transduced HEK293T::hB1R cells because of the in vivo instability of the native kallidin sequence. Subsequently, we replaced Pro4 and Phe6 with hydroxyproline (Hyp) and cyclohexylalanine (Cha), respectively, and the resulting 68Ga-P03034 (23) greatly enhanced visualization of B1R expression in vivo despite reduced binding affinity (Ki = 16 nM, Table 1). To further improve imaging contrasts, we prepared 2 novel B1R-targeting tracers, 68Ga-P04158 and 68Ga-Z02090 (Table 1; Fig. 1), that were derived from potent B1R antagonists B9858 (24) and B9958 (25), respectively. Herein, we present the synthesis and characterization of these B1R-targeting tracers and comparative studies of 68Ga-P03034, 68Ga-P04158, and 68Ga-Z02090 for in vivo imaging of B1R expression with PET.
Amino Acid Sequences and B1R Binding Affinities of BK and Related Peptides

Chemical structures of P03034, P04158, and Z02090. Unnatural amino acids in receptor-binding domain are shown in brown.
MATERIALS AND METHODS
Information for reagents, instruments, peptide synthesis, in vitro B1R binding assays, radiochemistry, LogD7.4 (D: distribution coefficient) measurement, and plasma stability studies are provided in the supplemental materials (available at http://jnm.snmjournals.org).
Biodistribution and PET/CT Imaging Studies
Male immunocompromised nonobese diabetic/severe combined immunodeficient IL2RKO mice were obtained from an in-house breeding colony at the Animal Resource Centre of the BC Cancer Research Centre. Mice were maintained and the experiments were conducted in accordance with the guidelines established by the Canadian Council on Animal Care and approved by the Animal Ethics Committee of the University of British Columbia. Wild-type HEK293T and HEK293T::hB1R tumors were inoculated by subcutaneous injection of 1 × 106 cells on each dorsal flank of the mice. Thus, each mouse had a positive (B1R+) and negative (B1R−) tumor. After 10–14 d, palpable tumors measuring approximately 7 mm in diameter were obtained. Mice were injected with approximately 3.7 MBq of 68Ga-labeled pep-tides. For blocking experiments, the radiotracer was coinjected with 100 μg of nonradioactive standard. After 60 min of uptake, the mice were anesthetized by isoflurane inhalation, followed by euthanization via CO2 inhalation. Blood was promptly withdrawn, and the organs/tissues of interest were harvested and weighed. The radioactivity of the collected mouse tissues was counted and expressed as the percentage injected dose per gram of tissue (%ID/g).
Mice bearing 2 tumors, HEK293T and HEK293T::hB1R as described above, were also used for imaging experiments. The mice were briefly sedated with 2% isoflurane inhalation for intravenous injection of the radiotracer with or without the presence of 100 μg of nonradioactive standard. After injection, the mice were allowed to recover and roam freely in their cages for 55 min. At that point, the mice were sedated with 2% isoflurane inhalation and placed in the scanner. A baseline CT scan was obtained for localization and attenuation correction. The mice were kept warm by a heating pad during acquisition. A single static emission scan was acquired for 10 min. Afterward, the mice were euthanized and the organs harvested for biodistribution.
Data Analysis
All statistics were performed using Excel software (Microsoft). P values were calculated using a Student t test, and values less than 0.05 were considered statistically significant. The unpaired 1-tailed test was used to compare B1R+ tumor uptake and B1R+ tumor-to-background ratios between the control and blocked mice, whereas a paired 1-tailed test was used to compare the uptake in B1R+ and B1R− tumors in the same mice. The comparison of uptake in B1R− tumors and other tissues between control and blocked mice was performed using an unpaired, 2-tailed test.
RESULTS
DOTA-dPEG2-B9858 and P04158 were prepared in 21% and 99% yields, respectively, whereas DOTA-dPEG2-B9958 and Z02090 were obtained in 13% and 89% yields, respectively. The identities of these 4 peptides were confirmed by mass spectrometry analysis. The radiolabeling data are summarized in Table 2. 68Ga-labeled P03034, P04158, and Z02090 were obtained with average decay-corrected radiochemical yields ranging from 67% to 76% and with a greater than 99% radiochemical purity. The average specific activities were 222 ± 37, 89 ± 22, and 100 ± 33 GBq/μmol for 68Ga-labeled P03034, P04158, and Z02090, respectively. These three 68Ga-labeled tracers are highly hydrophilic, with an average LogD7.4 in the range between −2.76 and −2.50.
Radiolabeling, LogD7.4, and Plasma Stability Data of 68Ga-Labeled [Des-Arg10]Kallidin Derivatives
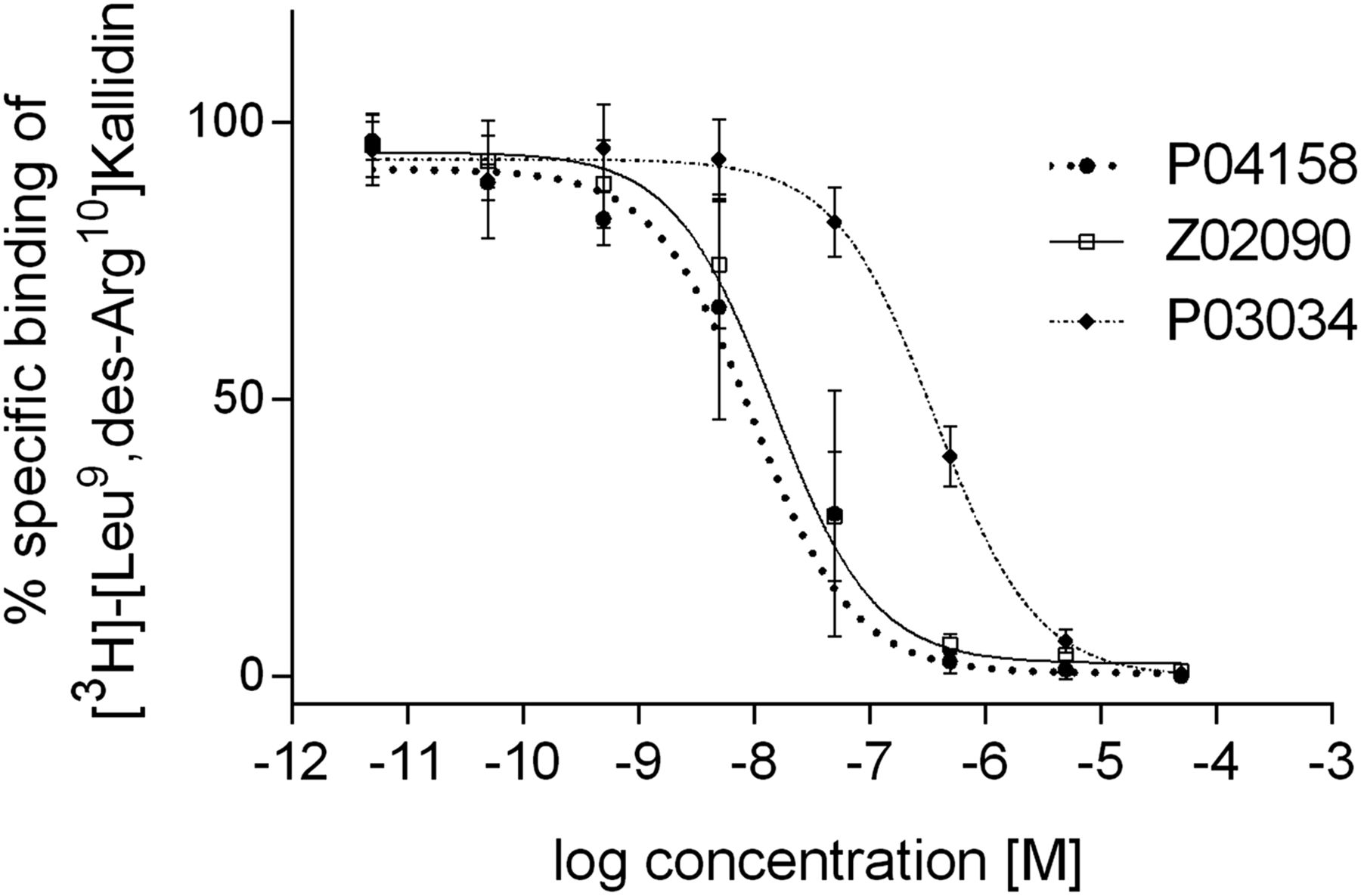
P03034, P04158, and Z02090 inhibited the binding of [3H]-[Leu9,des-Arg10]kallidin to hB1R-expressing Chinese hamster ovary-K1 cell membranes in a dose-dependent manner (Fig. 2), and their corresponding Ki values were 16.0 ± 2.9, 1.5 ± 1.9, and 1.1 ± 0.8 nM (Table 1), respectively. 68Ga-labeled P03034 and P04158 were stable in mouse plasma, with more than 90% of the tracers remaining intact after 1-h incubation at 37°C (Table 2). Under the same conditions, only 81% of 68Ga-Z02090 remained intact.
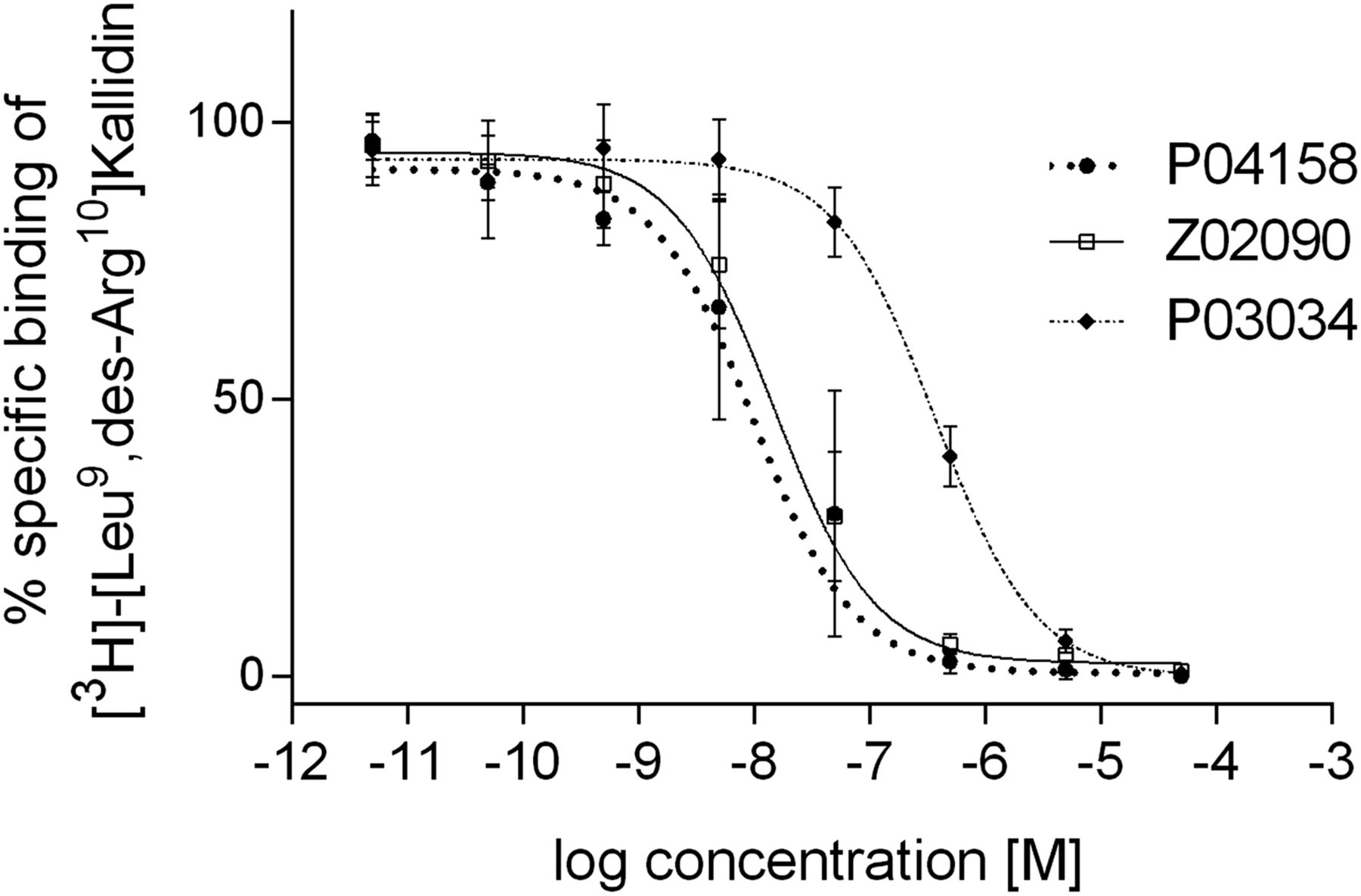
Representative displacement curves of [3H]-[Leu9,des-Arg10]Kallidin by P03034, P04158, and Z02090.
The 1-h biodistribution data are summarized in Table 3. All tracers were excreted mainly through the renal pathway because low uptake was observed in liver and intestines. The background level was lowest when 68Ga-P03034 was used, followed by 68Ga-Z02090, whereas 68Ga-P04158 exhibited the highest background level. The uptake in B1R− tumors was similar to or less than that in blood for all tracers. Higher uptake values were observed in the kidneys and B1R+ tumors: 4.50 ± 2.17 and 2.17 ± 0.49 %ID/g for 68Ga-P03034; 69.2 ± 7.39 and 19.6 ± 4.50 %ID/g for 68Ga-P04158; and 50.1 ± 9.68 and 14.4 ± 1.63 %ID/g for 68Ga-Z02090, respectively. Coinjection with 100 μg of cold standard significantly reduced the uptake of 68Ga-P03034, 68Ga-P04158, and 68Ga-PZ0290 in B1R+ tumors to 0.41 ± 0.08, 1.44 ± 0.12, and 0.85 ± 0.17 %ID/g, respectively, but showed no effect on the uptake of other organs/tissues.
Biodistribution Data of 68Ga-Labeled [Des-Arg10]Kallidin Derivatives at 1 Hour After Injection in Tumor-Bearing Mice
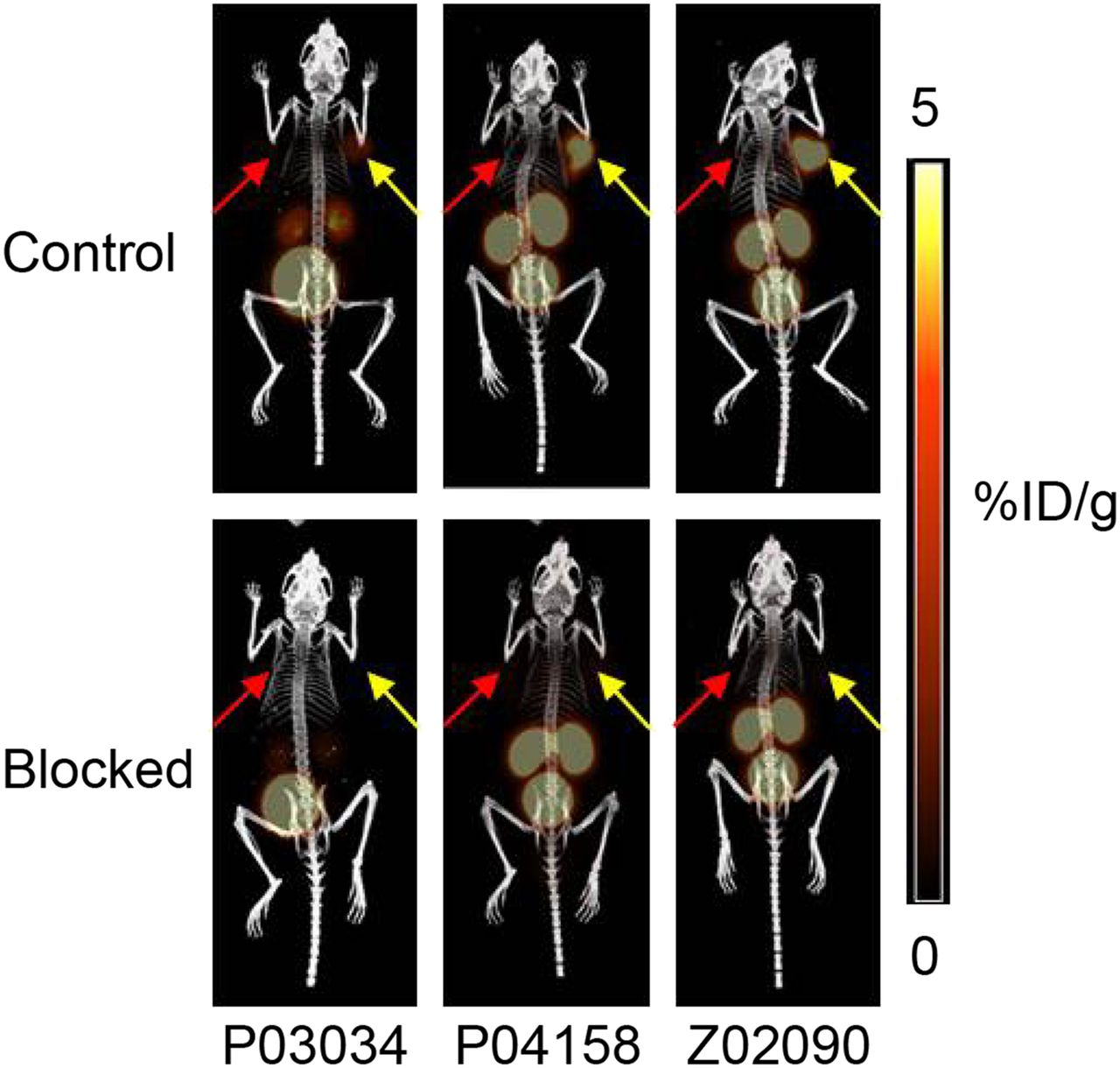
PET/CT images (Fig. 3) obtained at 1 h after injection were consistent with the biodistribution data, with low background uptake, and only B1R+ tumors, kidneys, and bladders were visualized in the images. Much higher uptake in the kidneys and B1R+ tumors was observed when 68Ga-P04158 and 68Ga-Z02090 were used, and the coinjection of cold standard effectively blocked the uptake of all three 68Ga-labeled tracers in B1R+ tumors.
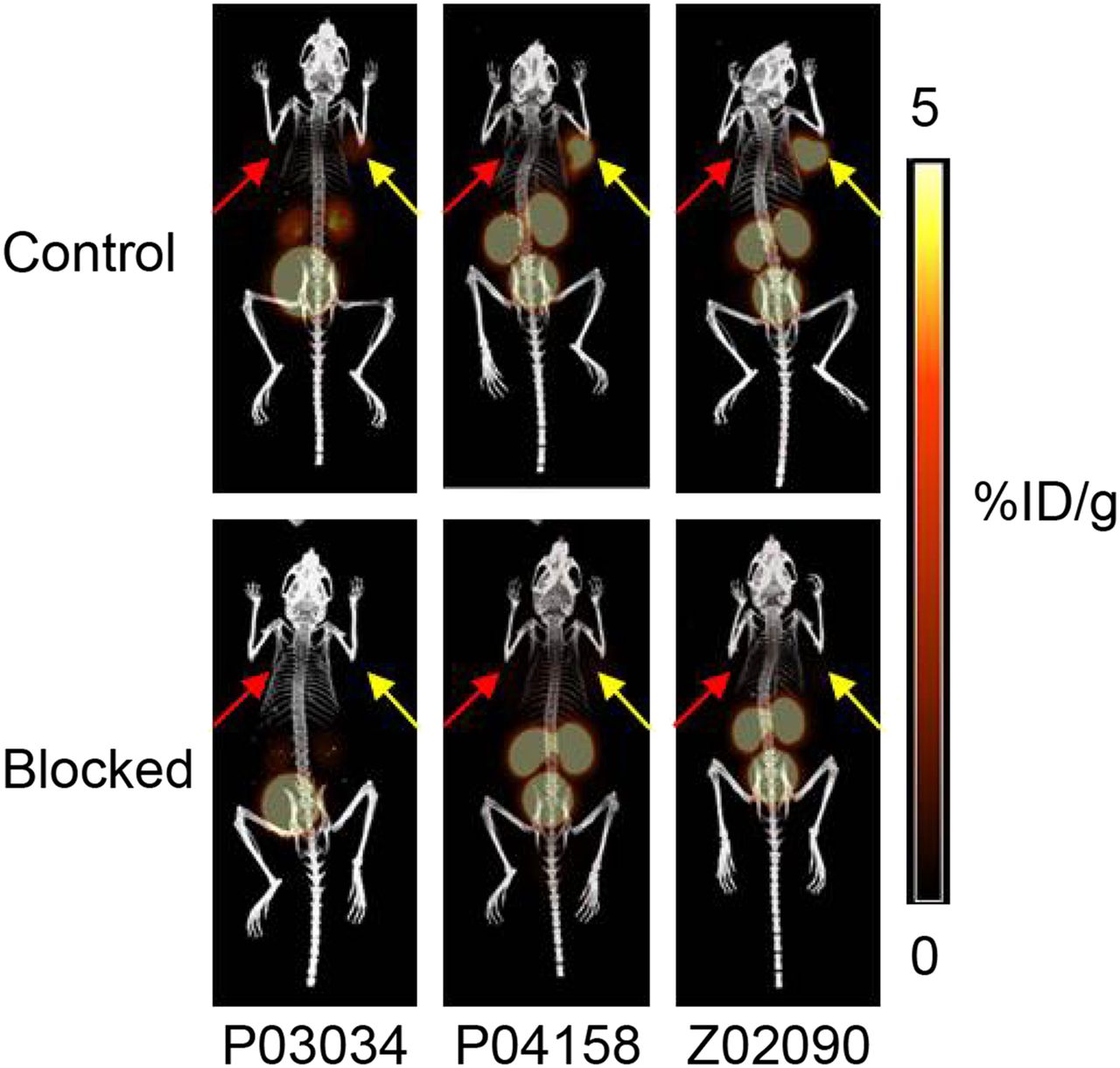
PET/CT images of 68Ga-labeled P03034, P04158, and Z02090 in mice bearing both B1R+ (yellow arrows) and B1R− (red arrows) tumors without (upper) and with (lower) coinjection of cold standard (100 μg).
DISCUSSION
Modifications of B1R agonists and antagonists at the N terminus have been shown to retain their binding affinity to B1R. This was previously demonstrated by B1R-targeting probes for receptor-binding assays and flow cytometry (26–28) as well as by B1R antagonists that are resistant to the cleavage by aminopeptidase N (25). On the basis of this finding, we initiated the development of PET probes for in vivo imaging of B1R expression. Our first tracer, 68Ga-P03083 (23), was derived from the potent B1R antagonist [Leu9,des-Arg10]kallidin (29) with the addition of 68Ga-DOTA-Ahx (Ahx: 6-aminohexanoic acid) to the N terminus. As expected, P03083 retained high binding affinity (Ki = 2.6 ± 0.7 nM) to B1R. However, biodistribution studies showed that uptake of 68Ga-P03083 in B1R+ HEK293T::hB1R tumors was suboptimal (0.79 ± 0.22 %ID/g) due to the fast in vivo degradation of 68Ga-P03083 by peptidases such as angiotensin-converting enzyme (24). Enhanced uptake (2.77 ± 1.22 %ID/g) in HEK293T::hB1R tumors was observed (23) by coinjecting 68Ga-P03083 with a peptidase inhibitor, phosphoramidon (30). Similar enhancement in tumor uptake was also observed using a more metabolically stable tracer, 68Ga-P03034, which has Pro4 and Phe6 substituted with unnatural amino acids Hyp and Cha, respectively (Table 1). Because the in vivo stability of B1R-targeting probes is essential for achieving higher tumor uptake, we started exploiting other reported metabolically stable B1R-targeting peptides that could be potentially modified for use as imaging agents.
B1R antagonists B9858 and B9958 (Table 1) were selected because of their reported metabolic stability and high binding affinity to B1R (24,25). Each peptide sequence contains 4 unnatural amino acids: Hyp4, Igl6 (2-indanylglycine), D-Igl8, and Oic9 (octahydroindolecarboxylic acid) for B9858; Hyp4, Cpg6 (cyclopentylglycine), D-Tic8 (tetrahydroisoquinoline-3-carboxylic acid), and Cpg9 for B9958 (Fig. 1). To perform a side-by-side comparison with 68Ga-P03034, 2 novel B1R-targeting probes, 68Ga-P04158 and 68Ga-Z02090, were prepared with the same 68Ga-DOTA complex and dPEG2 linker conjugated to the N terminus of B9858 and B9958, respectively. As expected, P04158 and Z02090 retained high binding affinity to B1R (Ki = 1.1–1.5 nM), further confirming that modifications at the N terminus of B1R-targeting peptides are tolerable.
In this study, we chose DOTA for 68Ga labeling as it is currently the most widely used chelator for the preparation of 68Ga-labeled tracers for PET imaging. 68Ga labeling was performed in N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) buffer via microwave heating with 100 μg of DOTA-dPEG2-B9858 or 40 μg of DOTA-dPEG2-B9958. Despite excellent radiolabeling yields (>90%), high-performance liquid chromatography purification was performed to achieve higher specific activity of 68Ga-P04158 and 68Ga-Z02090. Reducing the amount of precursors for radiolabeling resulted in suboptimal yields (data not shown). Recently, triazacyclononane-phosphinate (TRAP) chelators were shown to be superior to traditional chelators, DOTA and NOTA (1,4,7-triazacyclononane-triacetic acid), for 68Ga labeling (31). As demonstrated by Notni et al. (31), specific activity values at approximately 5,000 GBq/μmol were routinely obtained using 1 GBq of 68Ga for the preparation of 68Ga-TRAP(RGD)3. Replacing the DOTA chelator of P04158 and Z02090 with a TRAP chelator could potentially generate new B1R-targeting probes that could be efficiently radiolabeled with 68Ga in high specific activity without tedious high-performance liquid chromatography purification.
The higher LogD7.4 value of 68Ga-labeled P04158 and Z02090 indicates that these 2 tracers are less hydrophilic than 68Ga-P03034. This reduced hydrophilicity is mainly due to the substitutions of Pro8 and Leu9 in P03034 with more lipophilic unnatural amino acids: D-Igl8 and Oic9 for P04158; D-Tic8 and Cpg9 for Z02090 (Fig. 1). The CLogP (calculated LogP; P: partition coefficient) values for Pro and Leu are −0.060 and 0.799, respectively, whereas the CLogP values for D-Igl, Oic, D-Tic, and Cpg are 1.361, 1.593, 1.302, and 0.871, respectively (data were obtained from Scifinder, a chemistry database provided by the American Chemical Society, and calculated using Advanced Chemistry Development software, version 11.02 [ACD/Labs] (32)).
Biodistribution (Table 3) and PET/CT images (Fig. 3) showed that all 3 tracers had fast renal excretion and minimal uptake in the liver, which are preferable features for cancer imaging agents to detect primary or metastatic lesions within the abdomen. Minimal brain uptake of these tracers indicated their inability to cross the blood–brain barrier. The uptake in blood and nontarget tissues or organs at 1 h after injection is positively correlated with their lipophilicity. 68Ga-P03034 (LogD7.4 = −2.76 ± 0.01) showed the lowest background uptake, whereas 68Ga-P04158 (LogD7.4 = −2.50 ± 0.16) had the highest background uptake. The higher kidney uptake with 68Ga-P04158 and 68Ga-Z02090 (69.2 ± 7.39 and 50.1 ± 9.68 %ID/g, respectively) than 68Ga-P03034 (4.50 ± 2.17 %ID/g) is believed to result from the additional lysine (Lys) added to the N terminus of P04158 and Z02090. This lysine contributes an additional +1 to the overall charge of the tracers (Table 2) and could potentially promote their reabsorption and retention in the kidneys (33). For potential clinical translation, it may be possible to reduce the kidney reabsorption or retention by incorporating negative-charged amino acids to the peptide sequences of B9858 and B9958 to balance the overall charge.
The uptake values in B1R+ tumors were 19.6 ± 4.50 and 14.4 ± 1.63 %ID/g for 68Ga-P04158 and 68Ga-Z02090, respectively, which were much higher than 2.17 ± 0.49 %ID/g obtained using 68Ga-P03034. We hypothesized that the significant increase in tumor uptake for 68Ga-P04158 and 68Ga-Z02090 was in part due to the improved in vivo stability (incorporation of more unnatural amino acids) and bioavailability. The in vitro plasma stability data (Table 2) showed that 68Ga-P04158 had a plasma stability similar to that of 68Ga-P03034, whereas 68Ga-Z02090 had slightly poorer plasma stability. Although commonly used to estimate in vivo metabolic stability of radiotracers, in vitro plasma stability assays might not be suitable for radiopeptides that are metabolized mainly by tissue peptidases rather than soluble circulating peptidases. It is known that BK and related peptides are metabolized mainly by angiotensin-converting enzyme, which is localized principally in the endothelium of the pulmonary vasculature (24). Further in vivo metabolite study is needed to verify whether 68Ga-P04158 and 68Ga-Z02090 indeed have higher in vivo stability than 68Ga-P03034.
The higher uptake of 68Ga-P04158 and 68Ga-Z02090 in B1R+ tumors might also be attributed to the improved binding affinities, which are approximately 10-fold better than that of 68Ga-P03034. To verify that these three 68Ga-labeled tracers could specifically target B1R in vivo, we performed baseline and blocking biodistribution and imaging studies in mice bearing both B1R+ HEK293T::hB1R tumors and B1R− tumors derived from the parent HEK293T cells. In the baseline biodistribution and imaging studies, significantly higher uptake in B1R+ tumors than B1R− tumors was observed (Table 3; Figs. 3 and 4). Furthermore, a coinjection of 100 μg of cold standard reduced approximately 80% uptake of 68Ga-P03034, and greater than 90% of 68Ga-P04158 and 68Ga-Z02090 in B1R+ tumors confirmed the specific uptake of these 3 tracers in B1R+ tumors. Interestingly, significantly higher uptake in B1R+ than B1R− tumors was observed for all tracers even in the blocked mice, as shown in Figure 4. The coinjection of 100 μg of nonradioactive standard presumably blocked most of B1R, and only small amounts of B1R in B1R+ tumors remained unbound and were available for targeting. The ability to differentiate B1R+ and B1R− tumors even in the blocked mice indicates that these tracers are highly sensitive for the in vivo detection of B1R expression.

Difference on uptake of 68Ga-labeled B1R-targeting tracers between B1R+ and B1R− tumors in mice in control (A) and blocked (B) groups. *P < 0.05. ***P < 0.001.
Besides cancers, the overexpression of B1R has also been found in other pathophysiologic conditions including infection (34–36) and atherosclerosis (37–39). 68Ga-P04158 and 68Ga-Z02090, which generated excellent B1R+ tumor-to-background contrasts, could be potentially used for imaging infection foci and atherosclerotic plaques as well.
CONCLUSION
We have synthesized and evaluated potent B1R-targeting peptides, 68Ga-P04158 and 68Ga-Z02090, and compared them with the previously reported 68Ga-P03034. All three 68Ga tracers generated specific and high-contrast images of B1R+ tumor xenografts in mice. With higher tumor uptake and target-to-nontarget ratios, 68Ga-labeled P04158 and Z02090 are superior to P03034 for in vivo imaging B1R expression with PET.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This work was supported by the Canadian Institutes of Health Research (MOP-126121) and the BC Leading Edge Endowment Fund. A provisional patent application was filed for some of the material presented in this article. No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Mar. 5, 2015.
- © 2015 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication November 25, 2014.
- Accepted for publication February 2, 2015.





{kind=link}
{kind=link}
{kind=link}
{kind=link}