


Abstract
Radiopeptides are powerful tools for diagnostic imaging and radionuclide therapy of various diseases. Since the introduction of the first radiopeptide into the clinical setting to diagnose neuroendocrine tumors about 25 y ago, many advances have been made in the field. This short review highlights novel strategies to improve the application of radiopeptides for imaging and therapy.
Radiopeptides targeting somatostatin receptor (SSTR) have contributed significantly to patient care. The SSTR agonist octreotide and modifications thereof ([Tyr3]octreotide [TOC], [Tyr3, Thr8]octreotide [TATE], and [1-NaI3]octreotide [NOC]) are widely used in routine clinical practice for diagnosis of neuroendocrine tumors (NETs). 111In-diethylenetriaminepentaacetic acid–octreotide (OctreoScan; Mallinckrodt Inc.) was the first registered radiopeptide, and it has become the gold standard for diagnosis and staging of SSTR-positive tumors using SPECT and SPECT/CT. After the successful application of 111In-octreotide for imaging of NETs in the clinic, somatostatin analogs radiolabeled with β-emitting isotopes (i.e., 177Lu-DOTATATE and 90Y-DOTATOC) were introduced as treatment. Peptide receptor radionuclide therapy (PRRT) using 177Lu-DOTATATE and 90Y-DOTATOC has given impressive results thus far.
The success of radiolabeled somatostatin analogs inspired the scientific community to develop radiopeptides targeting different receptor families, including gastrin-releasing peptide (GRP), cholecystokinin-2/gastrin (CCK-2/CCK-B), glucagonlike peptide-1 (GLP-1), ανβ3-integrin, neurokinin-1 (NK-1), melanocyte-stimulating hormone (MSH), neuropeptide Y (NPY), luteinizing hormone–releasing hormone (LHRH), and chemokine receptor-4 (CXCR4) (1). Examples of radiopeptide families and their applications for tumor imaging and therapy are shown in Figure 1. Radiopeptides have established themselves as important agents for imaging, but also for PRRT. For medical imaging, peptides can be radiolabeled with radionuclides such as 99mTc, 111In, 67Ga, or 123I for SPECT and 18F, 68Ga, 64Cu, 86Y, or 124I for PET. For therapeutic purposes, peptides can be labeled with β-emitters such as 177Lu, 90Y, or 67Cu and α-emitters such as 213Bi or 225Ac. Current applications of clinical radiopeptides for imaging and therapy have been described in detail by Ambrosini et al. (2). Approaches intending to improve the performance of radiopeptides for imaging and therapy and thereby widen their scope of application are discussed below.
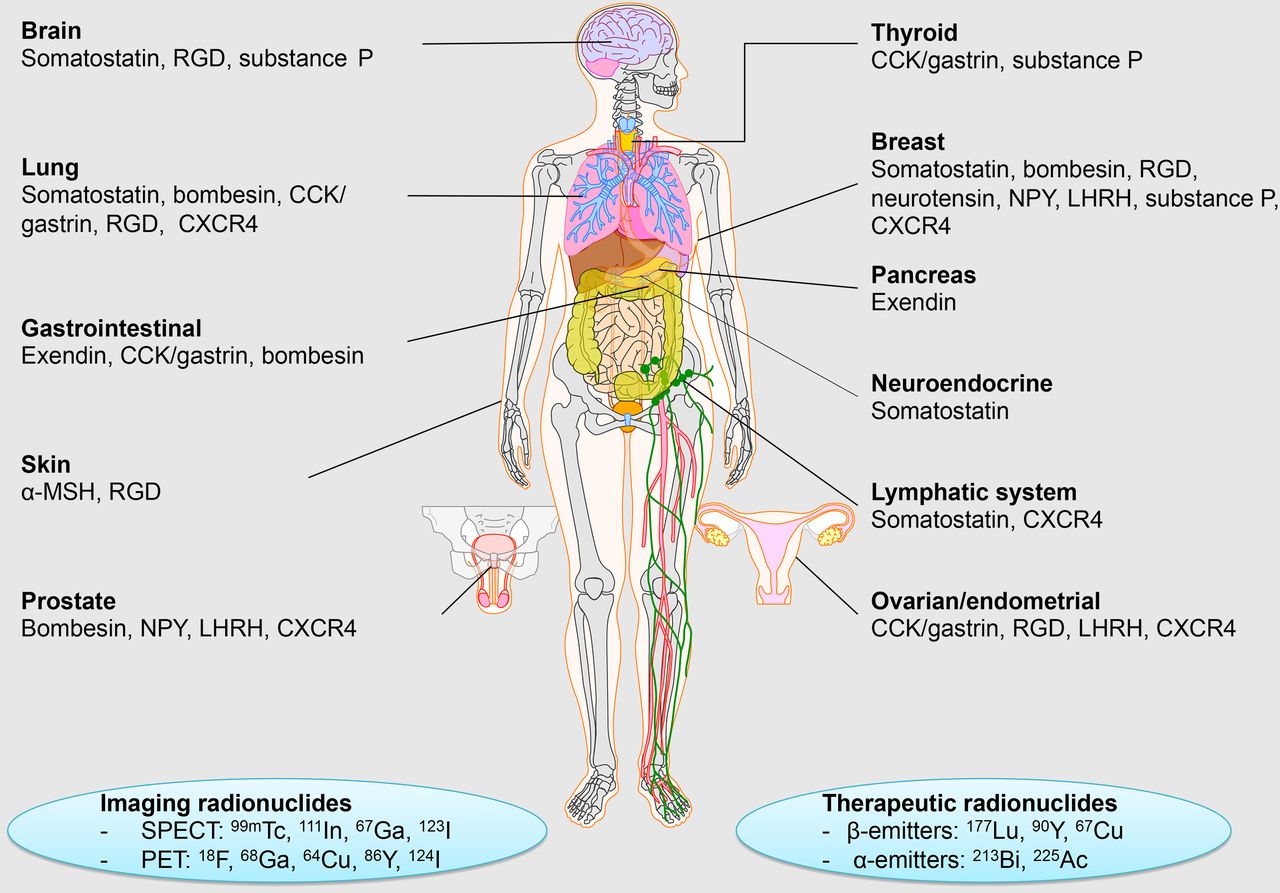
Examples of radiopeptide families, radionuclides, and their applications for imaging of tumors. RGD = arginylglycylaspartic acid.
NOVEL PERSPECTIVES FOR IMAGING
Intensive research efforts have improved the pharmacokinetics and in vivo stability of radiopeptides, as well as boosting image quality by identifying radionuclides with more suitable nuclear physical properties or by combining nuclear imaging modalities with complementary modalities. Some examples of novel approaches for radionuclide therapy are shown in Figure 2.

(A) PET/CT (top) of insulinoma patient showing focal 68Ga-DOTA-exendin-4 uptake in distal pancreatic tail (consistent with surgically removed insulinoma) that was not shown by SPECT/CT (bottom) with 111In-DOTA-exendin-4. (B) SPECT/CT images of mice with GRPR-expressing PC-3 tumors (arrows) 4 h after injection of bombesin analog 111In-DOTA-PEG2-dTyr-Gln-Trp-Ala-Val-βAla-His-Phe-Nle-NH2 without (left) or with (right) coinjection of neutral endopeptidase inhibitor phosphoramidon (300 μg).
Change of Paradigm Using Receptor Antagonists
An important eye-opener in the field of radiopeptides has been the recent introduction of SSTR antagonists, which showed more favorable pharmacokinetics and better tumor visualization than agonists did, despite their poor internalization rate. In a clinical study, the SSTR antagonist 111In-DOTA-BASS (111In-DOTA-pNO2-Phe-c(dCys-Tyr-dTrp-Lys-Thr-Cys)dTyrNH2) resulted in higher tumor uptake and better visualization of metastatic NETs than did the SSTR agonist 111In-diethylenetriaminepentaacetic acid-octreotide (3).
Subsequently, radiolabeled GRPR-targeted antagonists also entered the arena, showing a more favorable pharmacokinetic profile and fewer side effects than agonists. GRPR-targeted antagonists have shown high potential in early clinical trials for prostate cancer imaging (4).
Improving Pharmacokinetics and In Vivo Stability
Many efforts have been made to increase the in vivo stability of radiopeptides by introducing structural modifications, although such modifications can cause undesired changes in pharmacokinetics or impaired receptor affinity. A remarkable advance in this area is the discovery of the in vivo enzyme inhibition concept, in which radiopeptides are administered together with an enzyme inhibitor as a safeguard against enzymatic degradation. It has recently been shown that coinjection of a neutral endopeptidase inhibitor, such as phosphoramidon, can stabilize radiolabeled bombesin, minigastrin, and somatostatin analogs in vivo, leading to enhanced tumor uptake and improved tumor visualization (5). We believe that this strategy will have a significant impact on patient care, as it may enhance the diagnostic sensitivity and therapeutic efficacy of radiopeptides. For that purpose, a neutral endopeptidase inhibitor already applied in patients (e.g., thiorphan) is the preferred choice.
68Ga Versus 18F
Recently, imaging quality has been boosted by selecting radionuclides with more suitable nuclear physical properties. For SPECT imaging, 99mTc is the radioisotope of choice. However, recent developments in PET/CT and PET/MR technologies are steering the field toward positron emitters.
68Ga-radiopeptides are useful tools for PET/CT or PET/MRI of diseases, as 68Ga can be obtained in-house from a 68Ge/68Ga generator. The introduction of 68Ga-labeled DOTA-conjugated somatostatin analogs (TOC, TATE, NOC) into the clinic, enabling PET/CT imaging of NETs, is an important advance in somatostatin-based imaging. The superior performance of imaging techniques using these tracers for detection of NET lesions was described by Ambrosini et al. (2).
Recently, 68Ga-labeled exendin-4 has been introduced in the clinic for the detection of GLP-1 receptor–positive insulinomas (6). 68Ga-DOTA-exendin-4 PET/CT is preferred over 111In-DOTA-exendin-4 SPECT/CT because of the higher spatial resolution and better quantification options obtained with PET/CT cameras.
18F-labeled peptides have some advantages over 68Ga-labeled peptides, such as a longer half-life. Moreover, the physical properties of 18F are more favorable for PET imaging than those of 68Ga. Indeed, an 18F-labeled GRPR antagonist, BAY 864367 (3-cyano-4-18F-fluorobenzoyl-Ala(SO3H)-Ala(SO3H)-Ava-Gln-Trp-Ala-Val-NMeGly-His-Sta-Leu-NH2), has recently been implemented in the clinical setting (4). A drawback yet to be overcome with 18F-labeled peptides is their higher lipophilicity, which gives rise to less favorable pharmacokinetics. An interesting approach consists of labeling peptides with Al18F via radiometalation chemistry. Several peptides have been labeled with Al18F, such as octreotide, E[c(RGDyK)]2, exendin-4, the bombesin analog NOTA-8-Aoc-BBN(7–14)NH2, and the GRPR antagonist JMV4168 (DOTA-βAla-βAla-JMV594) (7,8). The development of a kit-based method for labeling peptides with 18F would make application of 18F-labeled peptides possible in a wider range of hospitals.
Nuclear/Optical Multimodality Imaging
Nuclear imaging technologies available in the clinic (SPECT/CT, PET/CT, and PET/MR) are superior to any other clinical imaging modality in terms of specificity and sensitivity. However, optical imaging offers interesting applications as well and could complement the nuclear medicine uses (9). An important development in the field of radiopeptides has been the introduction of hybrid derivatives, containing both a fluorescent and a radioactive label, as these have significant implications in the field of imaging-guided surgery. Several hybrid radiopeptides have been developed, including agents targeting SSTR, GRPR, interleukin-11 receptor-α, ανβ3-integrin, matrix metalloproteinase, CXCR4, and GLP-1R (10–12). Because of the size and lipophilicity of common fluorescent dyes, the main challenge lies in optimizing the chemical structures of hybrid tracers to preserve receptor affinity and biodistribution patterns while maximizing the range of tissue penetration.
NOVEL PERSPECTIVES FOR THERAPY
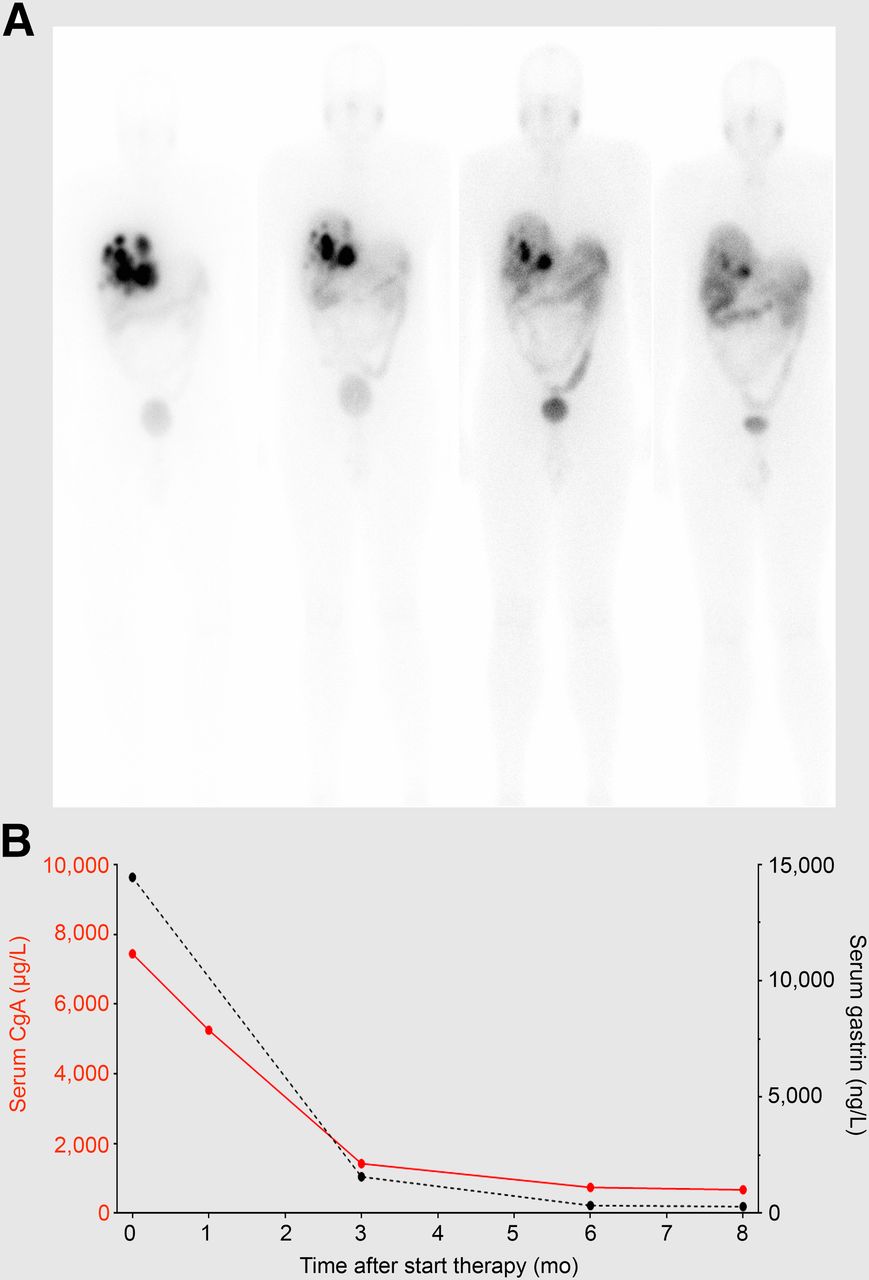
Novel strategies might improve the outcome of PRRT. These include the improvement of pharmacokinetics and in vivo stability, the combination of radionuclides, the combination of PRRT with chemotherapy, the use of α-emitting radionuclides, and the use of novel methods to predict therapy response. Recent developments to increase the therapeutic effects of PRRT have been described in detail by Bison et al. (13). Some examples of novel approaches for radionuclide therapy are shown in Figure 3.

(A) Total-body images after each cycle of PRRT with 7.4 GBq of 177Lu-octreotate in combination with low-dose capecitabine as chemosensitizer in patient with liver metastases of gastrinoma. Decreasing uptake in liver metastases indicates and preludes tumor response, which was later confirmed with CT. (B) Course of tumor markers chromogranin-A (CgA) and gastrin.
Improving Pharmacokinetics and In Vivo Stability
The success of PRRT would presumably be improved by enhancing the dose delivered to the tumor or limiting the dose delivered to normal healthy tissues. The use of SSTR antagonists has enhanced tumor targeting and prolonged tumor retention compared with SSTR agonists, resulting in an enhanced tumor radiation dose during PRRT. In a clinical study, the SSTR antagonist 177Lu-DOTA-JR11 exhibited higher tumor uptake and a longer intratumoral residence time than the SSTR agonist 177Lu-DOTATATE (14). This study was performed on only a few patients, and more systematic clinical studies are needed now to fully evaluate the role of 177Lu-DOTA-JR11 for therapy of NETs.
Patients with advanced prostate cancer could also benefit from PRRT, as treatment options for this patient group are limited. 177Lu-GRPR antagonists have not been tested in clinical studies thus far, even though they have shown promising results in preclinical studies (15). Radionuclide therapy using PSMA-targeted tracers has been more widely explored, as PSMA expression has been validated in advanced prostate cancer in several reports. Extensive comparative studies showing target expression of PSMA and GRPR in advanced prostate cancer would guide the choice of therapeutic options for those patients.
Combination Therapies
In the search for increased therapeutic efficacy of PRRT, therapeutic radionuclides with different physical characteristics have been combined to target a wider range of tumor lesions. 90Y and 177Lu β-emitting isotopes suitable for treatment of large and small tumors have been combined in simultaneous or sequential treatment. A recent retrospective study (16) described the benefits of the combination of 90Y-DOTATOC and 177Lu-DOTATOC over treatment with either radiopeptide alone, with increased survival after the combination treatment. However, patient selection bias cannot be excluded; therefore, prospective, randomized trials are needed before firm conclusions can be drawn.
PRRT can also be combined with other therapies to increase tumor response. Chemotherapeutics can sensitize tumor cells to PRRT by modulating DNA damage and repair mechanisms, apoptosis, cell proliferation, cell-cycle synchronization, and tumor cell oxygenation. However, careful design of such combination studies is needed, as chemotherapeutics can also alter receptor expression and tumor vascularization. For instance, the DNA alkylating drug temozolomide increased tumor perfusion in a preclinical study (13). Clinical studies combining PRRT using 177Lu-DOTATATE with several chemotherapeutics have been reported. Capecitabine and temozolomide both increased the response rate of 177Lu-DOTATATE in comparison with 177Lu-DOTATATE alone (17). A recent proof-of-concept study described the combination of PRRT with everolimus (18). It was shown that everolimus can safely be combined with 177Lu-octreotate to treat low-grade NETs.
Introduction of Powerful α-Emitters
An important development in the field of radionuclide therapy has been the introduction of α-particle–emitting radionuclides. A first-in-human study showed that 213Bi-DOTATOC can eradicate neuroendocrine liver metastases pretreated with 90Y/177Lu-DOTATOC. Only moderate acute and midterm hematologic and renal toxicity was observed at effective therapeutic doses (19). Nonoperable and critically located gliomas have been treated with α-targeted therapy, by local injection of 213Bi-DOTA-substance P, providing local irradiation of the tumor. The short tissue range of 213Bi prevents damage to adjacent brain areas. Up till now, this therapy has proven to be feasible and safe, with only mild adverse effects observed (20). Despite the promising preliminary results obtained with 213Bi-DOTATOC and 213Bi-substance P in patients, the short half-life of 213Bi (45.6 min) and the costs and limited availability of 225Ac/213Bi generators limit clinical use. The recently described accelerator-driven production of 225Ac may allow it to be produced in quantities sufficient to supply centers for clinical trials and make the treatment available to a wider range of centers (21).
Innovative Methods as Predictors of Therapeutic Response
There is a clear need for personalization and standardization of PRRT to optimize efficacy and minimize long-term toxicity, as emphasized in a recent review of Bodei et al. (22). They recommended collecting data on several parameters to construct a patient-specific treatment plan, taking into account various patient-specific characteristics to predict the probability of response and predisposition to toxicity. Dosimetry can be used as a predictor of therapy response and toxicity; a relationship between tumor-absorbed dose and response has already been demonstrated for 90Y-DOTATOC (23).
Markers of DNA damage and repair, such as phosphorylated variant H2A histone family member X (γ-H2AX), may predict tumor and normal-organ radiosensitivities. A recent paper described the use of a γ-H2AX-foci assay as a predictor of normal-tissue toxicity after PRRT (24). Genetic components can also predict disease stage and response to therapy (25).
CONCLUSION
Derived from natural peptide ligands, radiopeptides can be tailored and optimized to provide personalized treatment for several types of diseases. In recent years, several methods have been developed to optimize radiopeptides for imaging and therapy; these efforts and achievements may bridge the gap between the exciting promises of radiopeptides and their implementation in the clinic. Today, complex multimodal ligands are being designed for theranostic applications, whereas simple radiopeptides combine all the key ingredients for effective theranostic applications, including easy manufacturing, fast clearance, low immunogenicity, and efficient targeting. We believe that the recent advances described in this article may lead the way to a radiant future for radiopeptides in the field of theranostics.
DISCLOSURE
Marion de Jong and Dik Kwekkeboom own stock in Advanced Accelerator Applications (AAA). No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Oct. 29, 2015.
- © 2015 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication August 7, 2015.
- Accepted for publication October 26, 2015.





{kind=link}
{kind=link}
{kind=link}